General
All reactions were performed under nitrogen. EPA ethyl ester was obtained from Pronova Biopharma, Sandefjord, Norway. All other reagents were used as purchased. The NMR spectra were recorded on a Varian Gemini spectrometer. MS (EI) spectra were recorded on an Autospec Ultima instrument and are presented as m/z (% relative intensities). HRMS were recorded on the same instrument. IR spectra were obtained on a reflectance cell on a Perkin Elmer FT-IR instrument. The syntheses of some of the compounds have been previously published. When included here, modifications and/or improvements of yields have been obtained.
5,6-Dihydroxy-(8Z,11Z,14Z,17Z)-icosatetraenoic acid (
11). A solution of the iodolactone (
7) [
3] (12.09 g, 28.2 mmol) and 5% LiOH
.H
2O in MeOH-H
2O (19:1, 120 mL) was refluxed for 6 h. Water (120 mL) was added and most of the methanol was removed under reduced pressure. The reaction mixture was cooled (ice bath) and acidified with dilute HCl. Solid NaCl was added to saturation and the reaction mixture extracted with EtOAc (3 × 50 mL). The organic extracts was washed with brine (2 × 50 mL), dried (Na
2SO
4) and solvent was removed under reduced pressure to obtain
11 (9.46 g; 99%) as a yellow oil. Occasionally small amounts of the corresponding hydoxylactone were seen in the NMR spectra. Spectral data were in agreement with those previously reported [
21].
3Z,6Z,9Z,12Z-pentadecatetraenal (
6). A mixture of the dihydroxy acid
11 (9.55 g, 28.3 mmol) and 5% LiOH
.H
2O in MeOH-H
2O (19:1) (90 mL) was cooled on an ice-bath and left stirring for 30 min. before water (90 mL) was added. A solution of saturated citric acid was added until pH 4 was attained in the reaction mixture. Solid NaIO
4 (7.5 g, 35 mmol) was added in one portion. The reaction mixture was left stirring at room temperature for 1 h. Solid NaCl was added to saturation and the product was extracted with hexane (3 × 50 mL). The extract was washed with brine (2 × 50 mL) and dried (MgSO
4). Evaporation of the solvent under reduced pressure gave the unstable aldehyde
6 (4.88 g; 80%) as a colorless oil. Spectral data were in agreement with those previously reported [
17].
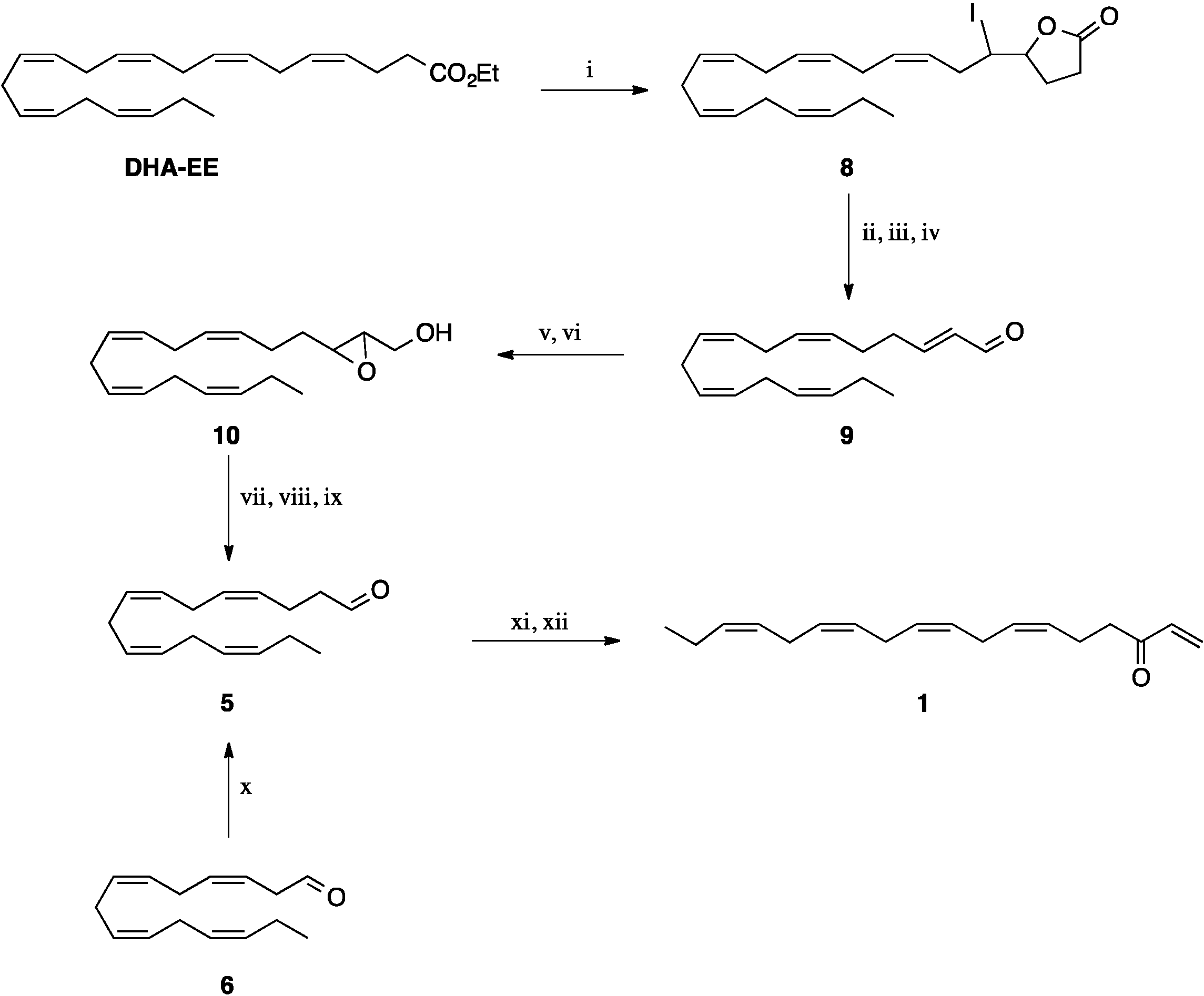
Dihydro-5-((3Z,6Z,9Z,12Z,15Z)-1-iodooctadecapentaenyl)furan-2(3H)-one (
8). A mixture of DHA ethyl ester (10.02 g, 28 mmol) and LiOH
.H
2O (5.8 g, 140 mmol) in EtOH-H
2O (1:1) (60 mL) was left stirring until all the DHA ethyl ester was converted (TLC, CH
2Cl
2). Water (90 mL) were added, the reaction flask was covered with aluminium-foil and cooled to 0 °C. Hydrogen iodide (57%; 20 mL) was added to the reaction mixture, followed successively by saturated KHCO
3 (10 mL) and dropwise addition of a solution of I
2 (21.32 g, 84 mmol) in EtOH (70 mL). The mixture was left stirring at 0–4 °C in the dark for 18 h. The reaction was quenched by adding a saturated aq. solution of Na
2S
2O
3 (100 mL). Solid NaCl was added to saturation and the product extracted with hexane (3 × 50 mL). The extract was washed with brine (2 × 50 mL), dried (Na
2SO
4) and evaporated under reduced pressure to give
8 (12.3 g; 97%) as pale yellow oil. Spectral data were in agreement with those previously reported [
17].
4,5-dihydroxydocosa-(7Z,10Z,13Z,16Z,19Z)-pentaenoic acid (
12). A solution of the iodolactone
8 (9.53 g, 21 mmol) in dry MeOH (110 mL) was cooled to 0 °C and K
2CO
3 (5.8 g, 40 mmol) was added. The mixture was left stirring overnight at room temperature. Water (12 mL) was added followed by a solution of 5% LiOH
.H
2O in MeOH-H
2O (19:1) (90 mL). The mixture was refluxed for 4 h, cooled in an ice bath and acidified with dilute HCl. Solid NaCl was added to saturation and the product extracted with EtOAc (3 × 50 mL). The extract was washed with brine (2 × 50 mL) and dried (Na
2SO
4). Evaporation of the solvent under reduced pressure gave
12 (6.46 g; 85%) as an oil. Spectral data were in accord with the literature [
21]. Small amounts of the corresponding hydroxylactone were inevitably present as shown by the NMR spectra [
18,
22].
2E,6Z,9Z,12Z,15Z-octadecapentaenal (
9). A mixture of the crude dihydroxy acid
12 (6.46 g, 18 mmol) and 5% LiOH
.H
2O in MeOH-H
2O (19:1) (60 mL) was cooled in ice and left stirring for 30 min. Water (60 mL) was added followed by a saturated solution of citric acid until pH 4 was attained. Solid NaIO
4 (5.56 g, 26 mmol) was added in one portion and the reaction mixture left stirring at room temperature for 1 h. Solid NaCl was added to saturation and the reaction mixture was extracted with hexane (3 × 50 mL) The extract was washed with brine (2 × 50 mL) and dried (Na
2SO
4). Evaporation under reduced pressure left a residue that was dissolved in ether (125 mL) and DBU (1 mL) was added. After stirring for 30 min, the organic phase was washed with water to neutral pH, then with brine (2 × 50 mL) and dried (Na
2SO
4). The solvent was removed under reduced pressure furnishing the aldehyde
9 (3.44 g; 74%) as a yellow oil. Spectral data were in agreement with those previously reported [
17].
2E,6Z,9Z,12Z,15Z-octadecapentaen-1-ol (
13) was prepared by reduction of
9 with NaBH
4 according to the literature [
20].
(3-((3Z,6Z,9Z,12Z)-pentadecatetraenyl)oxiran-2-yl)methanol (
10). A solution of the alcohol
13 (2.68 g, 10 mmol) in CH
2Cl
2 (5 mL) was added to solution of Ti(
O-
i-Pr)
4 (3.55 mL, 12 mmol,) in CH
2Cl
2 (15 mL) precooled to −25 °C. The solution was allowed to stir for 20 min at −25 °C before
t-BuOOH (8.72 mL, 30 mmol) was added. The reaction was left stirring overnight and quenched with the addition of 10 % aq. tartaric acid (5 mL) at 0 °C. After stirring for 30 min the solution was filtered trough a short pad of Celite. The reaction mixture was extracted with CHCl
3 (3 × 25 mL). The organic extracts were washed with water (2 × 25 mL) and dried (Na
2SO
4). The residue was passed through a short pad of silica gel via reduced pressure eluting the epoxy alcohol with EtOAc. The epoxy alcohol
10 (1.74 g; 64%) was obtained as a pale yellow oil. The spectral data for
10 were in agreement with those previously reported [
20].
(3-((3Z,6Z,9Z,12Z)-pentadecatetraenyl)oxiran-2-yl)methyl methanesulfonate (14). To an ice cooled solution of the epoxyalcohol 10 (1.71 g, 6 mmol) and 2,6-lutidine (2.1 mL, 18 mmol) in dry CH2Cl2(30 mL) MsCl (1.4 mL, 18 mmol) was added with stirring. The reaction mixture was left stirring for 2 h at room temperature. Brine was added and most of the CH2Cl2 was removed by evaporation under reduced pressure. The residue was extracted with EtOAc (3 × 25 mL). The extract was washed with water (2 × 25 mL), sat. NaHCO3 (2 × 25 mL), brine (2 × 25 mL) and dried (Na2SO4). Evaporation of the solvent under reduced pressure gave a residue that was passed through a short pad of SiO2 and K2CO3 with CH2Cl2 as eluent to give 14 (1.43 g; 68%) as a pale yellow oil. 1H-NMR (300 MHz, CDCl3) δ 5.23ߝ5.47 (m, 8H), 4.46 (dd, J = 3 Hz, J = 12 Hz, 1H), 4.08 (dd, J = 6 Hz, J = 12 Hz, 1H), 3.05 (s, 3H), 2.91(m, 1H), 2.73-2.86 (m, 7H), 2.17–2.26 (q, J = 6 Hz 2H), 2.06 (p, J = 9 Hz, 2H), 1.55–1.76 (m, 2H), 0.95 (t, J = 9 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 132.29 (CH), 129.42 (CH), 128.84 (CH), 128.60 (CH), 128.43 (CH), 128.10 (CH), 128.00 (CH), 127.19 (CH), 70.06 (CH2), 56.36 (CH), 55.20 (CH), 38.07 (CH3), 31.52 (CH2), 25.84 (CH2), 25.80 (CH2), 25.75 (CH2), 23.78 (CH2), 20.78 (CH2), 14.52 (CH3). IR: 3011, 1652 cm−1. HRMS(EI). Calculated for C19H30O4S: 354.1865; Found 354.1896.
(6Z,9Z,12Z,15Z)-2,3-dihydroxyoctadeca-6,9,12,15-tetraenyl methanesulfonate (15). To a solution of the mesylate 14 (1.34 g, 3.8 mmol) in THF (30 mL) a 10% aqueous solution of HClO4 (24 mL) was added dropwise. The mixture was left stirring overnight at room temperature and quenched with a pH 7 phosphate buffer (1 M, 100 mL). The reaction mixture was saturated with solid NaCl and extracted with EtOAc (3 × 25 mL). The combined organic layers was washed with brine (2 × 25 mL) and dried (MgSO4). The residue obtained by evaporation of the solvent under reduced pressure was purified by flash chromatography SiO2, hexane-EtOAc (50:50) furnishing the diol mesylate 15 (0.69 g; 49%) as a pale yellow oil. 1H-NMR (300 MHz, CDCl3) δ 5.25–5.46 (m, 8H), 4.34–4.38 (m, 2H), 3.75–3.83 (m, 1H), 3.67–3.75 (m, 1H), 3.05 (s, 3H), 2.57–2.85 (m, 6H), 2.11–2.40 (m, 3H), 1.97–2.11 (m, 2H), 1.46–1.71 (m, 3H), 0.95 (t, J = 9 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 132.31 (CH), 129.39 (CH), 129.21 (CH), 128.87 (CH), 128.58 (CH), 128.26 (CH), 128.03 (CH), 127.22 (CH), 72.81 (CH), 71.83 (CH), 71.14 (CH), 37.72 (CH3), 32.61 (CH2), 25.84 (CH2), 25.77 (2×CH2), 23.68 (CH2), 20.78 (CH2), 14.49 (CH3). IR: 3511, 3011, 1650 cm−1. MS (EI) m/z (rel. %): 130 (12), 118 (25), 107 (33), 92 (44), 90 (62), 78 (100), 66 (44), 54 (94). HRMS (EI). Calculated for C19H32O5S: 372.1970; Found 372.1990.
4Z,7Z,10Z,13Z-hexadecatetraenal (
5)—Method 1: To a solution of 5% LiOH in MeOH-H
2O (1:1) (20 mL), acidified with saturated citric acid to pH 4, a solution of the diol mesylate
15 (0.64 g, 1.7 mmol,) in MeOH (2 mL) was added. To the reaction mixture, cooled in ice, NaIO
4 (0.45 g, 2.1 mmol) was added and the mixture was left stirring at room temperature for 1 h. After saturation with solid NaCl the reaction mixture was extracted with hexane (3 × 25 mL). The extract was washed with water (2 × 25 mL), brine (2 × 25 mL) and dried (MgSO
4). Evaporation under reduced pressure gave the aldehyde
5 (0.2 g; 50%) as a colorless oil. Spectral data were in agreement with those reported [
20].
4Z,7Z,10Z,13Z-hexadecatetraenal (
5)—Method 2:
t-BuOK (5.5 g, 49 mmol) was added portionwise to an ice-cooled suspension of (methoxymethyl)triphenylphosphonium chloride (17.83 g, 52 mmol) in dry THF (100 mL). After stirring for 15 min at 0 °C, a solution of the aldehyde
6 (5.67 g, 26 mmol) in dry ether (50 mL) was added. The mixture was left stirring at 4 °C overnight. Water (100 mL) was added and volatile compounds were removed under reduced pressure. The reaction mixture was extracted with ether (3 × 50 mL) and the extract was concentrated under reduced pressure. The residue was dissolved in dioxane (225 mL) and cooled to 0 °C. Aq. formic acid (225 mL; 80%) was added and the mixture was left stirring overnight at room temperature. Water (100 mL) was added and volatile compounds were removed under reduced pressure. The reaction mixture was extracted with hexane (3 × 50 mL) and the extract was washed successively with aq. NaHCO
3 (2 × 50 mL), water (2 × 50 mL) and brine (50 mL) and dried (MgSO
4). Evaporation of solvents under reduced pressure followed by flash column chromatography (SiO
2, CH
2Cl
2) gave the aldehyde
5 (3.31 g; 38%) as a colorless oil. Spectral data were in agreement with those previously reported [
20].
1,6Z,9Z,12Z,15Z-octadecapentaen-3-ol (3). A solution of the aldehyde 5 (2.31 g, 10 mmol) in dry Et2O (20 mL) was added dropwise to a solution of vinyl magnesium bromide in THF (20 mL, 1M, 20 mmol) at 0 °C. The mixture was stirred for 1 h at 0 °C and quenched with saturated aq. NH4Cl (30 mL). The product was extracted with Et2O (3 × 25 mL) and the extract dried (MgSO4). Evaporation under reduced pressure gave a residue that was purified by flash chromatography (SiO2, hexane-EtOAc (80:20)) to give the alcohol 3 (4.23 g; 63%) as a colorless oil. 1H-NMR (300 MHz, CDCl3) δ 5.86 (ddd, J = 6 Hz, J = 12 Hz, J = 18 Hz, 1H), 5.27–5.44 (m, 8H), 5.22 (d, J = 18 Hz, 1H), 5.10 (d, J = 9 Hz, 1H), 4.11 (q, J = 6 Hz, 1H), 2.69–2.88 (m, 6H), 1.99–2.21 (m, 4H), 1.47–1.64 (m, 3H) 0.96 (t, J = 6 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 141.29 (CH), 132.29 (CH), 129.66 (CH), 128.79 (CH), 128.74 (CH), 128.48 (CH), 128.41 (CH), 128.15 (CH), 127.26 (CH), 114.99 (CH2), 72.93 (CH), 36.99 (CH2), 25.87(CH2), 25.78 (2×CH2), 23.46 (CH2), 20.79 (CH2), 14.51 (CH3). IR: 3347, 3011, 1645 cm−1. MS (EI) m/z (rel. %): 130 (12), 116 (20), 107(28), 104 (32), 92(40), 90(65), 78(100), 66 (51), 54 (38), 40(52). HRMS (EI). Calculated for C18H28O: 260.2140; Found 260.2141.
1,6Z,9Z,12Z,15Z-octadecapentaen-3-one (
1). A solution of the alcohol
3 (0.31 g, 1.2 mmol) in CH
2Cl
2 (5 mL) was added to a suspension of Dess-Martin periodane (DMP) (0.64 g, 1.5 mmol) in CH
2Cl
2 (30 mL) at room temperature. The mixture was stirred for 1 h at room temperature. Saturated aq. KHCO
3 (30 mL) and 10% saturated aq. Na
2S
2O
3 (30 mL) were added, and the product was extracted with CH
2Cl
2 (3 × 10 mL). The extract was washed successively with water (2 × 10 mL) and brine (2 × 10 mL) and dried (MgSO
4). Evaporation of the solvent under reduced pressure gave a residue that was purified by flash chromatography (SiO
2, hexane-EtOAc (80:20)) to give
1 (0.27 g; 89%) as a colorless oil.
1H-NMR (300 MHz, CDCl
3) δ 6.36 (dd,
J = 12 Hz,
J = 18 Hz, 1H), 6.21 (dd,
J = 3 Hz,
J = 18 Hz, 1H
trans), 5.83 (dd,
J = 3 Hz,
J = 9 Hz, 1H
cis), 5.24–5.43 (m, 8H), 2.75–2.87 (m, 6H), 2.63 (t,
J = 9 Hz, 2H), 2.33–2.43 (m, 2H), 2.00–2.17 (m, 2H), 0.95 (t,
J = 9 Hz, 3H).
13C-NMR (75 MHz, CDCl
3) δ 200.25 (C=O), 136.72 (CH), 132.29 (CH), 129.24 (CH), 128.78 (CH), 128.48 (2×CH), 128.26 (2×CH), 128.05 (CH), 127.21 (CH), 39.56 (CH
2), 25.82 (CH
2), 25.78 (CH
2), 25.74 (CH
2), 21.92 (CH
2), 20.75 (CH
2), 14.46 (CH
3). IR: 3011, 1702, 1682, 1615 cm
−1. MS (EI)
m/z (rel. %): 130 (14), 118 (28), 107 (39), 92 (48), 90 (66), 78 (100), 66 (45), 54 (95). HRMS (EI) Calculated for C
18H
26O: 258.1984; Found 258.1995. Spectral data were in accord with those reported for the natural product [
2].


{kind=link}
{kind=link}
{kind=link}