Roles of Intramolecular and Intermolecular Hydrogen Bonding in a Three-Water-Assisted Mechanism of Succinimide Formation from Aspartic Acid Residues

Abstract
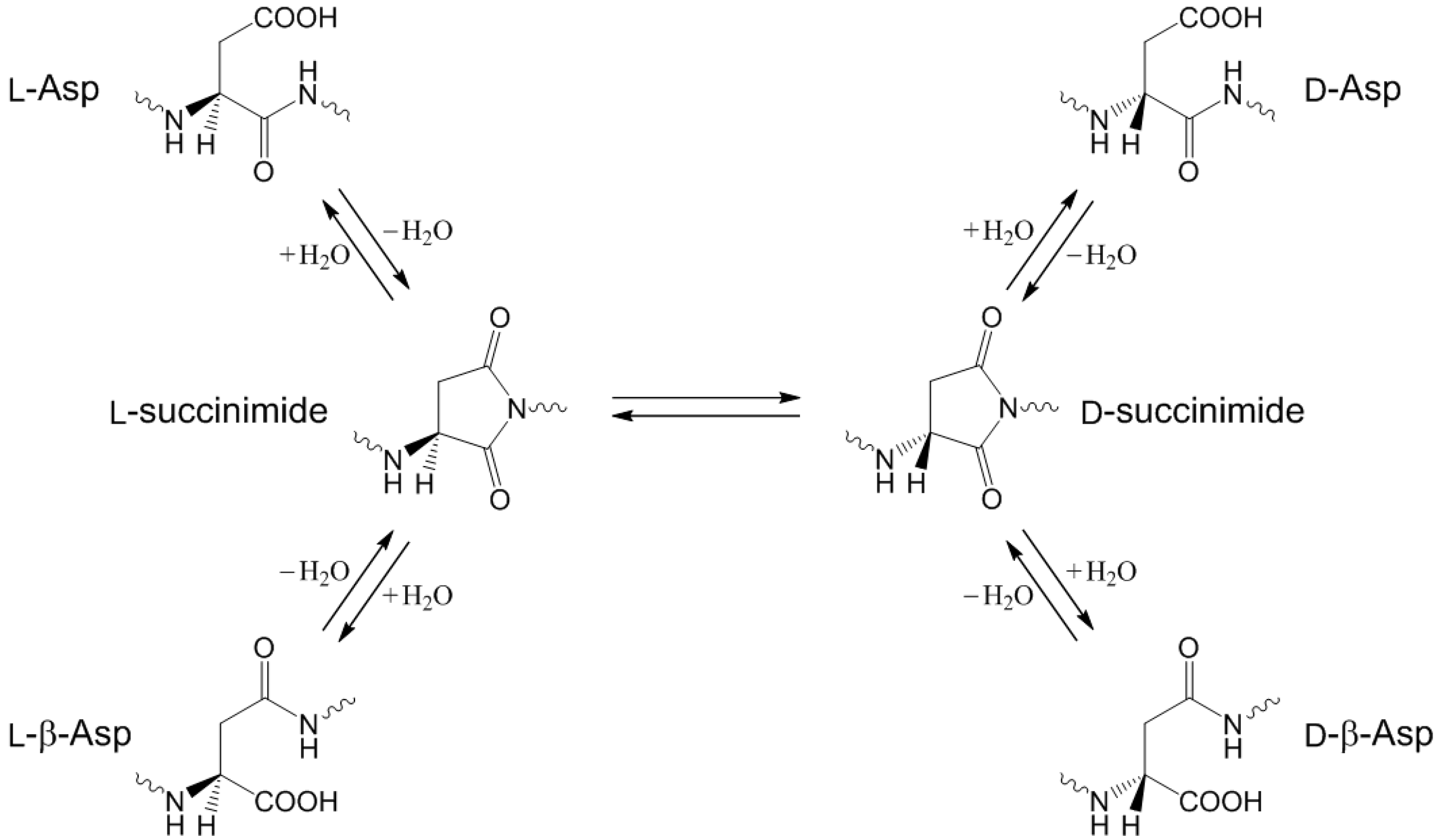
:1. Introduction
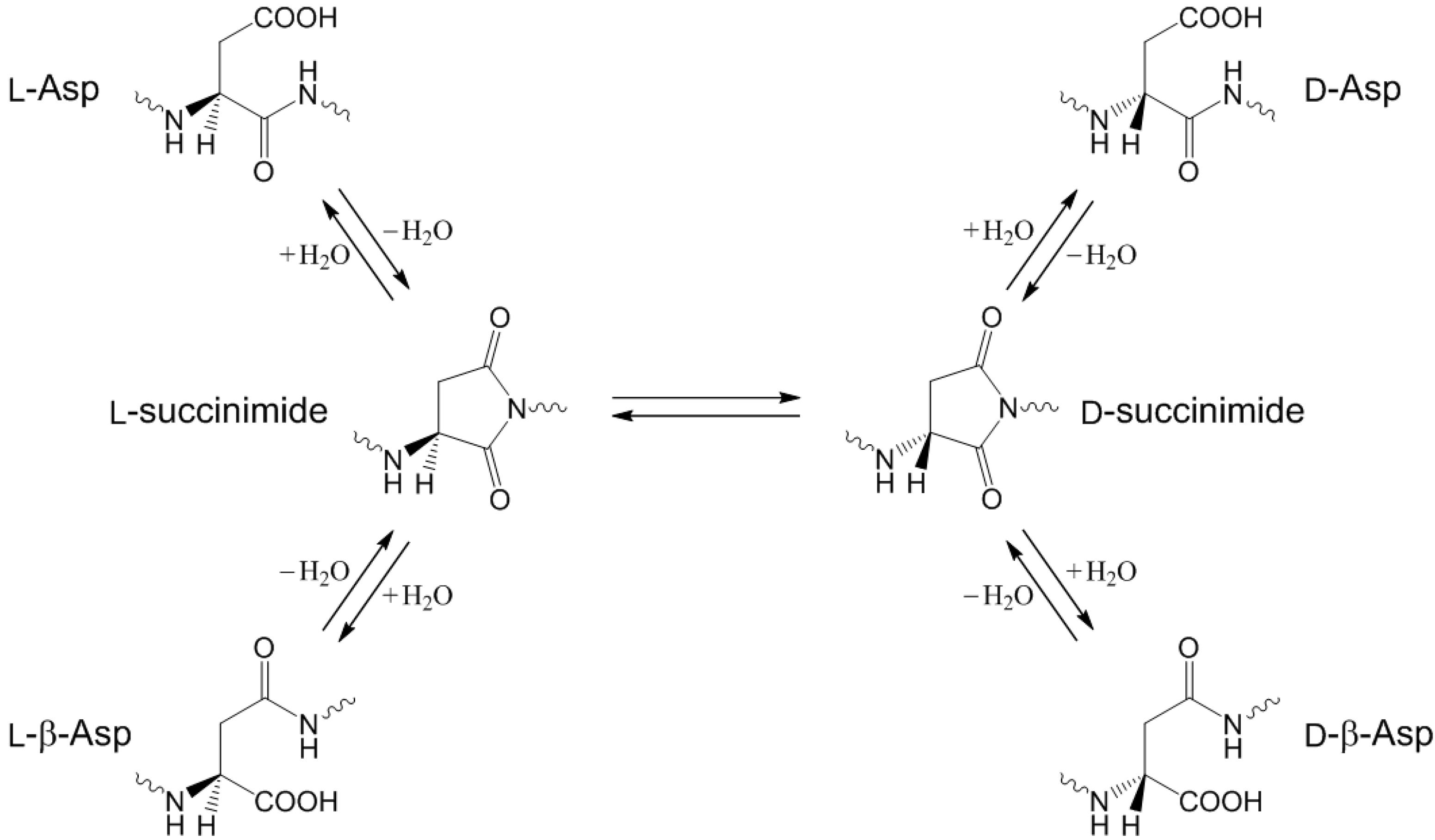
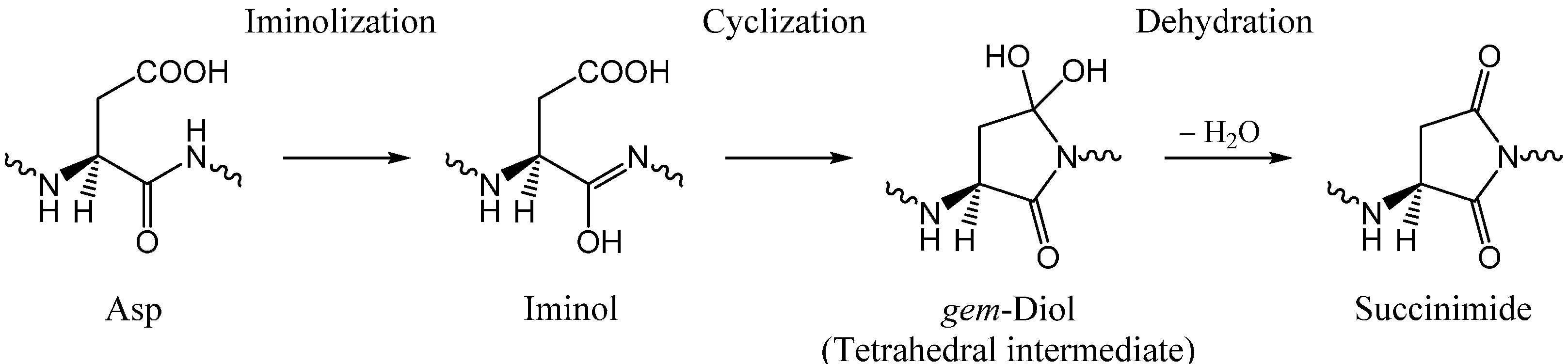
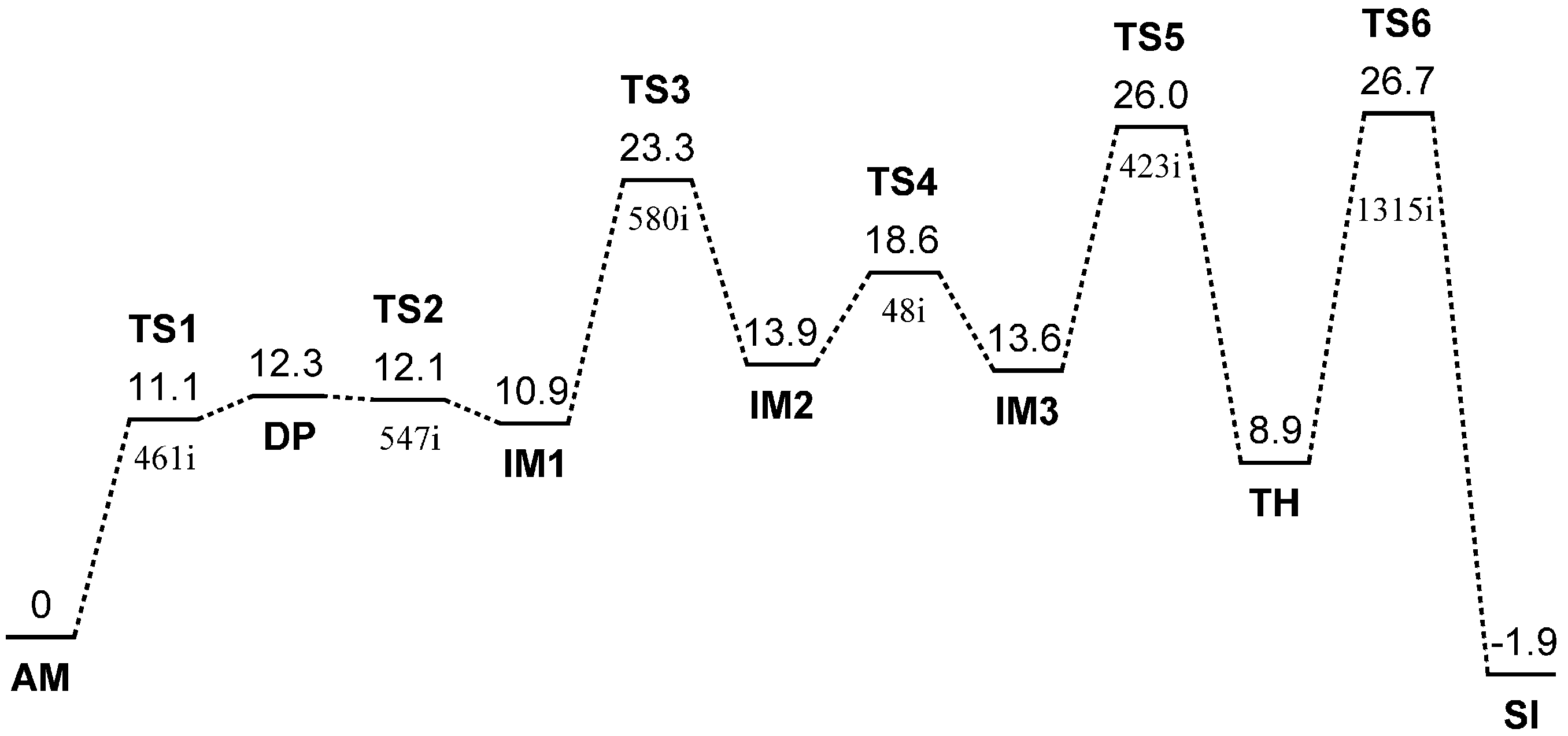
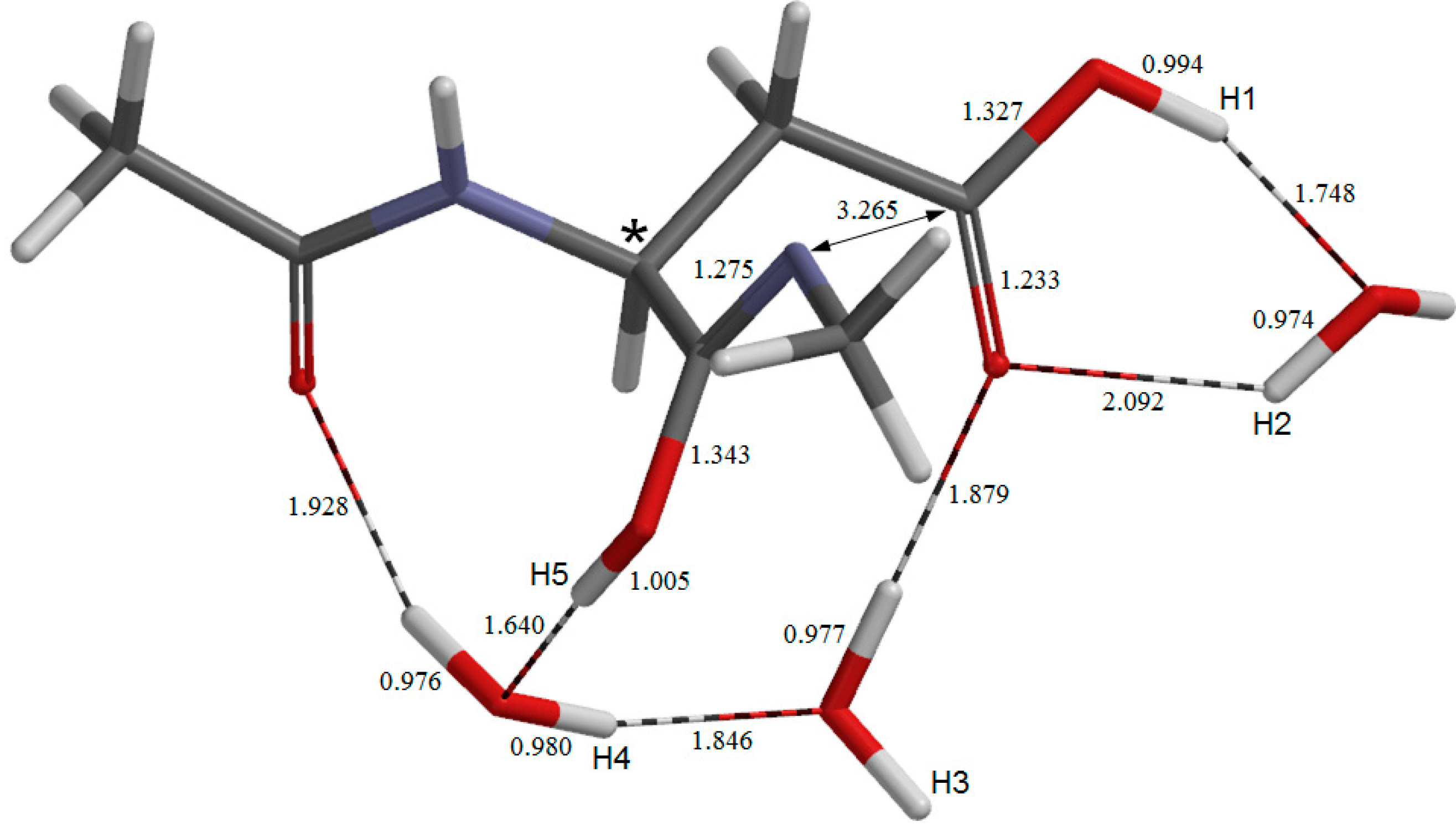
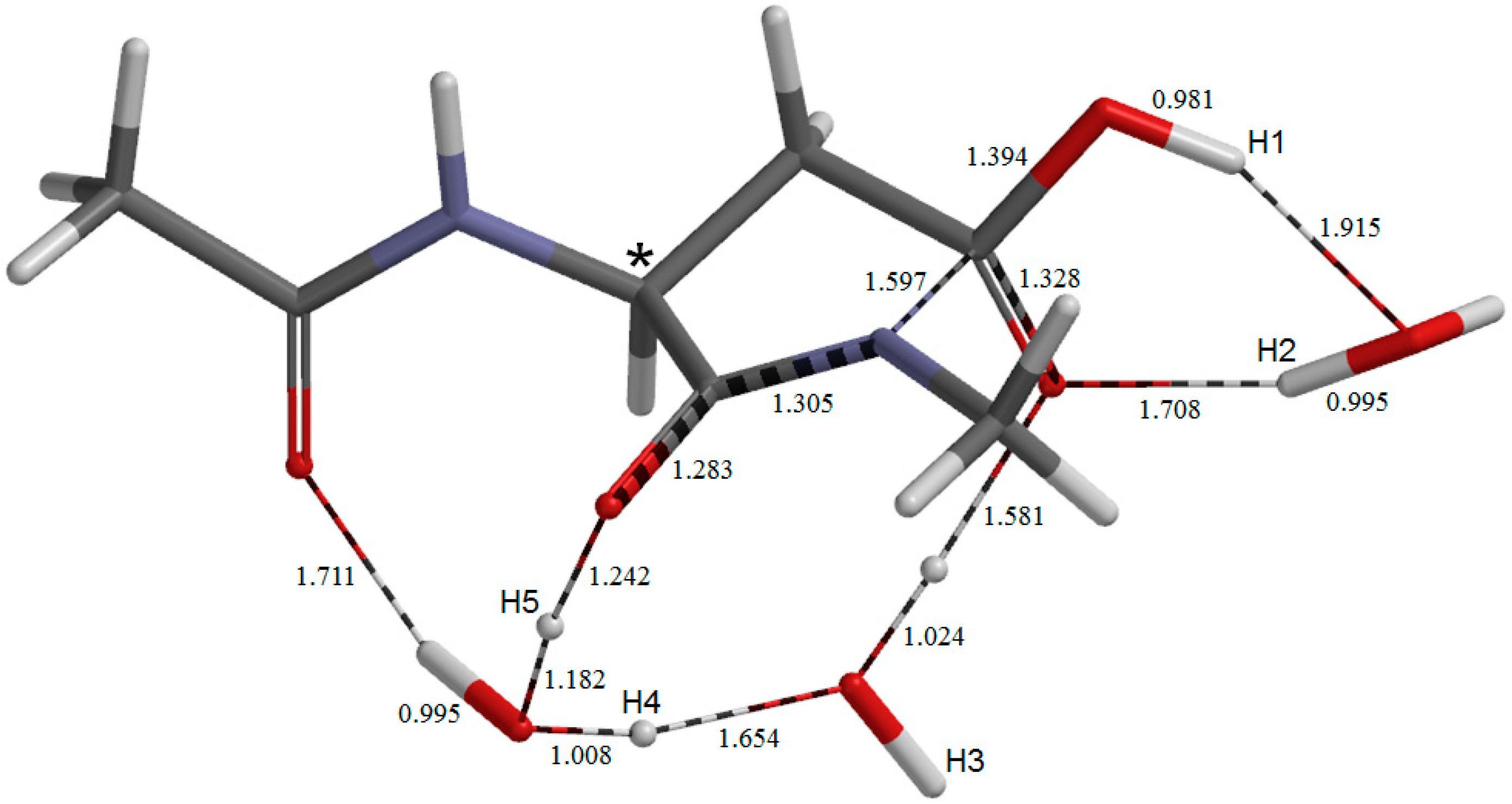
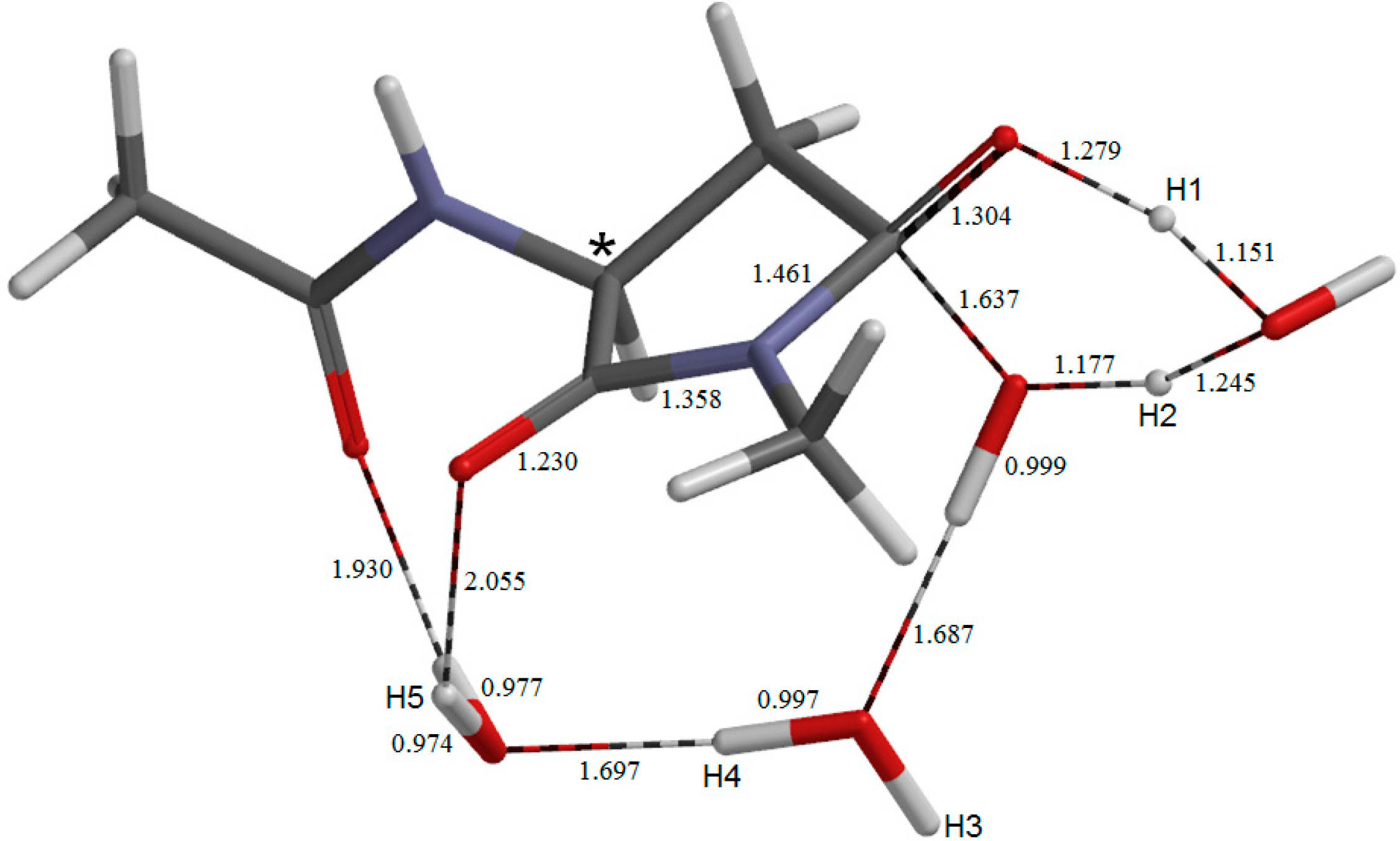
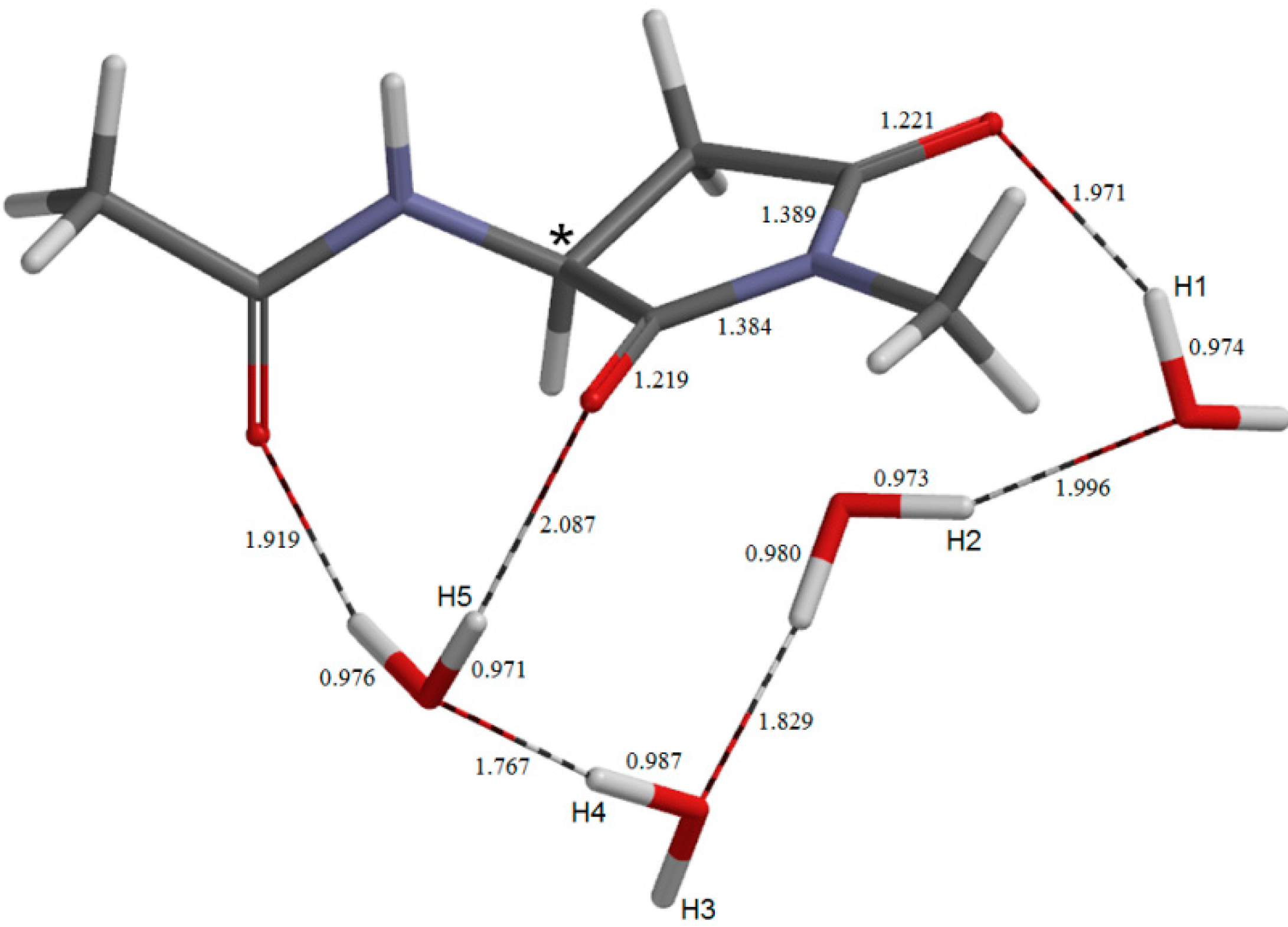
2. Results and Discussion
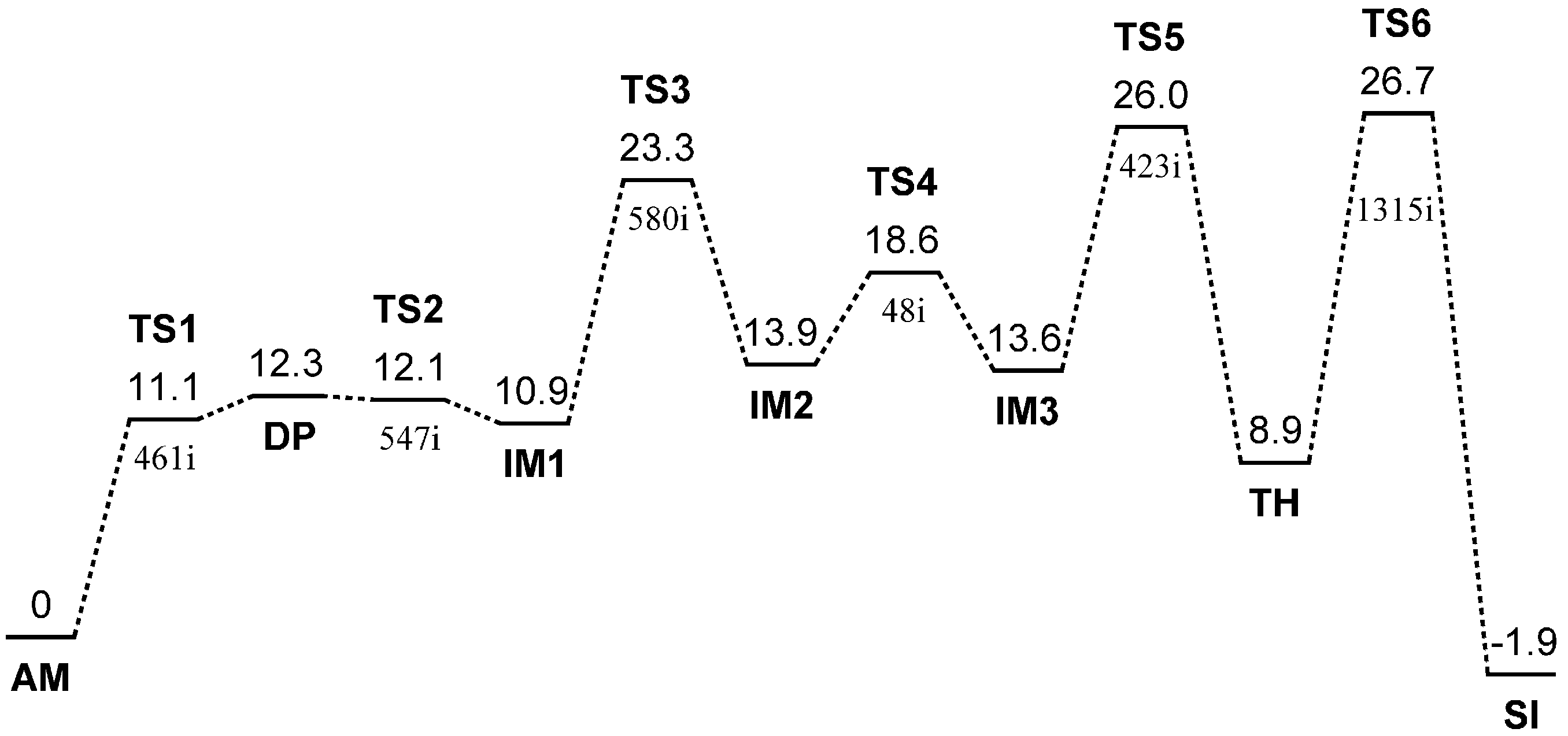
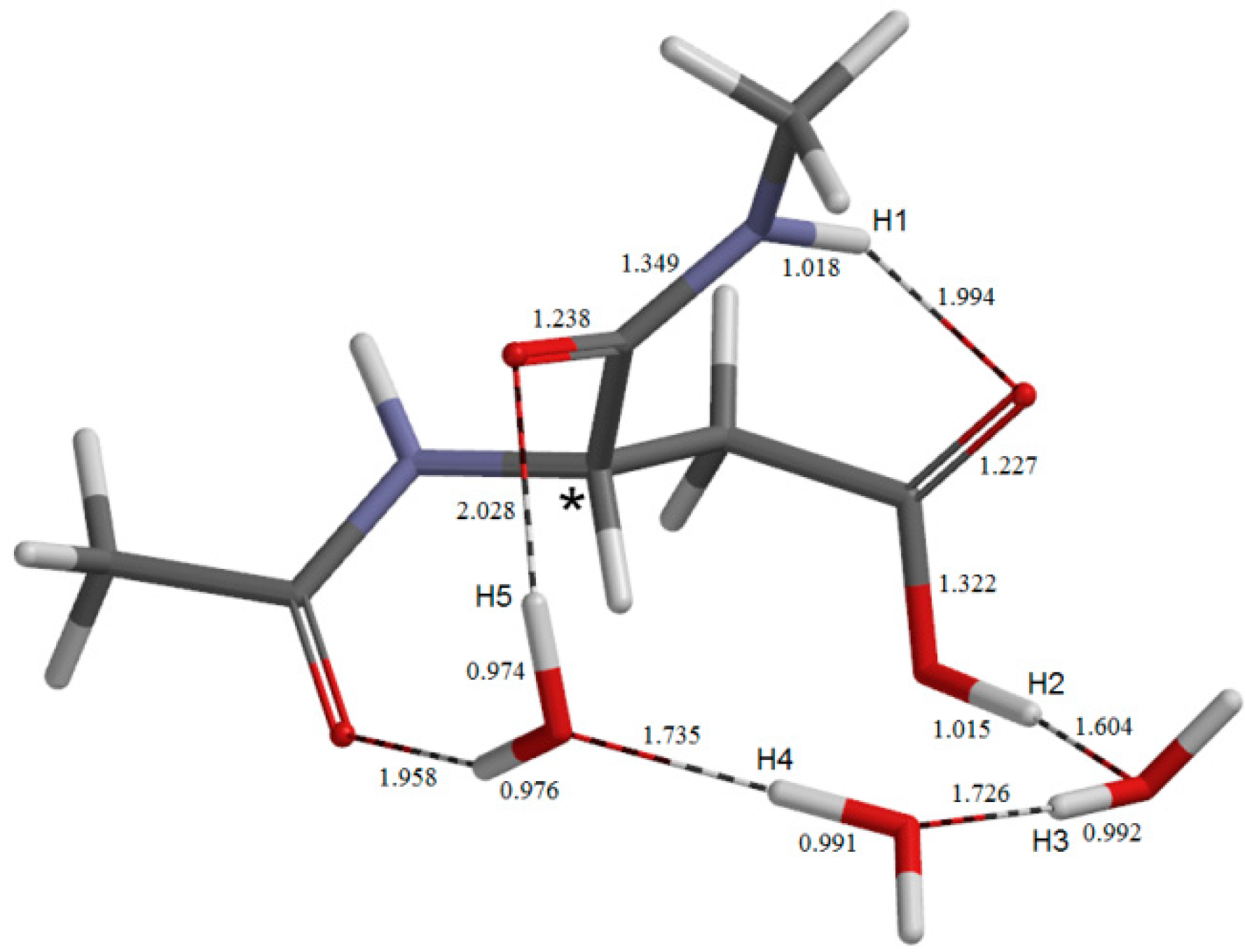


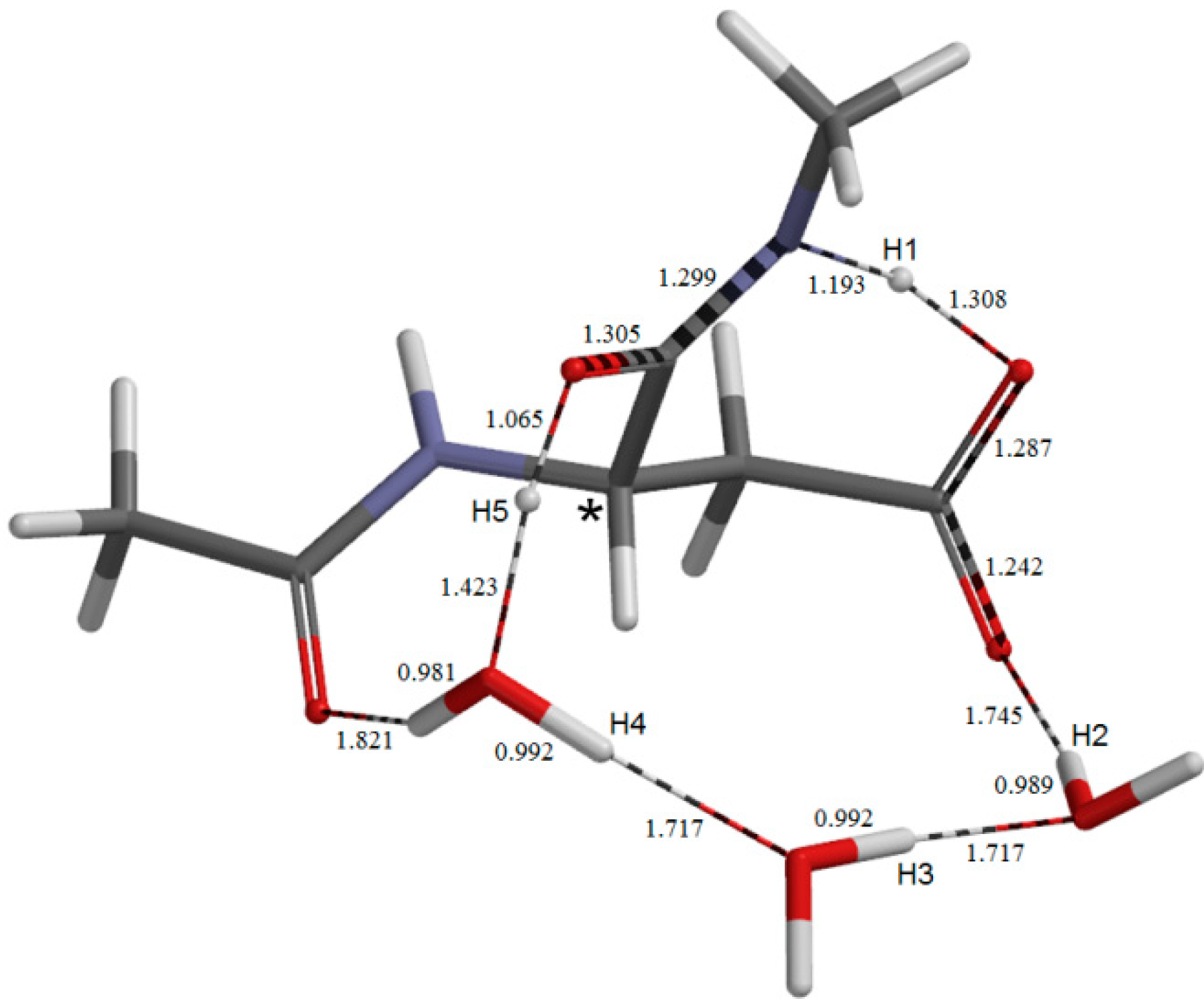
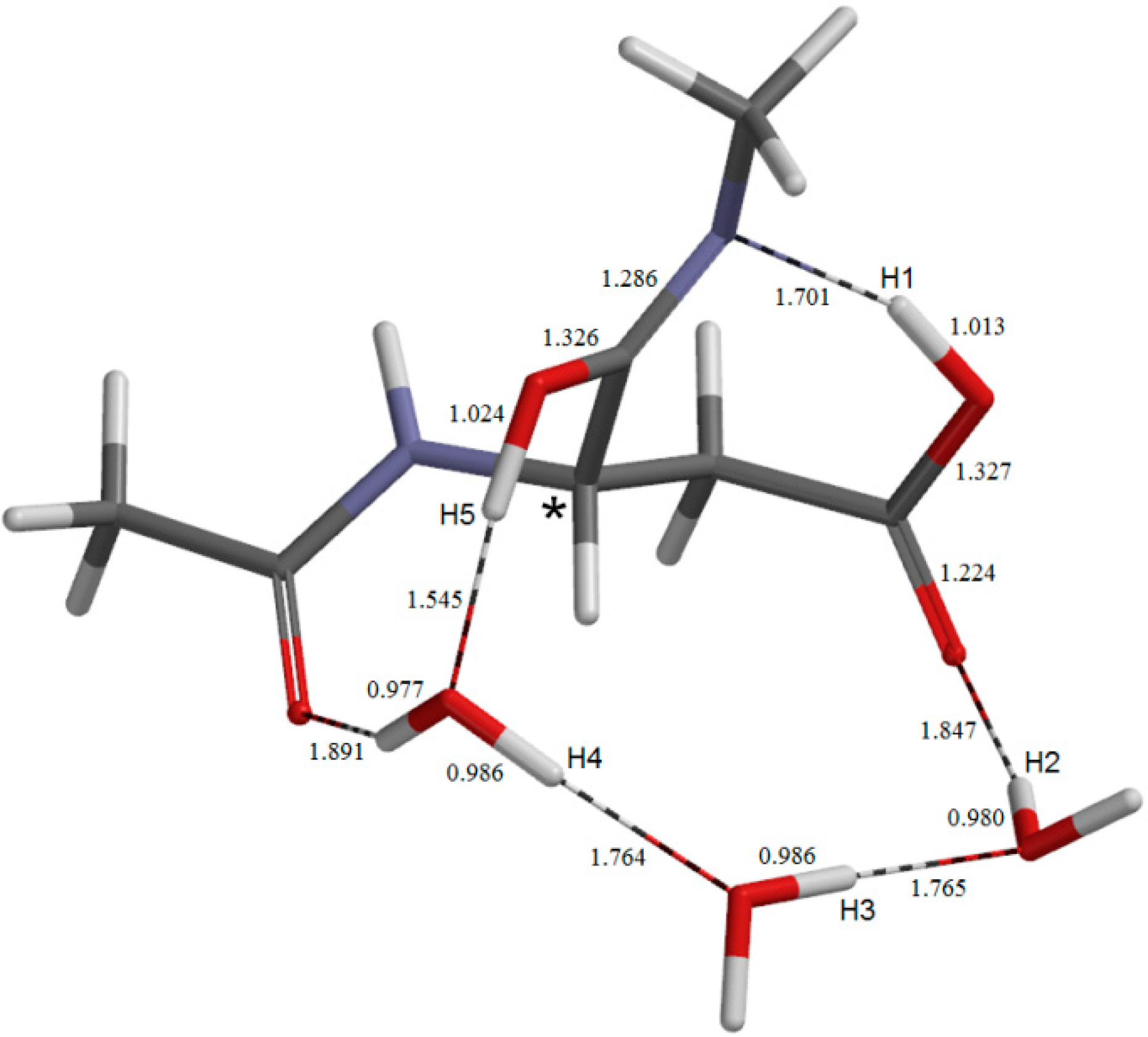
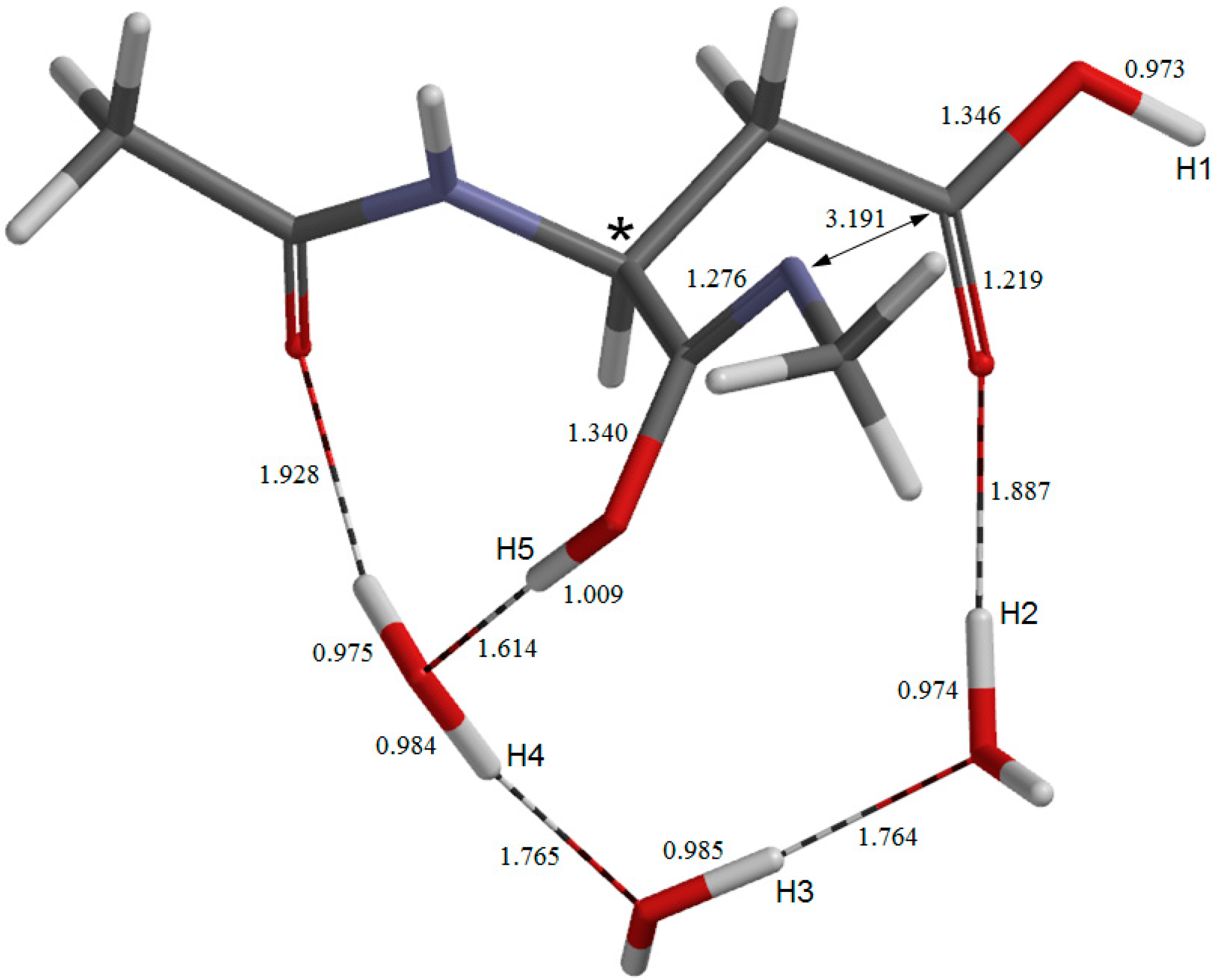

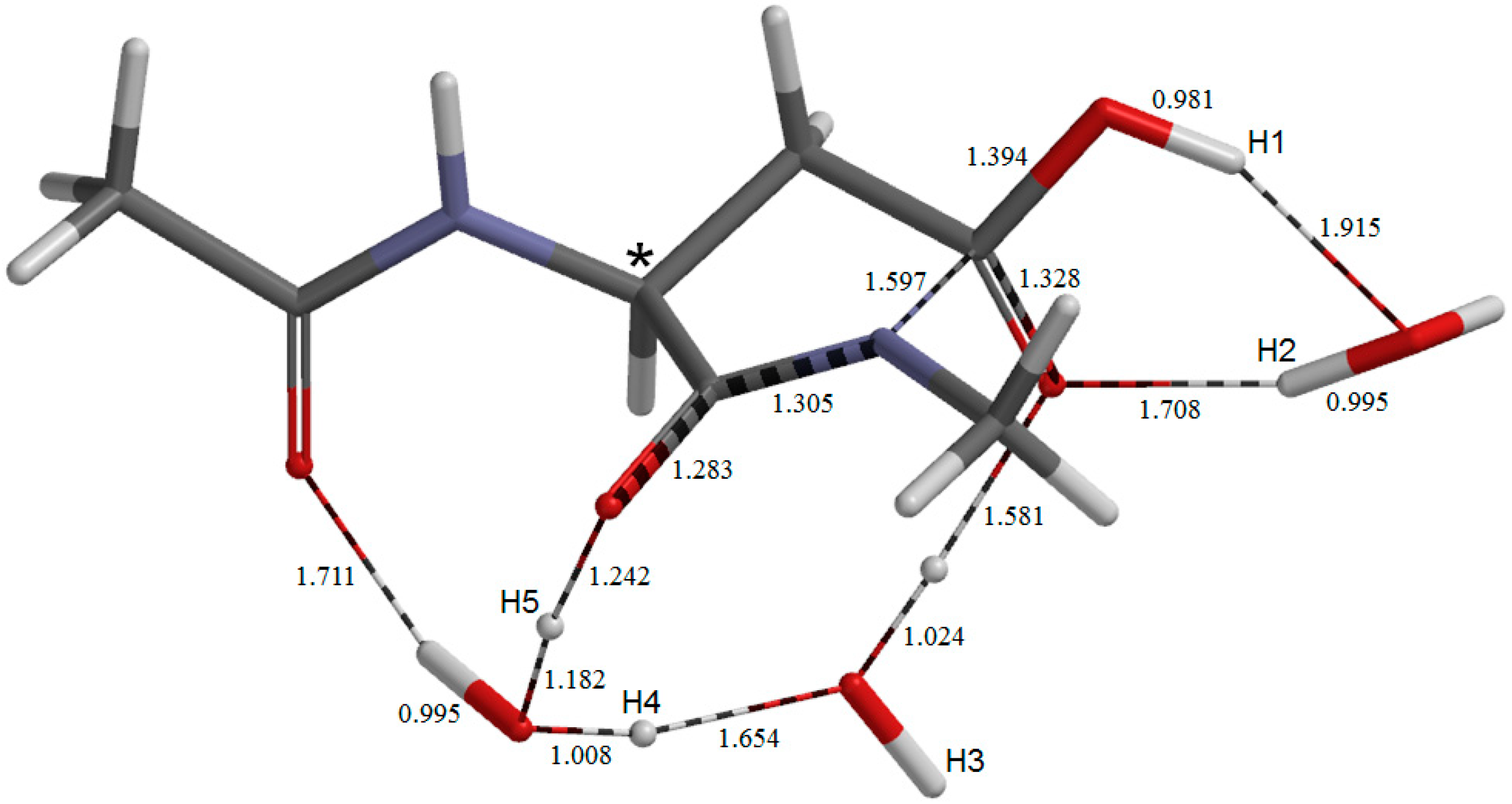

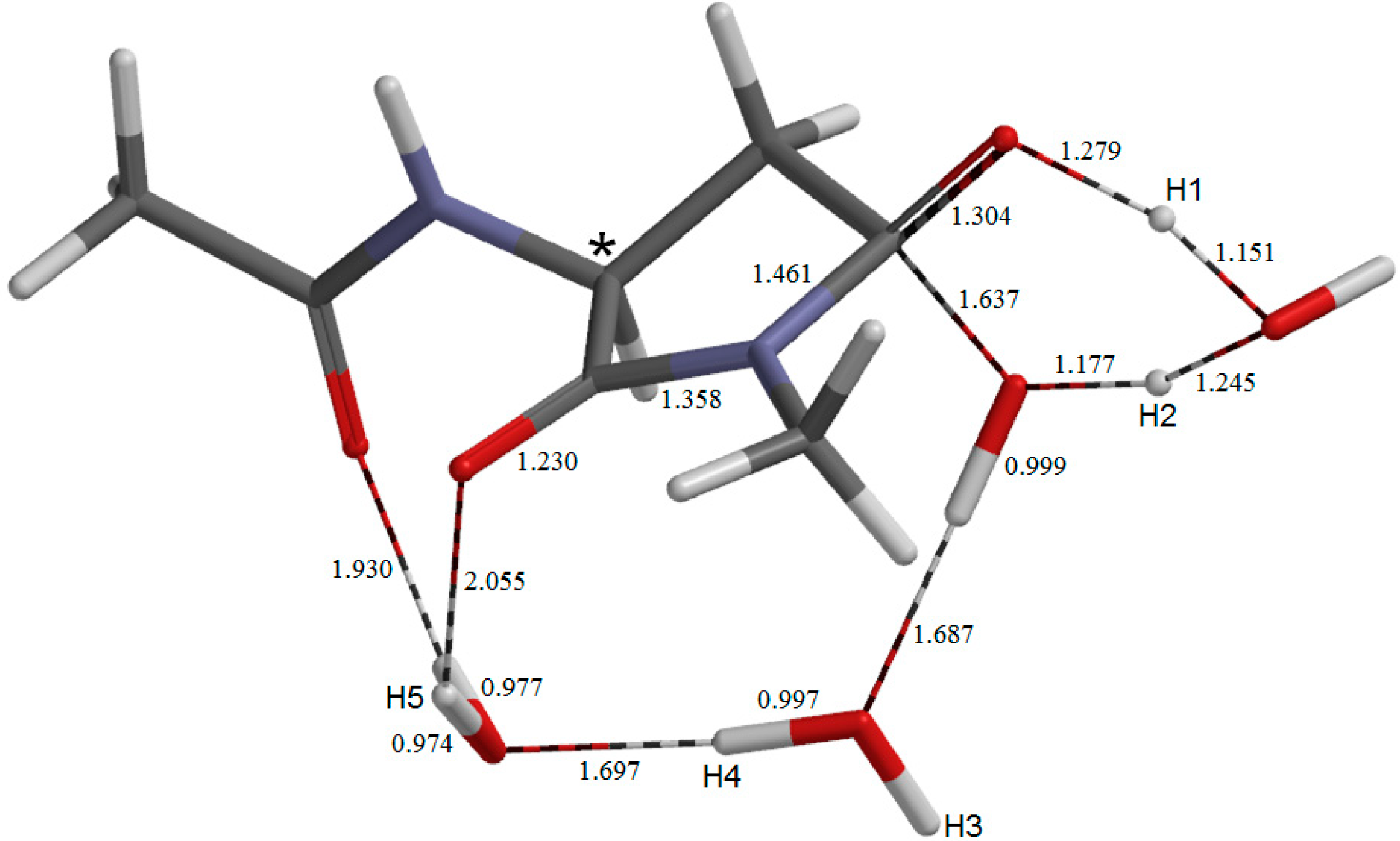


{kind=link}
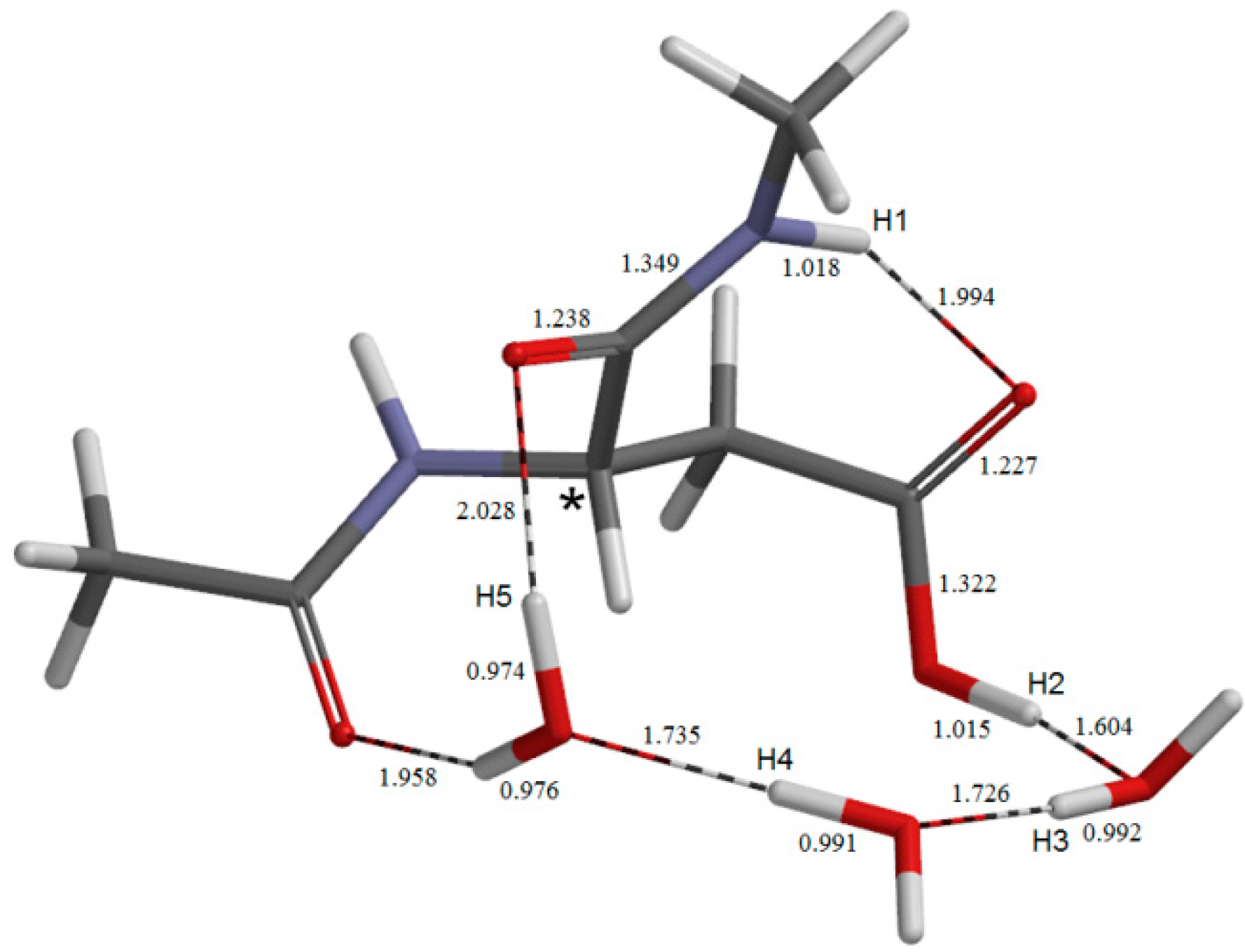{kind=link}
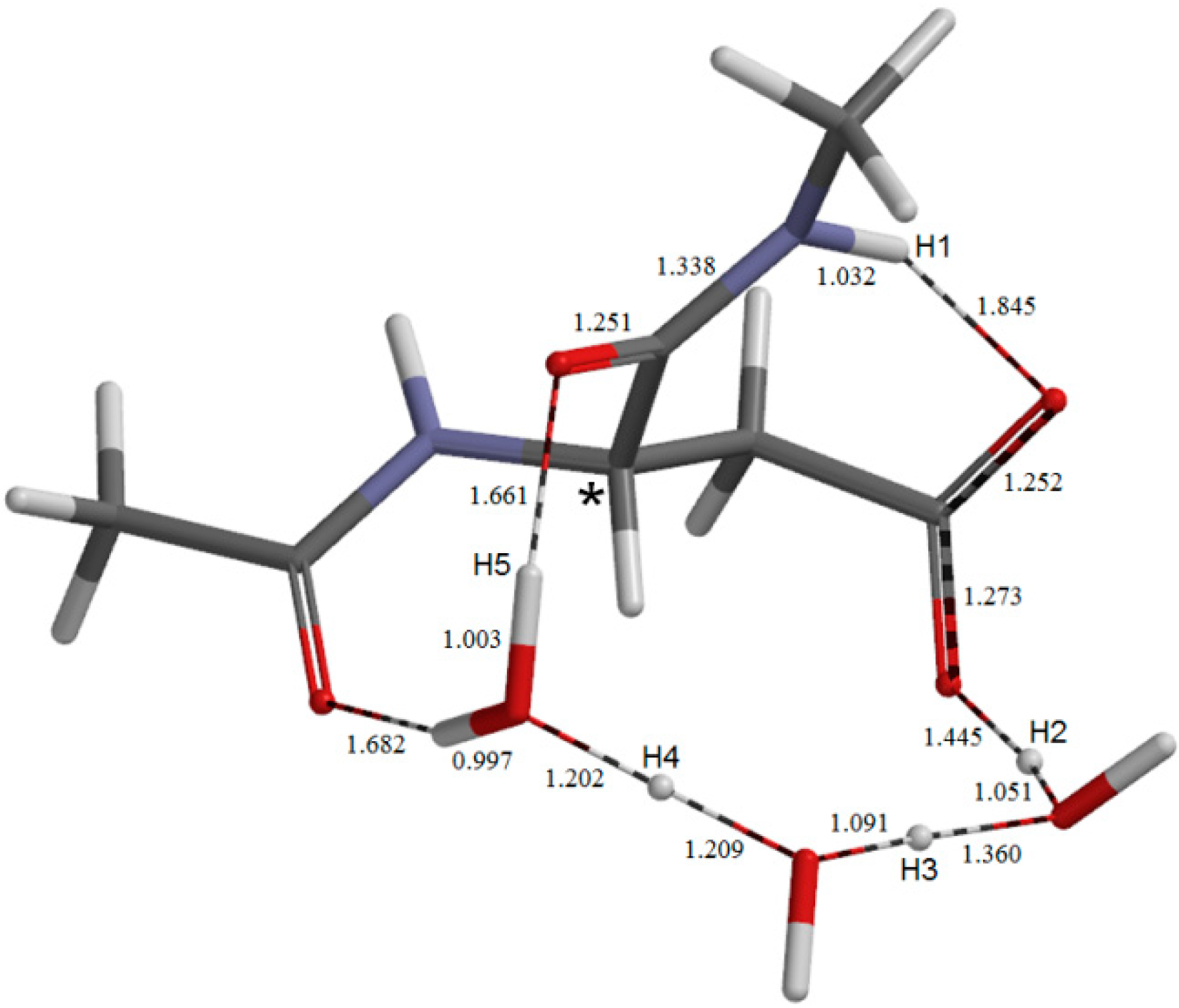{kind=link}
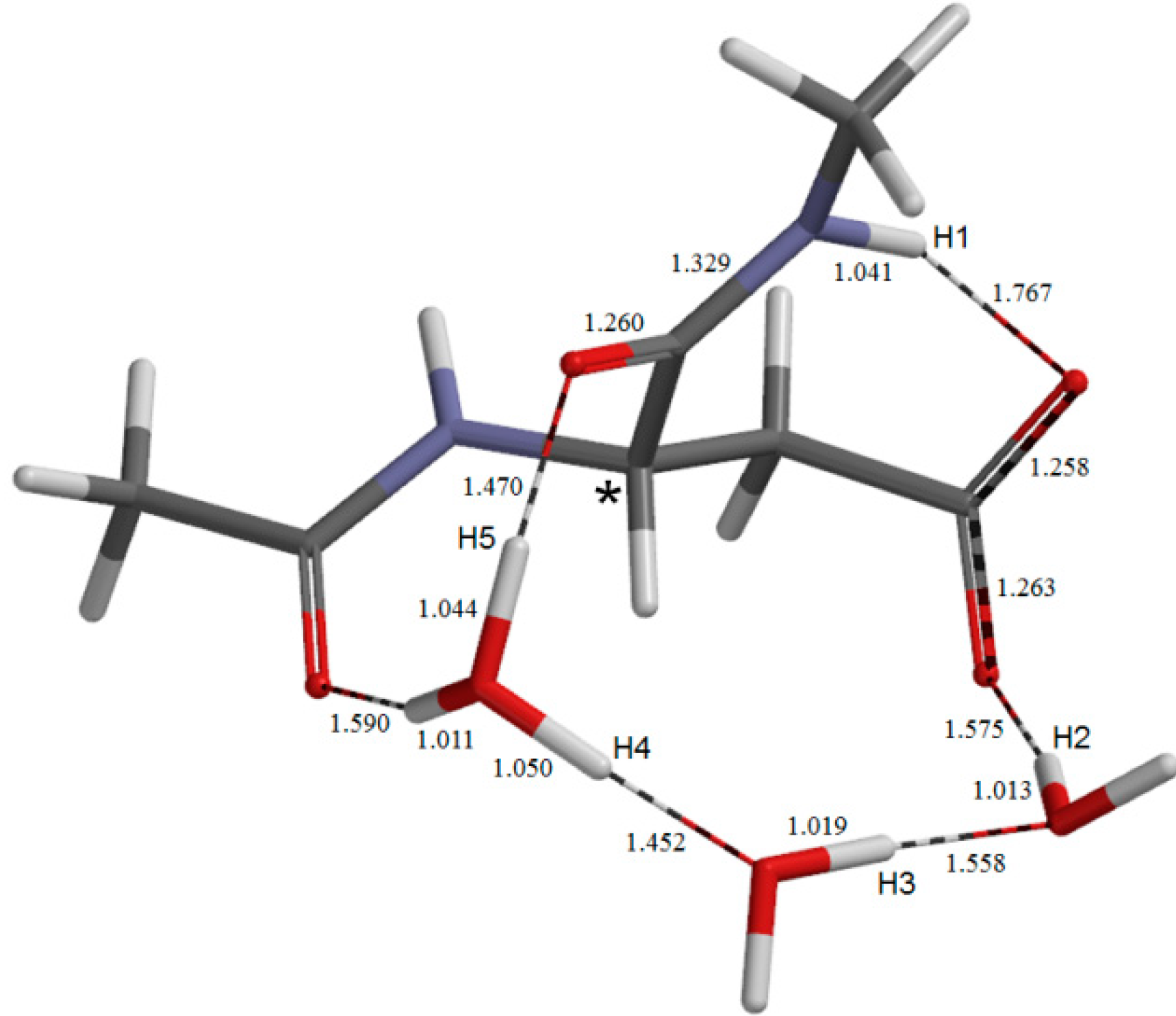{kind=link}
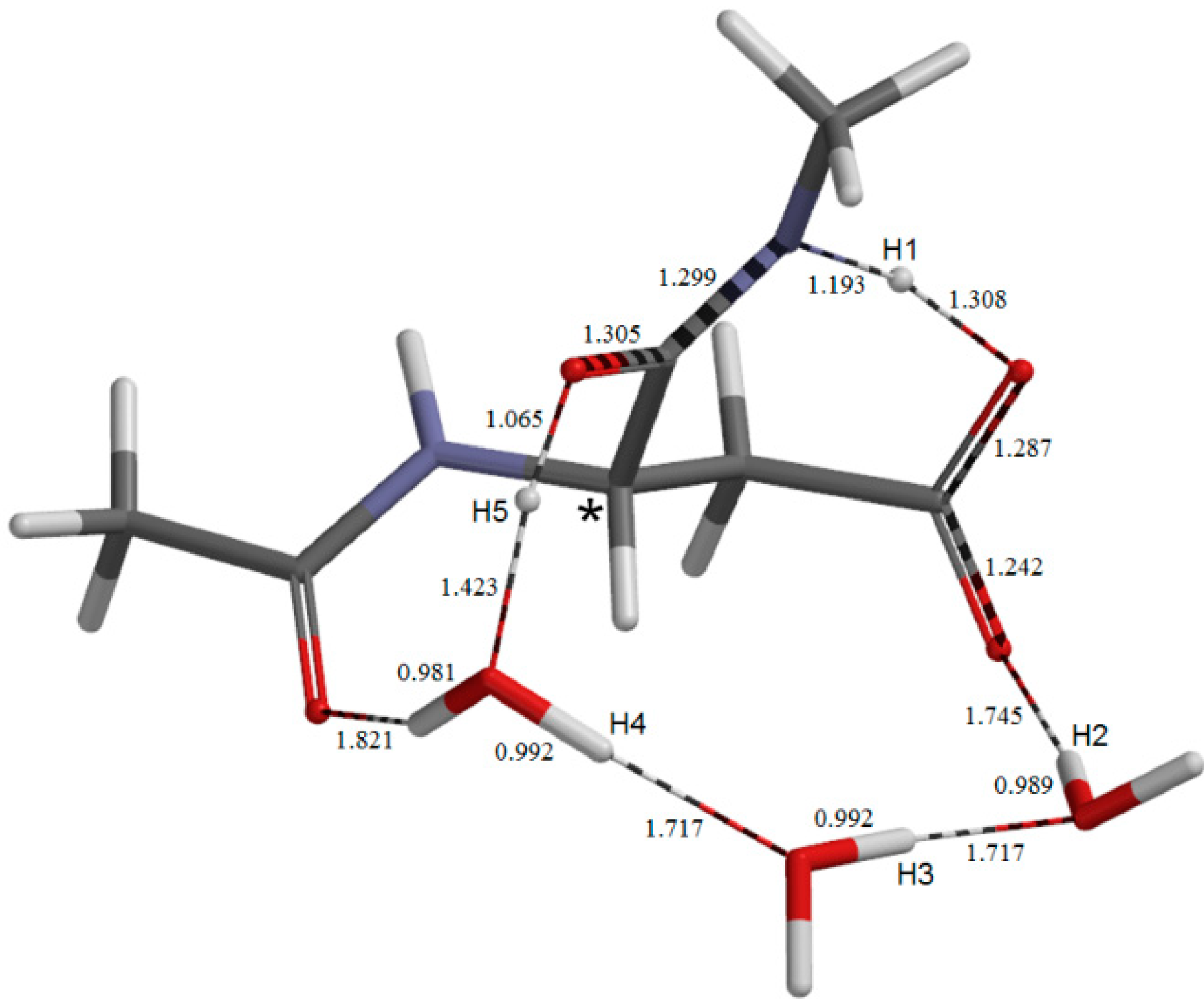{kind=link}
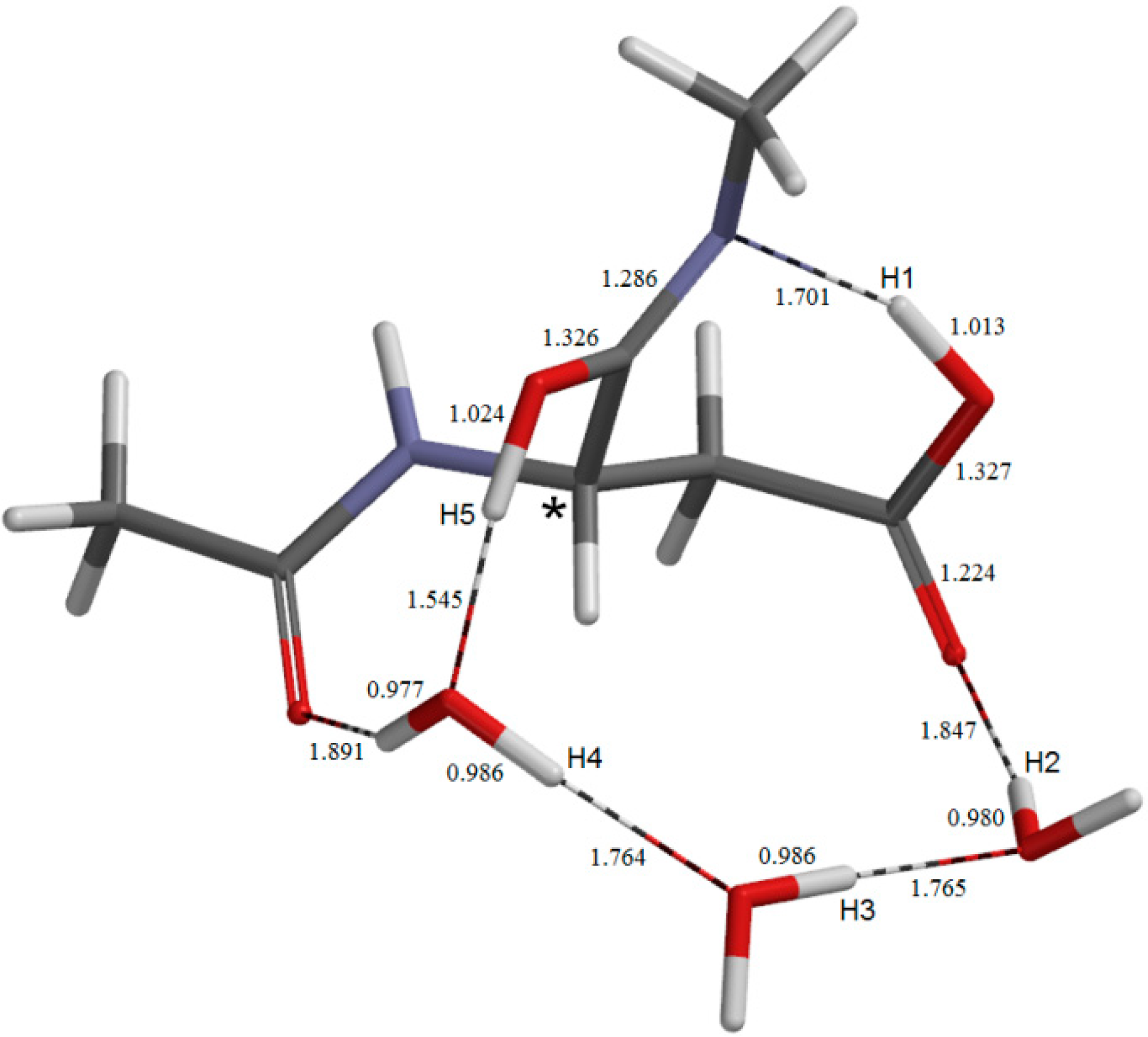{kind=link}
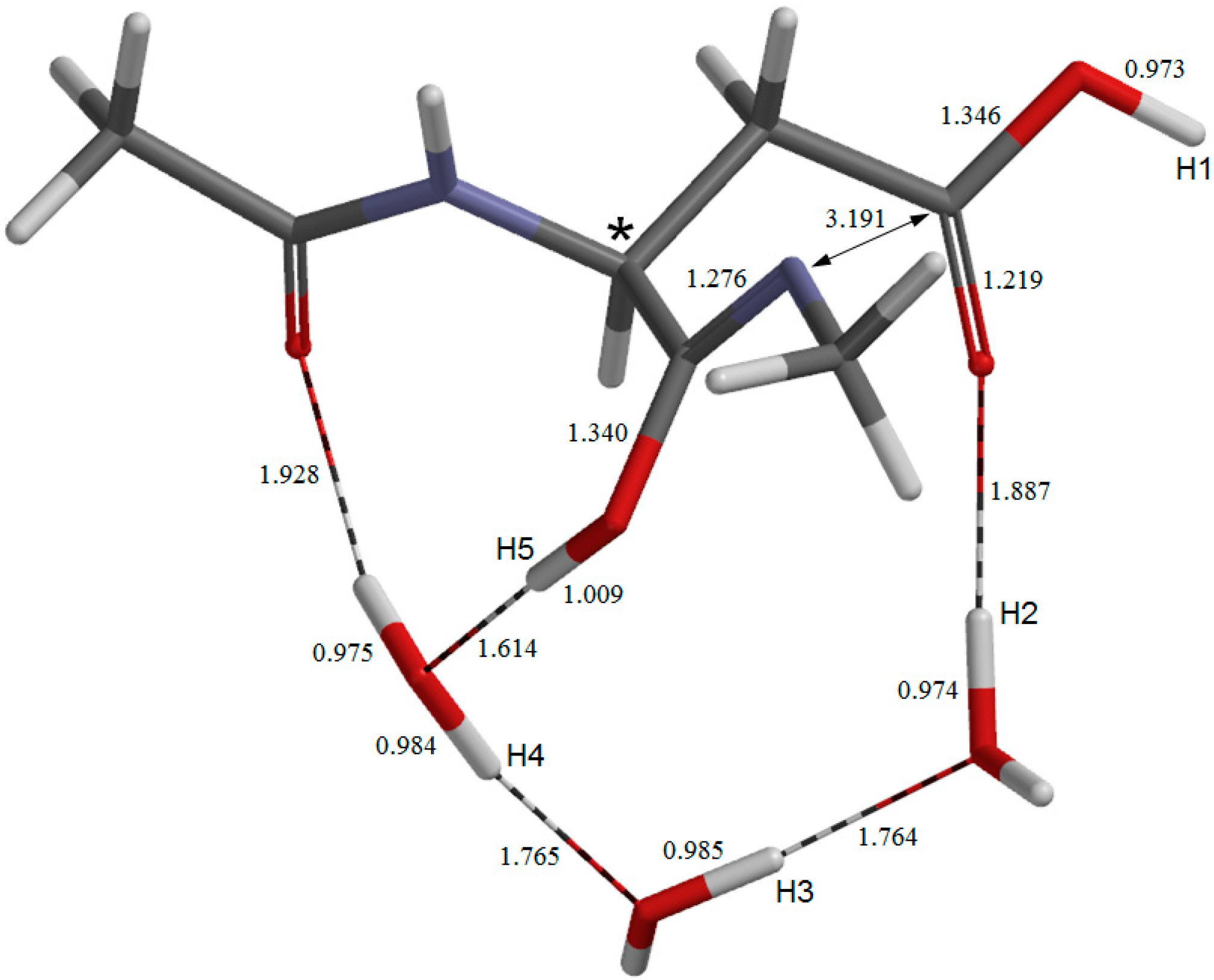{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
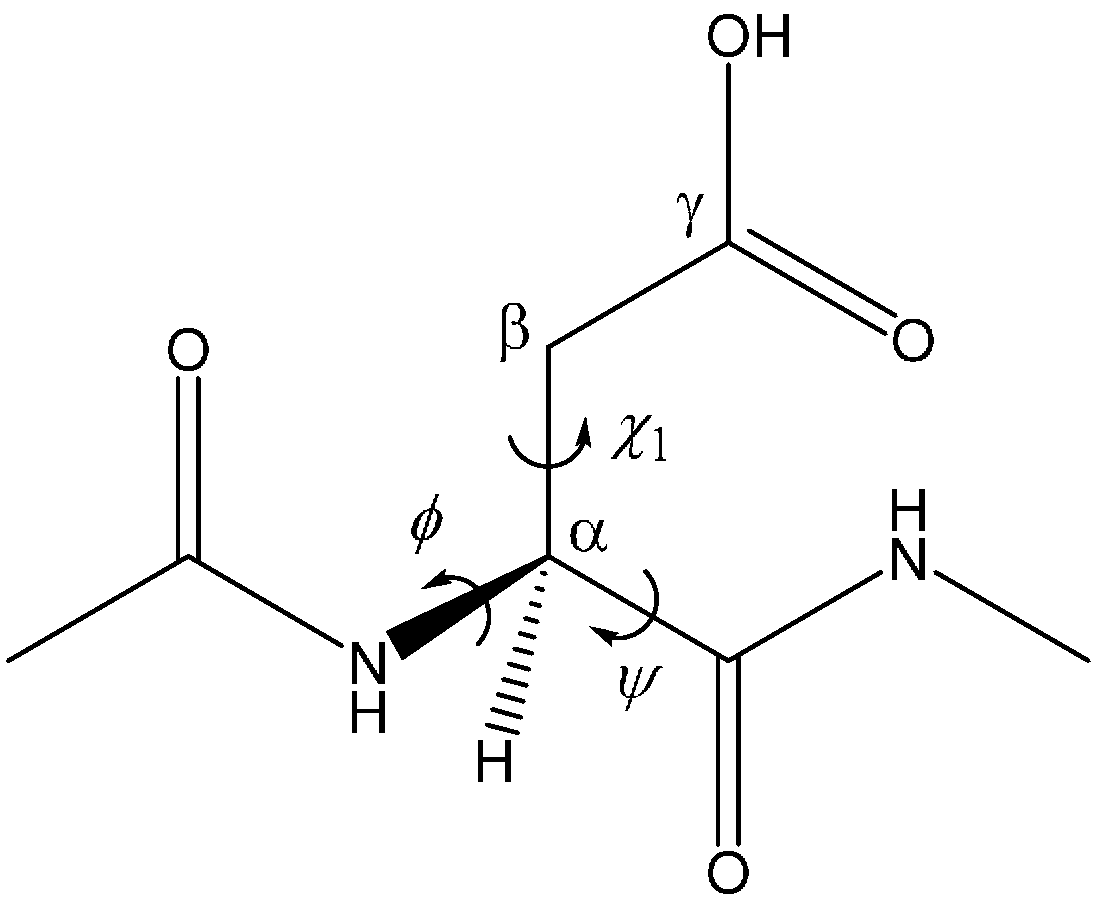
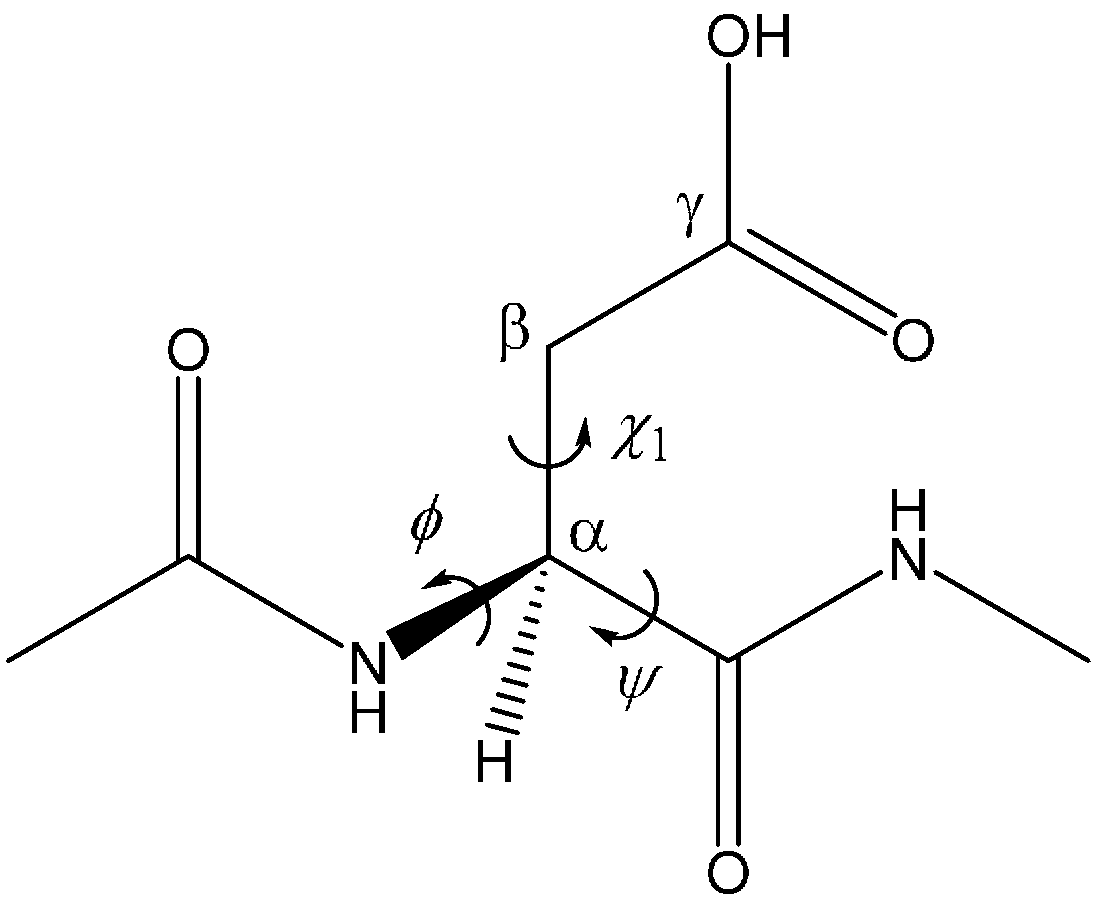
| Geometry | φ | ψ | χ1 |
|---|---|---|---|
| AM | −101 | −133 | −162 |
| TS1 | −102 | −126 | −166 |
| DP | −103 | −121 | −171 |
| TS2 | −112 | −118 | −179 |
| IM1 | −116 | −109 | −173 |
| TS3 | −121 | −110 | 173 |
| IM2 | −122 | −98 | −173 |
| TS4 | −115 | −98 | −174 |
| IM3 | −114 | −93 | −173 |
| TS5 | −97 | −136 | 155 |
| TH | −97 | −142 | 151 |
| TS6 | −96 | −146 | 153 |
| SI | −100 | −134 | 136 |
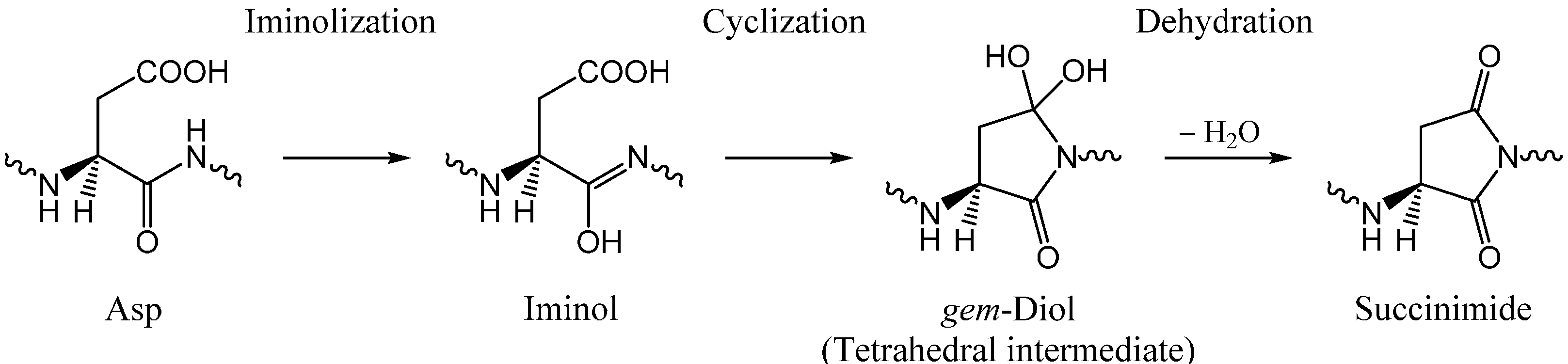
2.1. Iminolization
2.2. Change in Conformation and Complexation Mode in the Iminol Form
2.3. Cyclization to Form the Tetrahedral Intermediate
2.4. Dehydration of the Tetrahedral Intermediate
3. Experimental Section
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar]
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Aging Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Clarke, S. Aging as war between chemical and biochemical processes: Protein methylation and the recognition of age-damaged proteins for repair. Aging Res. Rev. 2003, 2, 263–285. [Google Scholar] [CrossRef]
- Fujii, N. d-Amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef]
- Truscott, R.J.W. Are ancient proteins responsible for the age-related decline in health and fitness? Rejuvenation Res. 2010, 13, 83–89. [Google Scholar] [CrossRef]
- Fujii, N.; Takemoto, L.J.; Momose, Y.; Matsumoto, S.; Hiroki, K.; Akaboshi, M. Formation of four isomers at the Asp-151 residue of aged human αA-crystallin by natural aging. Biochem. Biophys. Res. Commun. 1999, 265, 746–751. [Google Scholar] [CrossRef]
- Fujii, N.; Matsumoto, S.; Hiroki, K.; Takemoto, L. Inversion and isomerization of Asp-58 residue in human αA-crystallin from normal aged lenses and cataractous lenses. Biochim. Biophys. Acta 2001, 1549, 179–187. [Google Scholar] [CrossRef]
- Fujii, N.; Kawaguchi, T.; Sasaki, H.; Fujii, N. Simultaneous stereoinversion and isomerization at the Asp-4 residue in βB2-crystallin from the aged human eye lenses. Biochemistry 2011, 50, 8628–8635. [Google Scholar] [CrossRef]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemization and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. Age 2011, 33, 131–141. [Google Scholar] [CrossRef]
- Takahashi, O.; Oda, A. Amide−iminol tautomerization of the C-terminal peptide groups of aspartic acid residues. Two-water-assisted mechanism, cyclization from the iminol tautomer leading to the tetrahedral intermediate of succinimide formation, and implication to peptide group hydrogen exchange. In Tyrosine and Aspartic Acid: Properties, Sources and Health Benefits; Jones, J.E., Morano, D.M., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 131–147. [Google Scholar]
- Takahashi, O.; Kirikoshi, R. Intramolecular cyclization of aspartic acid residues assisted by three water molecules: A density functional theory study. Comput. Sci. Discov. 2014, 7. [Google Scholar] [CrossRef]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanism. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar]
- Schweiger, S.; Rauhut, G. Double proton transfer reactions with plateau-like transition state regions: Pyrazole–trifluoroacetic acid clusters. J. Phys. Chem. A 2006, 110, 2816–2820. [Google Scholar]
- Aki, K.; Fujii, N.; Fujii, N. Kinetics of isomerization and inversion of aspartate 58 of αA-crystallin peptide mimics under physiological conditions. PLoS One 2013, 8, e58515. [Google Scholar]
- Dunitz, J.D. Win some, lose some: Enthalpy–entropy compensation in weak intermolecular interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef]
- Capasso, S. Thermodynamic parameters of the reversible isomerization of aspartic residues via a succinimide derivative. Thermochim. Acta 1996, 286, 41–50. [Google Scholar] [CrossRef]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Reaction mechanism of deamidation of asparaginyl residues in peptides: Effect of solvent molecules. J. Phys. Chem. A 2006, 110, 8354–8365. [Google Scholar]
- Yi, L.; Beckley, N.; Gikanga, B.; Zhang, J.; Wang, Y.J.; Chih, H.; Sharma, V.K. Isomerization of Asp–Asp motif in model peptides and a monoclonal antibody Fab fragment. J. Pharm. Sci. 2013, 102, 947–959. [Google Scholar] [CrossRef]
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins. A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
- Spartan’08; Wavefunction, Inc.: Irvine, CA, USA, 2008.
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takahashi, O.; Kirikoshi, R.; Manabe, N. Roles of Intramolecular and Intermolecular Hydrogen Bonding in a Three-Water-Assisted Mechanism of Succinimide Formation from Aspartic Acid Residues. Molecules 2014, 19, 11440-11452. https://doi.org/10.3390/molecules190811440
Takahashi O, Kirikoshi R, Manabe N. Roles of Intramolecular and Intermolecular Hydrogen Bonding in a Three-Water-Assisted Mechanism of Succinimide Formation from Aspartic Acid Residues. Molecules. 2014; 19(8):11440-11452. https://doi.org/10.3390/molecules190811440
Chicago/Turabian StyleTakahashi, Ohgi, Ryota Kirikoshi, and Noriyoshi Manabe. 2014. "Roles of Intramolecular and Intermolecular Hydrogen Bonding in a Three-Water-Assisted Mechanism of Succinimide Formation from Aspartic Acid Residues" Molecules 19, no. 8: 11440-11452. https://doi.org/10.3390/molecules190811440
APA StyleTakahashi, O., Kirikoshi, R., & Manabe, N. (2014). Roles of Intramolecular and Intermolecular Hydrogen Bonding in a Three-Water-Assisted Mechanism of Succinimide Formation from Aspartic Acid Residues. Molecules, 19(8), 11440-11452. https://doi.org/10.3390/molecules190811440