Polyhydroyxalkanoate Synthase Fusions as a Strategy for Oriented Enzyme Immobilisation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Oriented Enzyme Immobilisation
2. Polyhydroxyalkanoate Biobeads
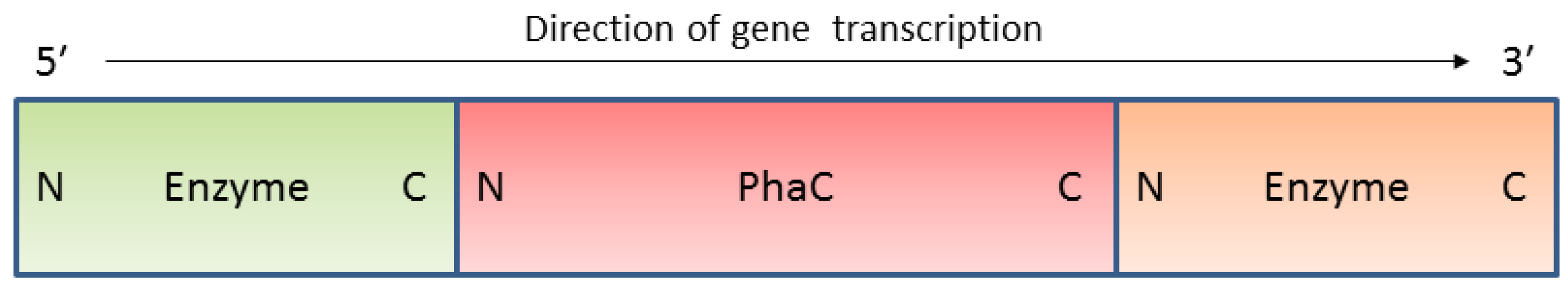
3. In Vivo Immobilisation and Surface Display
4.In Vitro Immobilisation and Surface Display
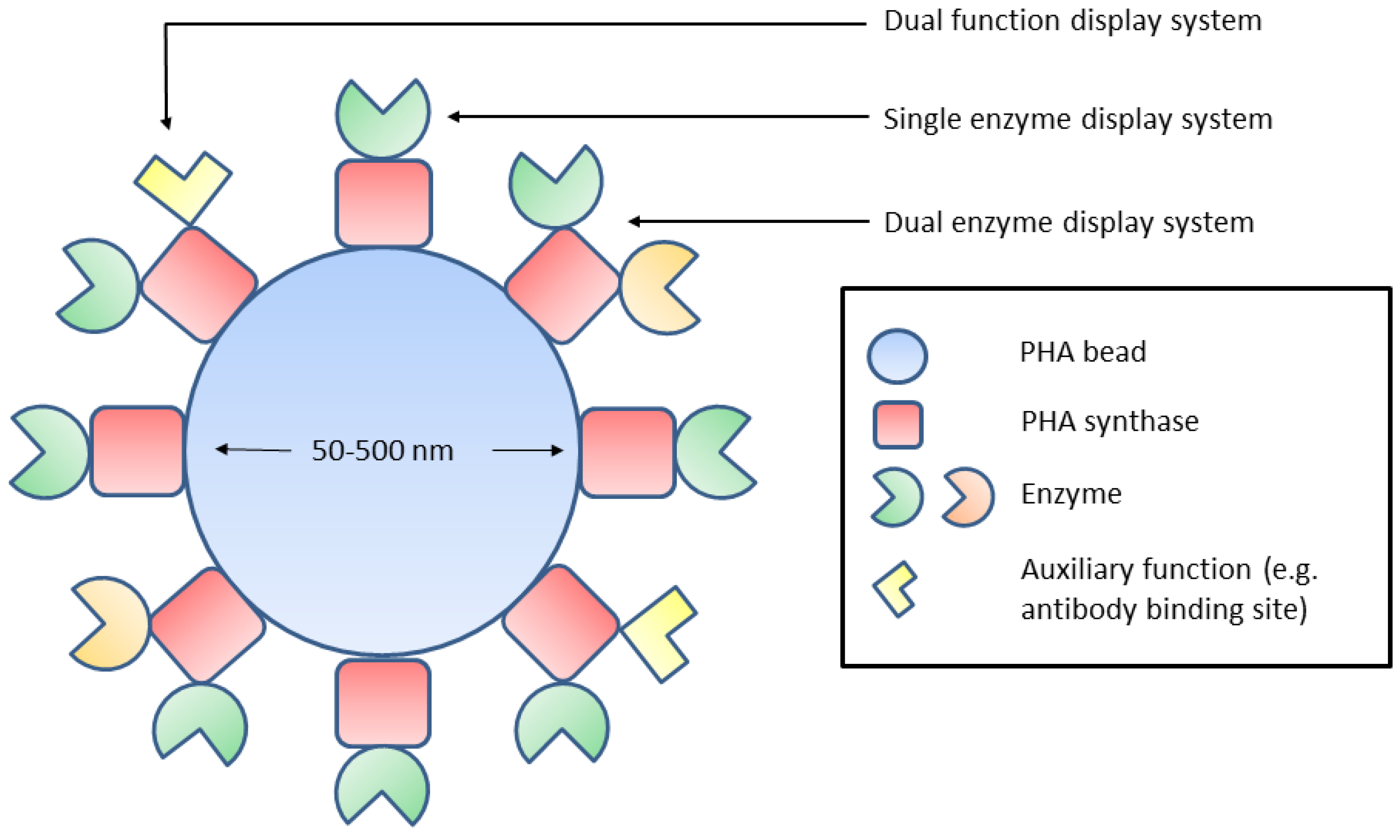
5. Orientation of Biobead Immobilised Enzymes

6. Quaternary Structures of Immobilised Enzymes
7. The Initial Proof of Concept of PHA Synthase Mediated Enzyme Immobilisation
8. Current Applications
9. Performance of PHA Bead Immobilized Enzymes
10. Potential Applications

11. Discussion
12. Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fernández-Fernández, M.; Sanromán, M.Á.; Moldes, D. Recent developments and applications of immobilized laccase. Biotechnol. Adv. 2013, 31, 1808–18025. [Google Scholar] [CrossRef]
- Muñoz Solano, D.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzyme Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Jia, F.; Narasimhan, B.; Mallapragada, S. Materials-based strategies for multi-enzyme immobilization and co-localization: A review. Biotechnol. Bioeng. 2013, 111, 209–222. [Google Scholar]
- Steen Redeker, E.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein engineering for directed immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef]
- Huang, W.; Wang, J.; Bhattacharyya, D.; Bachas, L.G. Improving the activity of immobilized subtilisin by site-specific attachment to surfaces. Anal. Chem. 1997, 69, 4601–4607. [Google Scholar] [CrossRef]
- Tan, G.-Y.; Chen, C.-L.; Li, L.; Ge, L.; Wang, L.; Razaad, I.; Li, Y.; Zhao, L.; Mo, Y.; Wang, J.-Y. Start a Research on Biopolymer Polyhydroxyalkanoate (PHA): A Review. Polymers (Basel) 2014, 6, 706–754. [Google Scholar] [CrossRef]
- Rehm, B.H.A. Polyester synthases: Natural catalysts for plastics. Biochem. J. 2003, 376, 15–33. [Google Scholar] [CrossRef]
- Rehm, B.H.A. Genetics and biochemistry of polyhydroxyalkanoate granule self-assembly: The key role of polyester synthases. Biotechnol. Lett. 2006, 28, 207–213. [Google Scholar] [CrossRef]
- Grage, K.; Jahns, A.C.; Parlane, N.; Palanisamy, R.; Rasiah, I.A.; Atwood, J.A.; Rehm, B.H.A. Bacterial polyhydroxyalkanoate granules: Biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 2009, 10, 660–669. [Google Scholar] [CrossRef]
- Normi, Y.M.; Hiraishi, T.; Taguchi, S.; Abe, H.; Sudesh, K.; Najimudin, N.; Doi, Y. Characterization and properties of G4X mutants of Ralstonia eutropha PHA synthase for poly(3-hydroxybutyrate) biosynthesis in Escherichia coli. Macromol. Biosci. 2005, 5, 197–206. [Google Scholar] [CrossRef]
- Satoh, Y.; Tajima, K.; Tannai, H.; Munekata, M. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis from acetate with CoA recycling and NADPH regeneration in vitro. J. Biosci. Bioeng. 2003, 95, 335–341. [Google Scholar] [CrossRef]
- Jo, S.-J.; Maeda, M.; Ooi, T.; Taguchi, S. Production system for biodegradable polyester polyhydroxybutyrate by Corynebacterium glutamicum. J. Biosci. Bioeng. 2006, 102, 233–236. [Google Scholar] [CrossRef]
- Mifune, J.; Grage, K.; Rehm, B.H.A. Production of functionalized biopolyester granules by recombinant Lactococcus lactis. Appl. Environ. Microbiol. 2009, 75, 4668–4675. [Google Scholar] [CrossRef]
- Valappil, S.P.; Boccaccini, A.R.; Bucke, C.; Roy, I. Polyhydroxyalkanoates in Gram-positive bacteria: Insights from the genera Bacillus and Streptomyces. Antonie Van Leeuwenhoek 2007, 91, 1–17. [Google Scholar]
- Hernandez, K.; Fernandez-Lafuente, R. Control of protein immobilization: Coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb. Technol. 2011, 48, 107–122. [Google Scholar] [CrossRef]
- Steinbuchel, A.; Aerts, K.; Babel, W.; Follner, C.; Liebergesell, M.; Madkour, M.H.; Mayer, F.; Pieper-Furst, U.; Pries, A.; Valentin, H.E. Considerations on the structure and biochemistry of bacterial polyhydroxyalkanoic acid inclusions. Can. J. Microbiol. 1995, 41 (Suppl. 1), 94–105. [Google Scholar] [CrossRef]
- Barnard, G.C.; McCool, J.D.; Wood, D.W.; Gerngross, T.U. Integrated recombinant protein expression and purification platform based on Ralstonia eutropha. Appl. Environ. Microbiol. 2005, 71, 5735–5742. [Google Scholar] [CrossRef]
- Geng, Y.; Wang, S.; Qi, Q. Expression of active recombinant human tissue-type plasminogen activator by using in vivo polyhydroxybutyrate granule display. Appl. Environ. Microbiol. 2010, 76, 7226–7230. [Google Scholar] [CrossRef]
- Peters, V.; Rehm, B.H.A. In vivo enzyme immobilization by use of engineered polyhydroxyalkanoate synthase. Appl. Environ. Microbiol. 2006, 72, 1777–1783. [Google Scholar] [CrossRef]
- Rasiah, I.A.; Rehm, B.H.A. One-step production of immobilized alpha-amylase in recombinant Escherichia coli. Appl. Environ. Microbiol. 2009, 75, 2012–2016. [Google Scholar] [CrossRef]
- Robins, K.J.; Hooks, D.O.; Rehm, B.H.A.; Ackerley, D.F. Escherichia coli NemA is an efficient chromate reductase that can be biologically immobilized to provide a cell free system for remediation of hexavalent chromium. PLoS One 2013, 8, e59200. [Google Scholar]
- Hooks, D.O.; Blatchford, P.A.; Rehm, B.H.A. Bioengineering of bacterial polymer inclusions catalyzing the synthesis of N-acetylneuraminic acid. Appl. Environ. Microbiol. 2013, 79, 3116–3121. [Google Scholar] [CrossRef]
- Blatchford, P.A.; Scott, C.; French, N.; Rehm, B.H.A. Immobilization of organophosphohydrolase OpdA from Agrobacterium radiobacter by overproduction at the surface of polyester inclusions inside engineered Escherichia coli. Biotechnol. Bioeng. 2012, 109, 1101–1108. [Google Scholar] [CrossRef]
- Juers, D.H.; Jacobson, R.H.; Wigley, D.; Zhang, X.J.; Huber, R.E.; Tronrud, D.E.; Matthews, B.W. High resolution refinement of beta-galactosidase in a new crystal form reveals multiple metal-binding sites and provides a structural basis for alpha-complementation. Protein Sci. 2000, 9, 1685–1699. [Google Scholar] [CrossRef]
- Benning, M.M.; Shim, H.; Raushel, F.M.; Holden, H.M. High resolution X-ray structures of different metal-substituted forms of phosphotriesterase from Pseudomonas diminuta. Biochemistry 2001, 40, 2712–2722. [Google Scholar] [CrossRef]
- Hwang, K.Y.; Song, H.K.; Chang, C.; Lee, J.; Lee, S.Y.; Kim, K.K.; Choe, S.; Sweet, R.M.; Suh, S.W. Crystal structure of thermostable alpha-amylase from Bacillus licheniformis refined at 1.7 A resolution. Mol. Cells 1997, 7, 251–258. [Google Scholar]
- Lee, Y.-C.; Wu, H.-M.; Chang, Y.-N.; Wang, W.-C.; Hsu, W.-H. The central cavity from the (alpha/alpha)6 barrel structure of Anabaena sp. CH1 N-acetyl-D-glucosamine 2-epimerase contains two key histidine residues for reversible conversion. J. Mol. Biol. 2007, 367, 895–908. [Google Scholar] [CrossRef]
- Izard, T.; Lawrence, M.C.; Malby, R.L.; Lilley, G.G.; Colman, P.M. The three-dimensional structure of N-acetylneuraminate lyase from Escherichia coli. Structure 1994, 2, 361–369. [Google Scholar] [CrossRef]
- Kim, H.-N.; Lee, J.; Kim, H.-Y.; Kim, Y.-R. Enzymatic synthesis of a drug delivery system based on polyhydroxyalkanoate-protein block copolymers. Chem. Commun. (Camb.) 2009, 7345, 7104–7106. [Google Scholar]
- Lee, J.; Jung, S.-G.; Park, C.-S.; Kim, H.-Y.; Batt, C.A.; Kim, Y.-R. Tumor-specific hybrid polyhydroxybutyrate nanoparticle: Surface modification of nanoparticle by enzymatically synthesized functional block copolymer. Bioorg. Med. Chem. Lett. 2011, 21, 2941–2944. [Google Scholar] [CrossRef]
- Derewenda, U.; Swenson, L.; Green, R.; Wei, Y.; Yamaguchi, S.; Joerger, R.; Haas, M.J.; Derewenda, Z.S. Current progress in crystallographic studies of new lipases from filamentous fungi. Protein Eng. 1994, 7, 551–557. [Google Scholar]
- Berg, O.G.; Cajal, Y.; Butterfoss, G.L.; Grey, R.L.; Alsina, M.A.; Yu, B.Z.; Jain, M.K. Interfacial activation of triglyceride lipase from Thermomyces (Humicola) lanuginosa: Kinetic parameters and a basis for control of the lid. Biochemistry 1998, 37, 6615–6627. [Google Scholar]
- Fernandez-Lafuente, R. Lipase from Thermomyces lanuginosus: Uses and prospects as an industrial biocatalyst. J. Mol. Catal. B Enzym. 2010, 62, 197–212. [Google Scholar] [CrossRef]
- Bastida, A.; Sabuquillo, P.; Armisen, P.; Fernandez-Lafuente, R.; Huguet, J.; Guisan, J. A single step purification, immobilization, and hyperactivation of lipases via interfacial adsorption on strongly hydrophobic supports. Biotechnol. Bioeng. 1998, 58, 486–493. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Armisén, P.; Sabuquillo, P.; Fernández-Lorente, G.; Guisán, J.M. Immobilization of lipases by selective adsorption on hydrophobic supports. Chem. Phys. Lipids 1998, 93, 185–197. [Google Scholar] [CrossRef]
- Bravo Rodríguez, V.; Jurado Alameda, E.; Martínez Gallegos, J.F.; Reyes Requena, A.; García López, A.I. Enzymatic hydrolysis of soluble starch with an alpha-amylase from Bacillus licheniformis. Biotechnol. Prog. 2006, 22, 718–722. [Google Scholar] [CrossRef]
- Shewale, S.D.; Pandit, A.B. Hydrolysis of soluble starch using Bacillus licheniformis alpha-amylase immobilized on superporous CELBEADS. Carbohydr. Res. 2007, 342, 997–1008. [Google Scholar] [CrossRef]
- Pandey, A.; Nigam, P.; Soccol, C.R.; Soccol, V.T.; Singh, D.; Mohan, R. Advances in microbial amylases. Biotechnol. Appl. Biochem. 2000, 31, 135–152. [Google Scholar]
- Riley, R.G.; Zachara, J.M.; Wobber, F.J. Chemical Contaminants on DOE Lands and Selection of Contaminant Mixtures for Subsurface Science Research; U.S. Department of Energy, Office of Energy Research, Subsurface Science Program; Available to the public from the National Technical Information Service, U.S. Department of Commerce: Washington, DC, USA; Springfield, VA, USA, 1992. [Google Scholar]
- Jeyaratnam, J. Acute pesticide poisoning: A major global health problem. World Health Stat. Q. 1990, 43, 139–144. [Google Scholar]
- Isbister, G.K.; Mills, K.; Friberg, L.E.; Hodge, M.; O’Connor, E.; Patel, R.; Abeyewardene, M.; Eddleston, M. Human methyl parathion poisoning. Clin. Toxicol. (Phila) 2007, 45, 956–960. [Google Scholar] [CrossRef]
- Von Itzstein, M. The war against influenza: Discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 2007, 6, 967–974. [Google Scholar] [CrossRef]
- Rodríguez-Aparicio, L.B.; Ferrero, M.A.; Reglero, A. N-acetyl-d-neuraminic acid synthesis in Escherichia coli K1 occurs through condensation of N-acetyl-d-mannosamine and pyruvate. Biochem. J. 1995, 308, 501–505. [Google Scholar]
- Luchansky, S.J.; Yarema, K.J.; Takahashi, S.; Bertozzi, C.R. GlcNAc 2-epimerase can serve a catabolic role in sialic acid metabolism. J. Biol. Chem. 2003, 278, 8035–8042. [Google Scholar] [CrossRef]
- Ball, J.C.; Puckett, L.G.; Bachas, L.G. Covalent immobilization of beta-galactosidase onto a gold-coated magnetoelastic transducer via a self-assembled monolayer: Toward a magnetoelastic biosensor. Anal. Chem. 2003, 75, 6932–6937. [Google Scholar] [CrossRef]
- Hu, S.; Chen, J.; Yang, Z.; Shao, L.; Bai, H.; Luo, J.; Jiang, W.; Yang, Y. Coupled bioconversion for preparation of N-acetyl-d-neuraminic acid using immobilized N-acetyl-d-glucosamine-2-epimerase and N-acetyl-d-neuraminic acid lyase. Appl. Microbiol. Biotechnol. 2010, 85, 1383–1391. [Google Scholar] [CrossRef]
- Wang, T.-H.; Chen, Y.-Y.; Pan, H.-H.; Wang, F.-P.; Cheng, C.-H.; Lee, W.-C. Production of N-acetyl-D-neuraminic acid using two sequential enzymes overexpressed as double-tagged fusion proteins. BMC Biotechnol. 2009, 9, 63. [Google Scholar]
- Venning-Slater, M.; Hooks, D.O.; Rehm, B.H.A. In vivo self-assembly of stable green fluorescent protein fusion particles and their uses in enzyme immobilization. Appl. Environ. Microbiol. 2014, 80, 3062–3071. [Google Scholar] [CrossRef]
- Jahns, A.C.; Rehm, B.H.A. Tolerance of the Ralstonia eutropha class I polyhydroxyalkanoate synthase for translational fusions to its C terminus reveals a new mode of functional display. Appl. Environ. Microbiol. 2009, 75, 5461–5466. [Google Scholar] [CrossRef]
- Pescador, P.; Katakis, I.; Toca-Herrera, J.L.; Donath, E. Efficiency of a bienzyme sequential reaction system immobilized on polyelectrolyte multilayer-coated colloids. Langmuir 2008, 24, 14108–14114. [Google Scholar] [CrossRef]
- Aranaz, I.; Ramos, V.; de La Escalera, S.; Heras, A. Co-immobilization of d-hydantoinase and d-carboamylase on Chitin: Application to the Synthesis of p-hydroxyphenylglycine. Biocatal. Biotransform. 2003, 21, 349–356. [Google Scholar] [CrossRef]
- Crestini, C.; Melone, F.; Saladino, R. Novel multienzyme oxidative biocatalyst for lignin bioprocessing. Bioorg. Med. Chem. 2011, 19, 5071–5078. [Google Scholar] [CrossRef]
- García-Fruitós, E.; Vázquez, E.; Díez-Gil, C.; Corchero, J.L.; Seras-Franzoso, J.; Ratera, I.; Veciana, J.; Villaverde, A. Bacterial inclusion bodies: Making gold from waste. Trends Biotechnol. 2012, 30, 65–70. [Google Scholar] [CrossRef]
- García-Fruitós, E.; Sabate, R.; de Groot, N.S.; Villaverde, A.; Ventura, S. Biological role of bacterial inclusion bodies: A model for amyloid aggregation. FEBS J. 2011, 278, 2419–2427. [Google Scholar] [CrossRef]
- Peternel, S.; Komel, R. Active Protein Aggregates Produced in Escherichia coli. Int. J. Mol. Sci. 2011, 12, 8275–8287. [Google Scholar] [CrossRef]
- Carrió, M.; González-Montalbán, N.; Vera, A.; Villaverde, A.; Ventura, S. Amyloid-like properties of bacterial inclusion bodies. J. Mol. Biol. 2005, 347, 1025–1037. [Google Scholar] [CrossRef]
- Jahns, A.C.; Maspolim, Y.; Chen, S.; Guthrie, J.M.; Blackwell, L.F.; Rehm, B.H.A. In Vivo Self-Assembly of Fluorescent Protein Microparticles Displaying Specific Binding Domains. Bioconjug. Chem. 2013, 24, 1314–1323. [Google Scholar] [CrossRef]
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hooks, D.O.; Venning-Slater, M.; Du, J.; Rehm, B.H.A. Polyhydroyxalkanoate Synthase Fusions as a Strategy for Oriented Enzyme Immobilisation. Molecules 2014, 19, 8629-8643. https://doi.org/10.3390/molecules19068629
Hooks DO, Venning-Slater M, Du J, Rehm BHA. Polyhydroyxalkanoate Synthase Fusions as a Strategy for Oriented Enzyme Immobilisation. Molecules. 2014; 19(6):8629-8643. https://doi.org/10.3390/molecules19068629
Chicago/Turabian StyleHooks, David O., Mark Venning-Slater, Jinping Du, and Bernd H. A. Rehm. 2014. "Polyhydroyxalkanoate Synthase Fusions as a Strategy for Oriented Enzyme Immobilisation" Molecules 19, no. 6: 8629-8643. https://doi.org/10.3390/molecules19068629
APA StyleHooks, D. O., Venning-Slater, M., Du, J., & Rehm, B. H. A. (2014). Polyhydroyxalkanoate Synthase Fusions as a Strategy for Oriented Enzyme Immobilisation. Molecules, 19(6), 8629-8643. https://doi.org/10.3390/molecules19068629