1. Introduction
Single-molecule experiments have a rich history in both life and physical science investigations [
1,
2,
3,
4,
5,
6,
7,
8,
9]. Experiments capable of quantifying molecular motion with nanoscale resolution continue to be of interest to scientists and engineers (as partially evident by this Special Issue). The ability to experimentally quantify the motion of single-molecules without ensemble averaging has enabled researchers to gain various new insights about the kinetics of molecular interactions [
10,
11]. Single-molecule experiments typically produce a collection of “trajectories” (a time ordered sequence of force or position measurements) containing rich amount of temporal and spatial multiscale information; the desire to extract reliable quantitative information from these trajectories has inspired a variety of new computational algorithms, e.g., [
12,
13,
14,
15,
16,
17,
18,
19,
20].
A surge of publications in optical microscopy techniques applied to monitor single-molecules in live cells [
11,
21,
22,
23,
24,
25,
26,
27,
28] has generated much excitement because recent advances in optical imaging allow researchers to (relatively) noninvasively monitor biological molecules in their native environment. With both
in vitro and
in vivo single-molecule measurements, researchers must account for various complex features including inherent thermal fluctuations, inter- and intra-trajectory “heterogeneity” (induced by unresolved conformational degrees of freedom and/or a time changing micro-environment [
29]), statistical artifacts introduced by the experimental apparatus, amongst other complications [
10,
11]. Quantifying the aforementioned “heterogeneity” to gain new insights on the system is often the motivation for carrying out a single-molecule study, but this feature of the data also severely complicates statistical analysis. For example, in current single-molecule studies, researchers typically only measure a point-like position of a fluorescently tagged molecule. Factors such as the molecule’s underlying conformation and/or if the tagged molecule is bound to another molecular complex in the cell tend to strongly influence the dynamics of position measurements, but these latent factors often cannot be directly observed in typical single-molecule experiments and need to be inferred from position versus time data (and the latent factors, or “kinetic states”, can vary substantially within and between trajectories).
In the earlier works, the spatial and temporal resolution afforded by the measurement device led researchers to focus mainly on Mean-Square-Displacement (MSD) type analyses to analyze single-molecule data [
2,
5,
30,
31]. MSD approaches have many undesirable features, namely they tend to introduce unnecessary temporal averaging (
i.e., they ignore the natural time ordering of the trajectory measurements) and they have a difficult time accounting for spatially varying forces (a common occurrence in live cells [
29]). Advances in spatial and temporal resolution have inspired many researchers to develop new techniques for reliably extracting single-molecule level information out of measurements [
12,
13,
14,
15,
16,
17,
18,
19,
20]. The previously cited works are most similar in spirit to the work presented, but all of the works encounter technical difficulties when there is an abrupt latent “state change” occurring in a molecule experiencing spatially dependent forces in a live cell environment (additional complications arise when position estimates are obscured by non-negligible “measurement noise”).
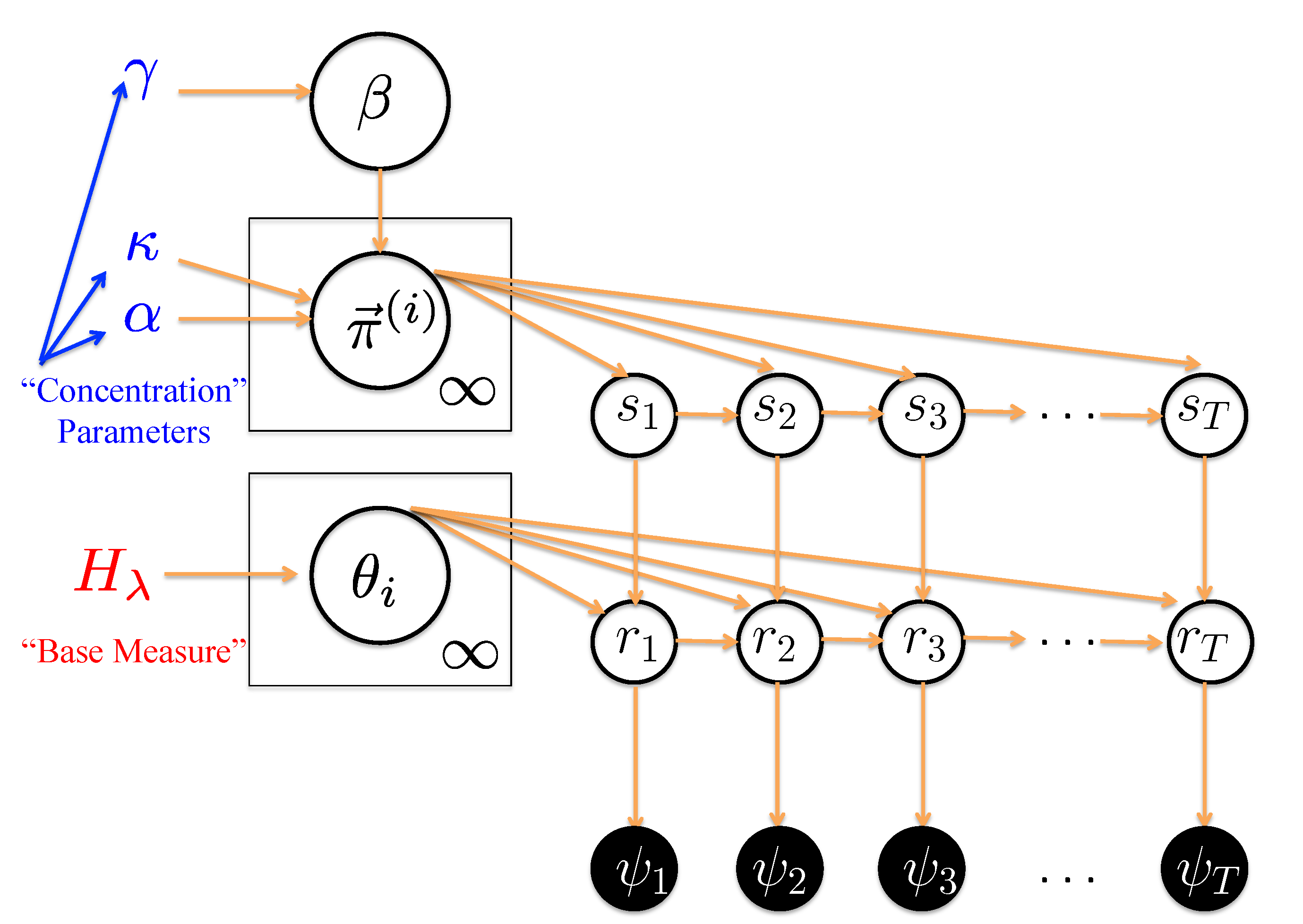
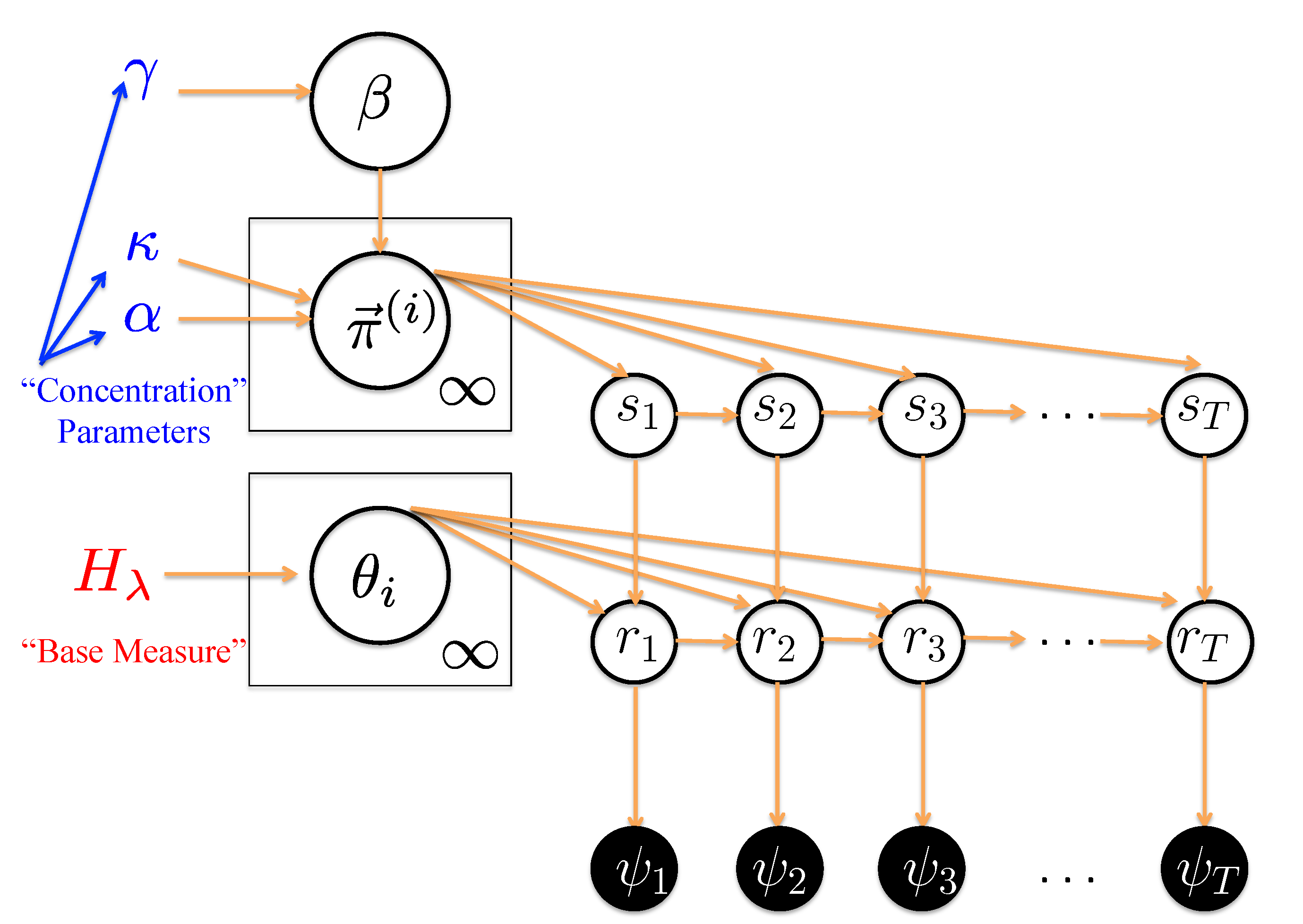
We demonstrate and discuss the utility of Hierarchical Dirichlet Process Switching Linear Dynamical System (HDP-SLDS) developed by Fox
et al. [
32] in identifying abrupt “state changes” where the number of states is unknown in advance, observations are corrupted by measurement noise, and the force or velocity field experienced by the molecule varies with position. An attractive novel feature of the HDP-SLDS approach is the joint estimation of the number of underlying latent states implied by the data along with kinetic parameter estimates. When estimating parameters, the likelihood function employed by the HDP-SLDS correctly accounts for the temporal and spatial statistical dependencies implied by a piecewise linear stochastic dynamical model. Other specific advantages over pre-existing approaches are discussed and illustrated through simulation examples motivated by Single Particle Tracking (SPT) experiments. Although we focus on simulations of SPT data, the basic idea behind the technique is anticipated to be applicable to a variety of single-molecule applications. In a companion paper, we illustrate how the technique can be applied to assist in the analysis of live yeast cells undergoing mitosis [
33].
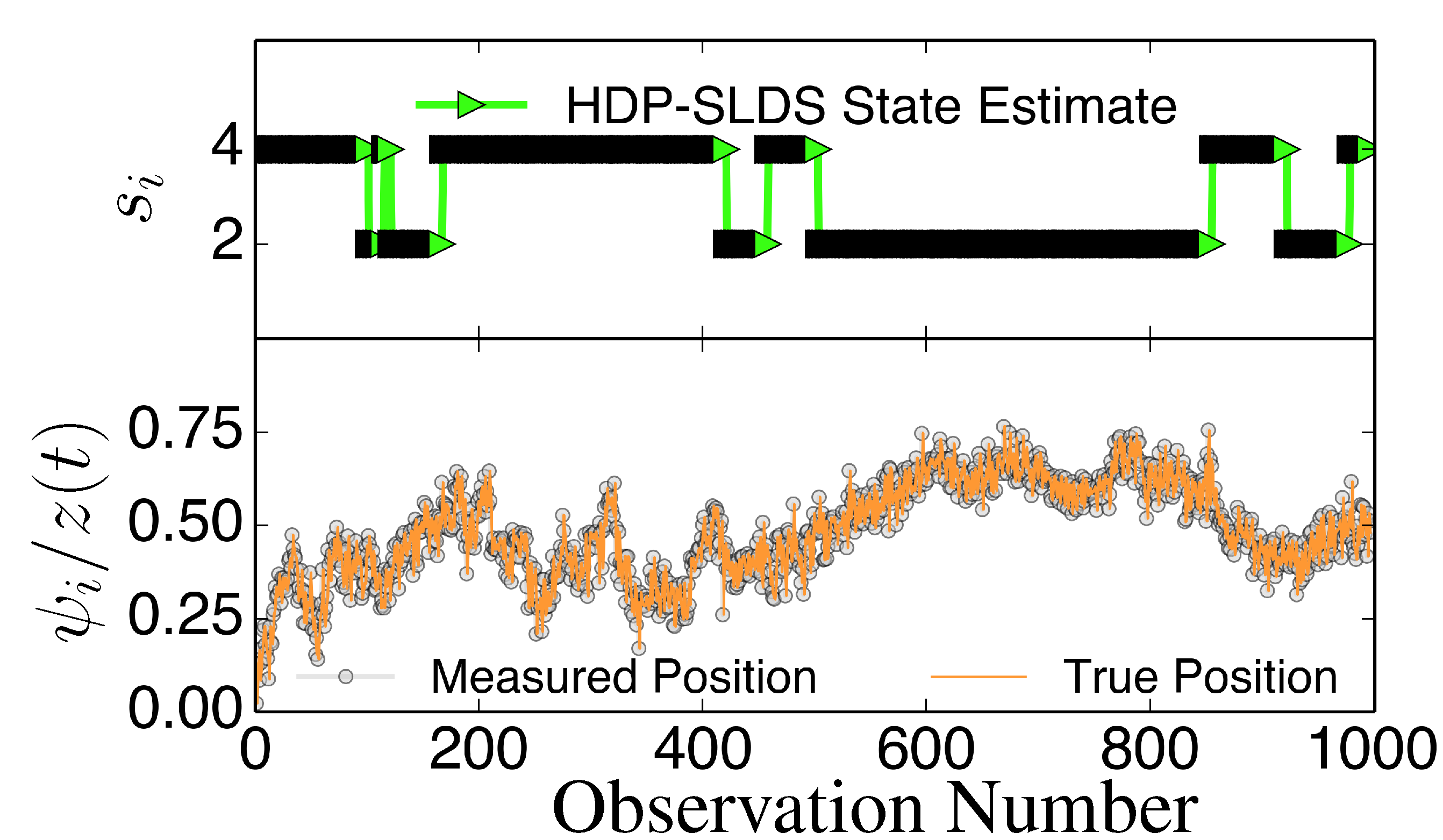
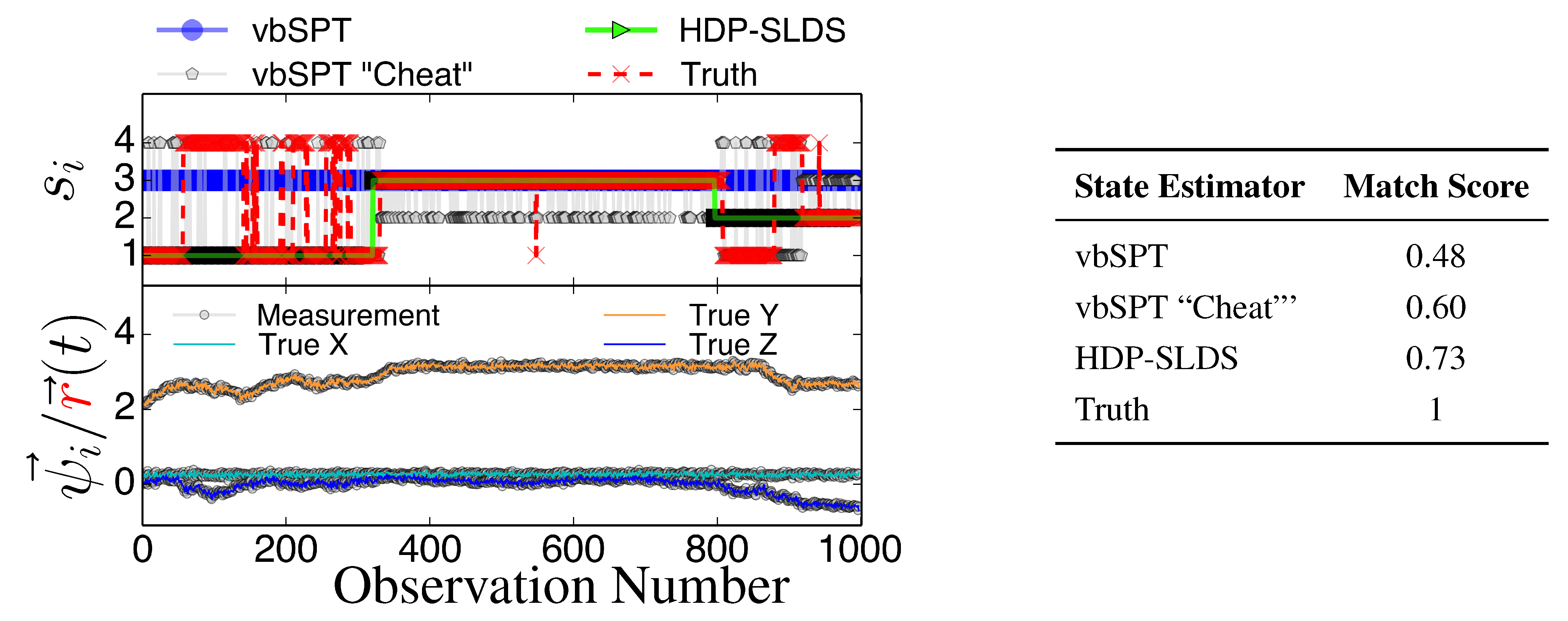
3. Results and Discussion
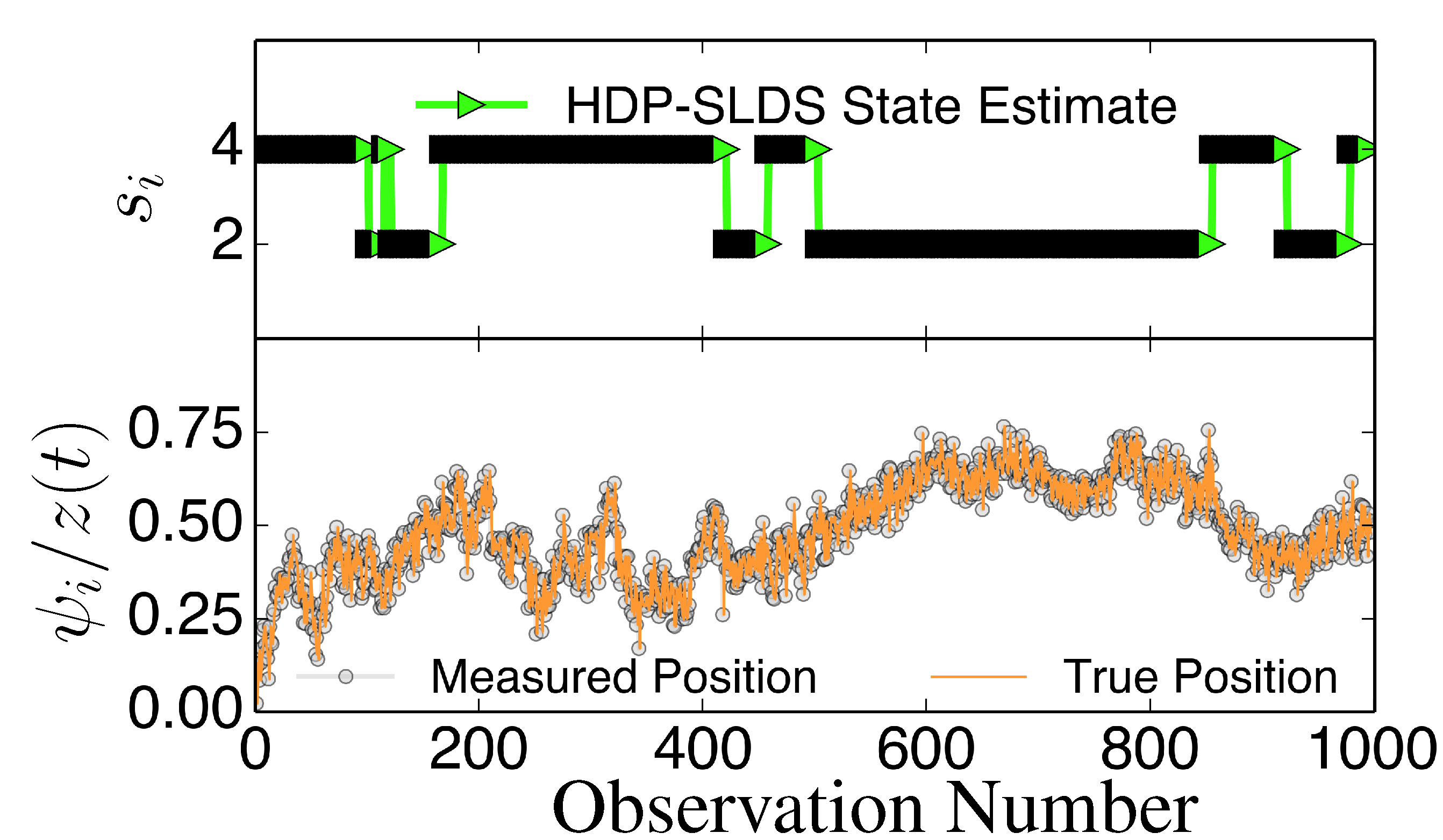
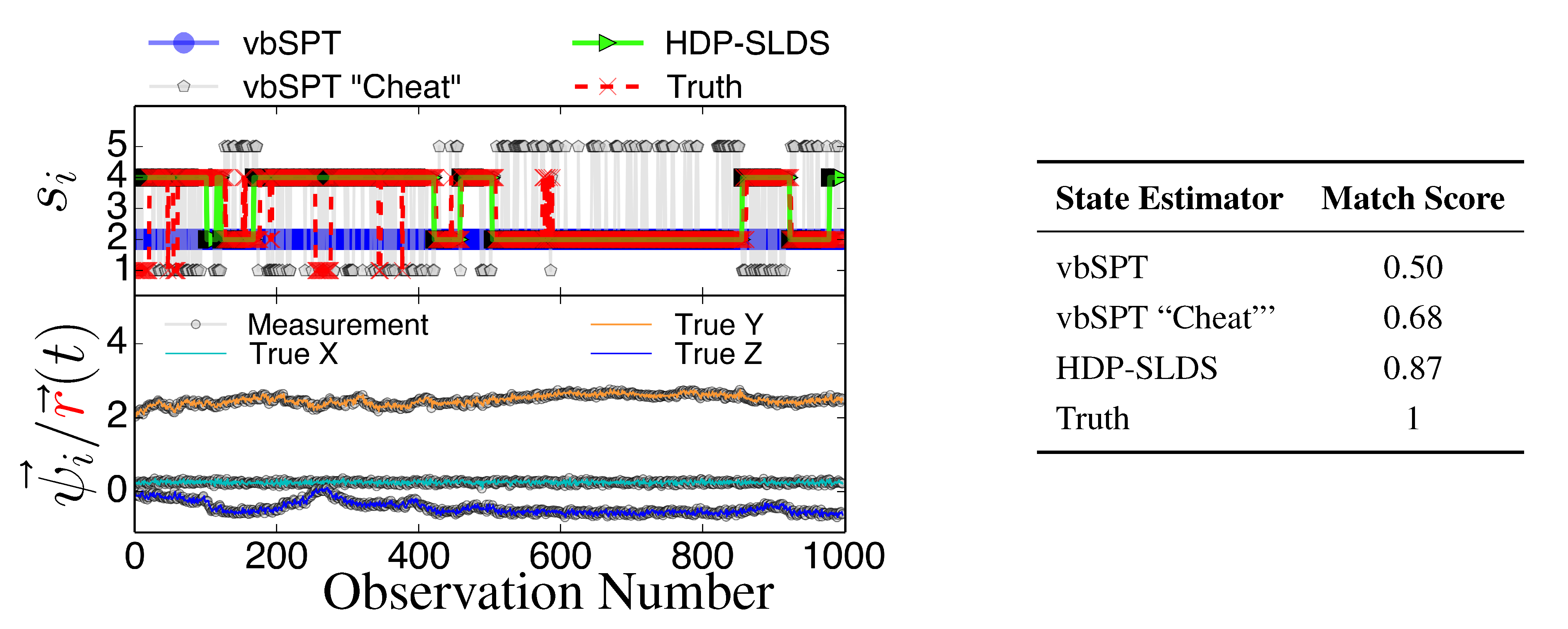
Figure 3 and
Figure 4 display two representative trajectories of
,
ψ⃗ (bottom panel) along with the true/estimated state sequence (top panel). A table is provided to the right of the trajectory plots where the “Match Score” quantifying the quality of the HDP-SLDS [
32] and vbSPT [
17] state estimators applied to the displayed trajectory is reported. The “Match Score” is defined as equal to one minus the average Hamming distance and a “Match Score” of 1 denotes perfect performance. The Hamming distance indicates the sum of the number of correct state assignments; the average Hamming distance divides the sum by the length of the time series. Hence an average Hamming distance of 0 denotes a situation where the algorithm matched states precisely and 1 denotes a situation where not a single state was matched correctly. Recall that the vbSPT technique is a variational approximation to a classic HMM model; also note that the current publicly available software implementation of vbSPT does not account for all statistical effects induced by Gaussian measurement noise and vbSPT relies on post analysis model selection criteria to select the number of hidden states.
Figure 3.
State estimates for three different state estimators (see text for description) along with the true state sequence (top panel), the 3D trajectory showing unobservable position and observable position (bottom panel), and the table quantifying the performance of the state estimators through the “Match Score”, which is defined as one minus the average Hamming distance for the trajectory.
Figure 3.
State estimates for three different state estimators (see text for description) along with the true state sequence (top panel), the 3D trajectory showing unobservable position and observable position (bottom panel), and the table quantifying the performance of the state estimators through the “Match Score”, which is defined as one minus the average Hamming distance for the trajectory.
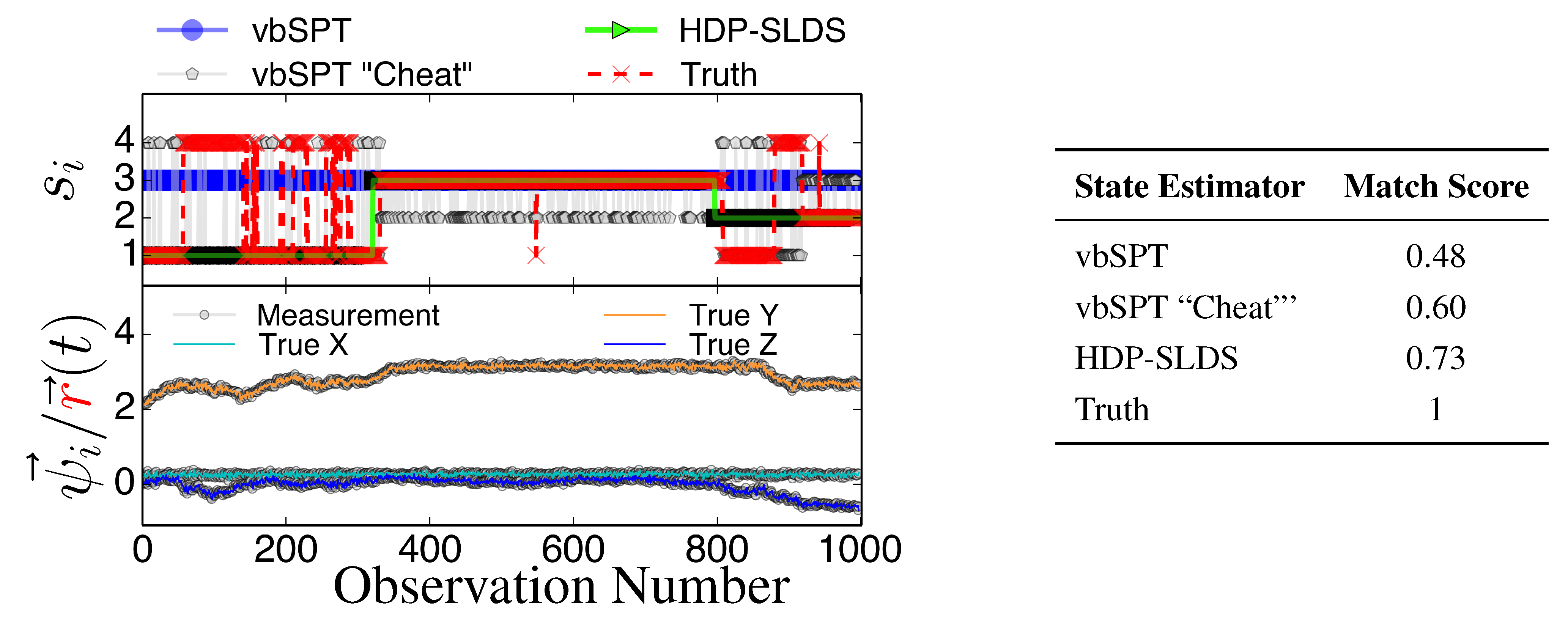
Figure 4.
Same as
Figure 3 except a new trajectory is analyzed where a different sequence of states is sampled.
Figure 4.
Same as
Figure 3 except a new trajectory is analyzed where a different sequence of states is sampled.
All estimators in
Figure 3 and
Figure 4 were provided priors having the mean diffusion coefficient and measurement noise parameters matching the DGP exactly. The technique labeled as “vbSPT” processed
ψ⃗ measurements directly (and used model selection criteria to find the best model containing 1–10 states) and that labeled “vbSPT Cheat” was carried out similarly, but the algorithm processed
directly (“Cheat” is used to label this estimator because in practice one cannot avoid measurement noise when analyzing laboratory data). The HDP-SLDS method estimated states by only analyzing a single long trajectory of
ψ⃗ containing 1000 observations. The vbSPT method was allowed to “pool” ten long trajectories (the collection provided an adequate representation of the four underlying states in the DGP) in an attempt to help this algorithm’s performance.
The HDP-SLDS method is able to quickly identify long lived state sequences, however it has the most difficultly in quickly identifying changes between State 1 and State 4 (where
changes abruptly). Large scale simulations shown later quantify the transition between the various states more precisely. The “vbSPT” case consistently only estimates one state. However, it should be emphasized that the approach advocated in [17] was not designed to explicitly account for measurement noise, changing
type parameters, or spatially varying forces. The vbSPT algorithm’s aim was to identify changes in diffusion coefficients in scenarios where measurement or localization effects are negligible in relation to the diffusion coefficient. The vbSPT technique was originally motivated to study a large collection of short SPT trajectories where it is not practical to estimate effective forces (in contrast to other SPT studies [
29,
33,
42]).
Figure 3 and
Figure 4 also illustrate how the approach labeled as “vbSPT Cheat” can identify the occurrence of state changes when measurement noise is removed in most situations, but in the situation studied, the “vbSPT Cheat” rapidly switches between two states for each single true state (rapid state switching is intentionally suppressed in the HDP-SLDS approach due to the use of “sticky” parameters [
32]). We elected to compare the HDP-SLDS approach to vbSPT because this approach was most similar in spirit to the HDP-SLDS; the latter is better suited to long trajectories and the former is tailored to simultaneously analyzing a large collection of short trajectories (note: when measurement noise is not subtracted, the vbSPT method consistently estimated only one state in the scenarios studied despite 2–4 states being present in each trajectory).
For the remainder of this paper, we focus almost exclusively on the HDP-SLDS results since we aim to show its utility in extracting detailed information out of states representative of classic modes of motion [
31,
43] (
i.e., “directed diffusion”, “confined diffusion”, “pure diffusion”). Note that the “pure diffusion” case is technically a stationary process with very weak mean reversion. All results that follow analyze a fixed collection of 500 trajectories each containing 1000 uniformly spaced observations. The HDP-SLDS is applied to single trajectories (
i.e., trajectories are not pooled). In each run, prior parameters are altered, but the same set of 500 trajectories are analyzed/re-analyzed under different HDP-SLDS “tuning parameters”.
Table 1 displays the average Hamming distance (recall this number is between 0 and 1, with 0 denoting a perfect fit) observed in the population of 500 trajectories obtained after 10,000 Markov Chain Monte Carlo (MCMC) draws were generated to make state assignments. The runs labeled as “Baseline” use the known diffusion coefficient and measurement noise of the DGP as the mean of the inverse Wishart prior parameters used in the HDP-SLDS analysis; the case labeled
D/4 divides the known average of the DGP and uses this as the average in the inverse Wishart prior over
D (similarly for the measurement noise covariance,
R). We also show the vbSPT results obtained when the exact DGP parameters are provided to the algorithm. (Recall that this algorithm was not tailored for this type of data and it consistently picks one state; however, the vbSPT technique is the most similar approach to the HDP-SLDS commonly currently used by the SPT community in the author’s opinion.) As can be readily observed (and as stated in [
36]), the base measure parameters can strongly influence the state segmentation inference and a “properly tuned” HDP-SLDS state estimator can have impressive performance in detecting subtle changes in trajectories containing spatially dependent forces, thermal noise, and measurement noise. Fortunately, tools exist for approximating trajectory-wise statistics on 2D and 3D trajectories [
29] (such tools can be used to construct data-driven priors and base measure parameters; however this topic is covered elsewhere [
33]).
Table 2 confirms that varying the primary “concentration parameters” associated with the HDP-SLDS [
32] has little effect on the state segmentation results.
Table 1.
Effects of misspecifying “Base Measure” parameters. The average Hamming distance (a number between 0 and 1, with 0 indicating a perfect match) measured over 500 trajectories each of length 1000 (empirical standard errors indicated in parenthesis). The cases in the leftmost column are described in the text.
Table 1.
Effects of misspecifying “Base Measure” parameters. The average Hamming distance (a number between 0 and 1, with 0 indicating a perfect match) measured over 500 trajectories each of length 1000 (empirical standard errors indicated in parenthesis). The cases in the leftmost column are described in the text.
| Case | Hamming Dist. |
|---|
| Baseline | 0.16 (0.03) |
| D/4 | 0.31 (0.04) |
| R/4 | 0.39(0.04) |
| vbSPT | 0.28 (0.04) |
Table 2.
Effects of misspecifying “Concentration Measure” parameters containing same information as in the previous table.
Table 2.
Effects of misspecifying “Concentration Measure” parameters containing same information as in the previous table.
| Case | Hamming Dist. |
|---|
| Baseline (
γb = 0.01; ρc= 25) | 0.16 (0.03) |
| γb = 0.001 | 0.17 (0.03) |
| γb = 0.1 | 0.15 (0.03) |
| ρc = 100 | 0.16 (0.03) |
| ρc = = 5 | 0.18 (0.03) |
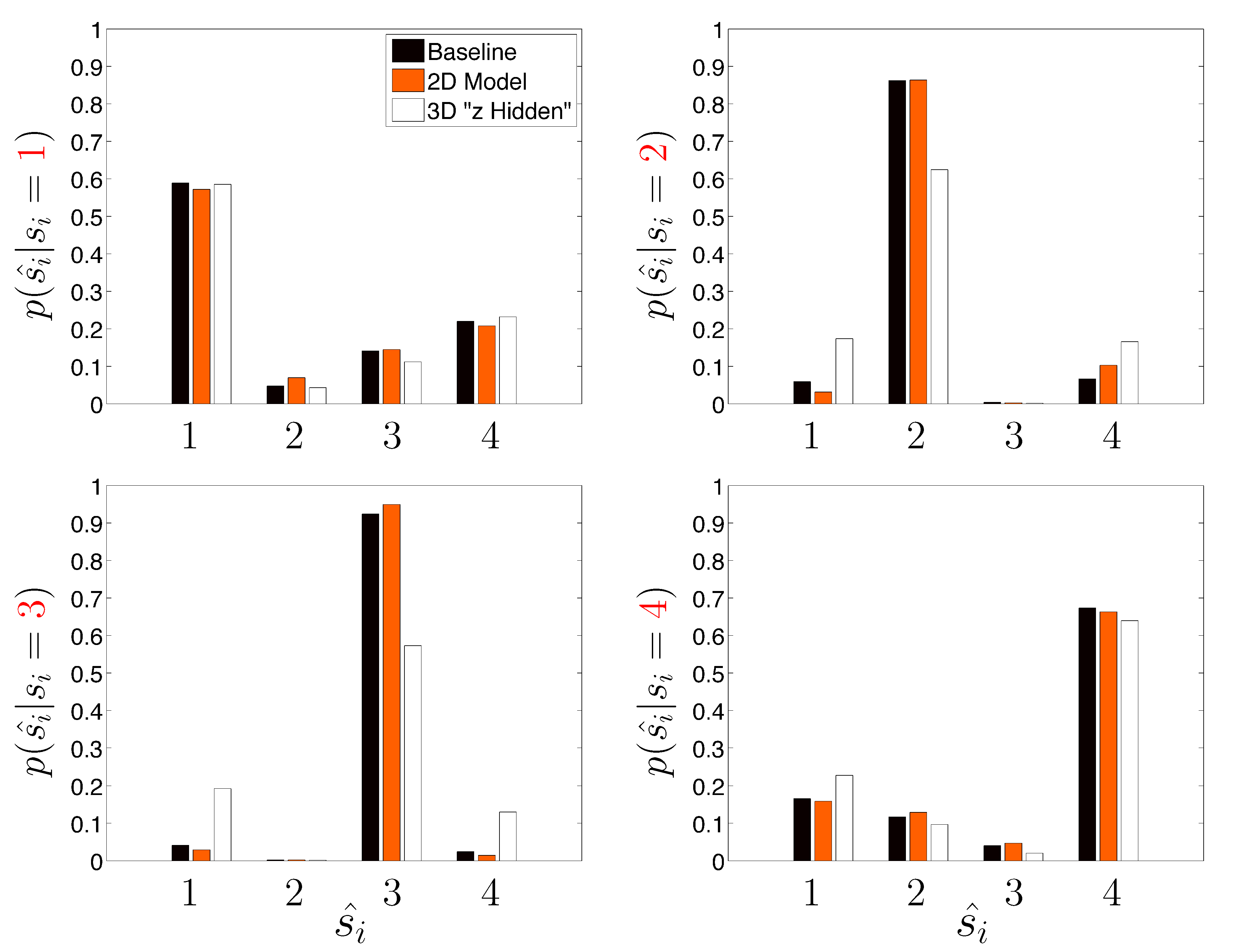
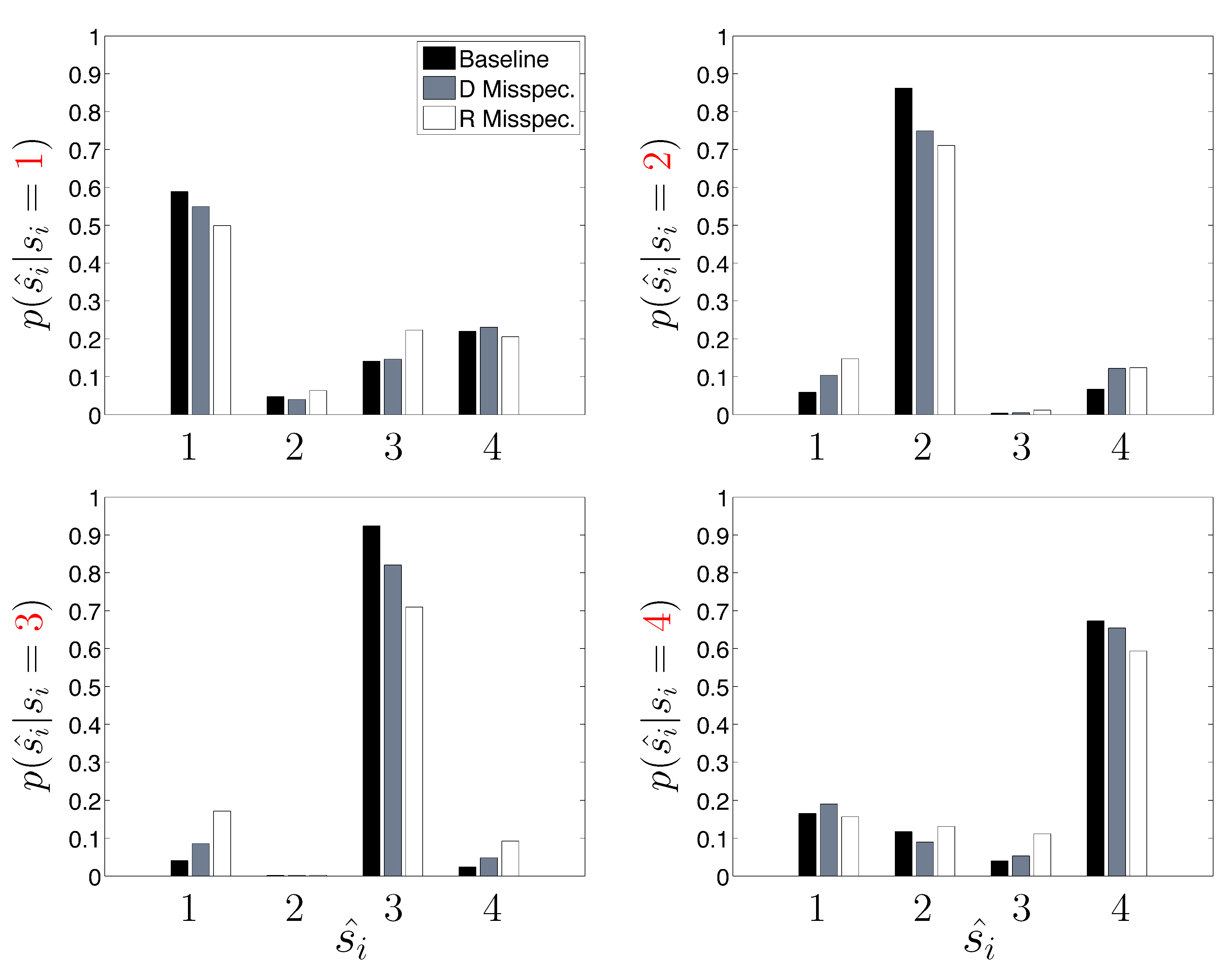
Next, we take a closer look at the error committed by the three HDP-SLDS analyses shown previously when trying to identify the four latent states used by the DGP (
Table 1 reported only the overall average Hamming distance). In
Figure 5, the empirical probability of state assignment (using the three HDP-SLDS methods used in
Table 1) is computed using the known underlying state of the DGP. Previously, in
Figure 4, we qualitatively demonstrated that abrupt and transient changes in
were difficult to identify (
i.e., see transitions from State 1 to State 4 and back occurring near observations 1–250). This is because the process mean changes quickly, but the position (and hence measurement) takes time to adjust to the new mean location (or the new “energy well minimum” if one wants to use the harmonic spring analogy) and the inference algorithm needs to accumulate sufficient evidence before it declares the existence of a new state.
Figure 5 quantifies this phenomenon more accurately using a large population of trajectories. Abrupt changes in the diffusion coefficient (State 3) and confinement parameters (State 4) are more readily correctly identified by the HDP-SLDS algorithm. This plot also gives a finer grained picture of how an “improperly tuned” prior quantitatively affects state estimation.
Figure 5.
A finer breakdown of the HDP-SLDS performance as a function of the known underlying state for three different conditions studied in
Table 1. The y-axis shows the empirical conditional probability of the state estimate
ŝi (x-axis) conditioned on the true (known) underlying state
si (the panels vary over the four truth states used by the simulated data generating process).
Figure 5.
A finer breakdown of the HDP-SLDS performance as a function of the known underlying state for three different conditions studied in
Table 1. The y-axis shows the empirical conditional probability of the state estimate
ŝi (x-axis) conditioned on the true (known) underlying state
si (the panels vary over the four truth states used by the simulated data generating process).
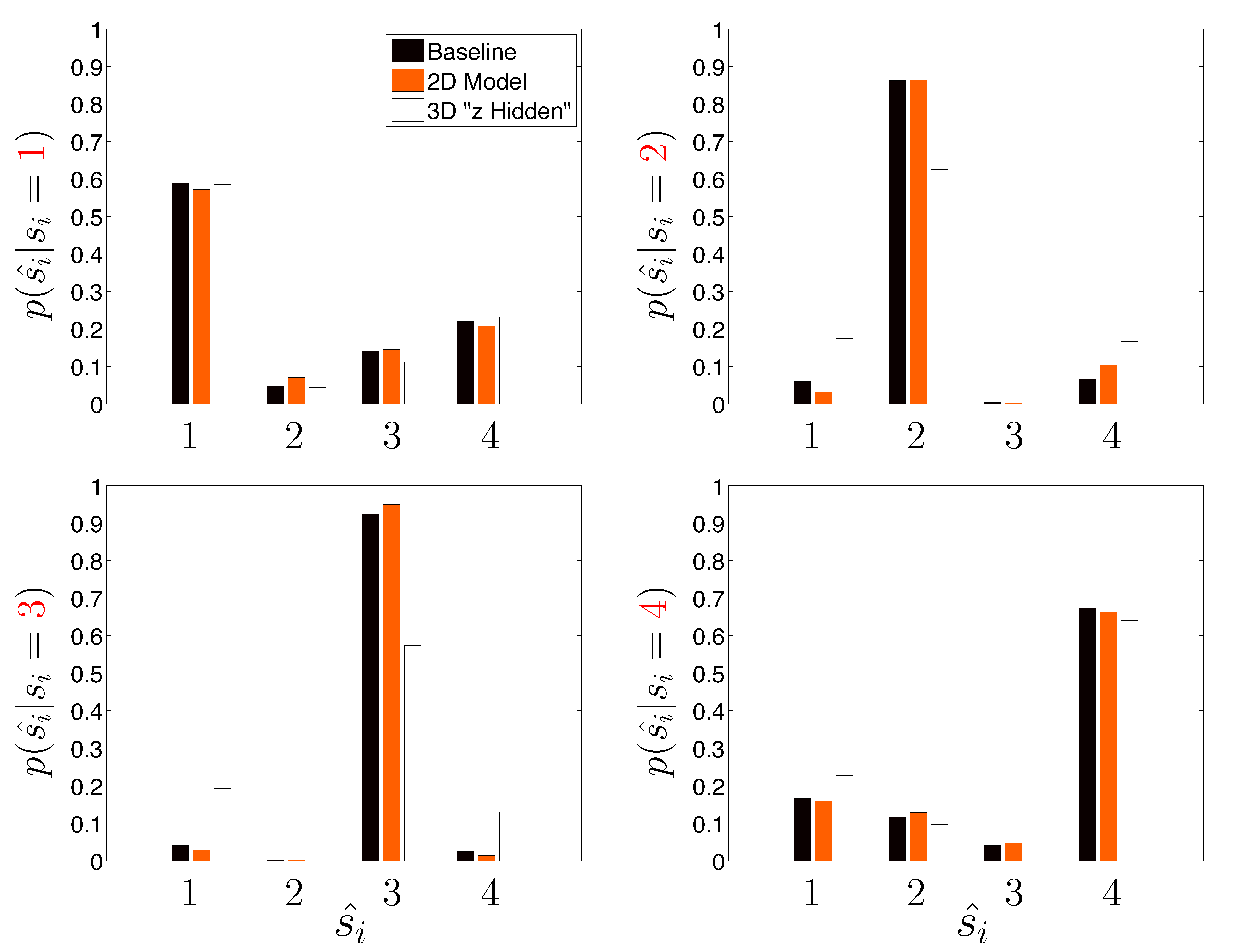
Table 3 assumes that the DGP used for the priors are known precisely (an admittedly unrealistic situation) and re-analyzes the same set of 500 trajectories, except this time the algorithm is only presented in
x and
y measurements. The axial
z dimension is considered unobserved (
i.e., the only available data is a time ordered sequence of paired
ψx and
ψy measurements); this situation is commonly encountered in SPT. However, recent advances in optical microscopy show promise in more accurately measuring long 3D trajectories [
21,
25,
27,
43]. The approach labeled “Naive 2D Model” considers the state to be a two-dimensional vector (
i.e., effects of
z are not explicitly computed in the likelihood function of the HDP-SLDS) and the approach labeled “3D Model (Hidden
z)” considers a Kalman filter where there is a three-dimensional state vector but the observation process is two-dimensional. Note how the “Naive 2D Model” slightly improves on the “Baseline” case in terms of the average Hamming distance. The reduction in dimension of the parameter vector characterizing the base measure governing the stochastic model improves the joint state and kinetic parameter inference in the scenario studied. A somewhat surprising result is the unambiguous statistically significant degradation in state segmentation obtained when the effects of
z were attempted to be accounted for in by the state space model. The fact that there were no off-diagonal terms in
and Σ account partially for the strength of the degradation, but we include this example to show that “more is not always better” (
i.e., attempting to explicitly model known, but unobservable, coordinates can be potentially detrimental to state segmentation results).
Figure 6 shows results analogous to
Figure 5 for the “Naive 2D Model” and “3D Model (Hidden
z)” model cases studied.
Table 3.
Effects of model’s state dimensionality. The average Hamming distance (a number between 0 and 1, with 0 indicating a perfect match) measured over 500 trajectories each of length 1000 (empirical standard errors indicated in parenthesis).
Table 3.
Effects of model’s state dimensionality. The average Hamming distance (a number between 0 and 1, with 0 indicating a perfect match) measured over 500 trajectories each of length 1000 (empirical standard errors indicated in parenthesis).
| Baseline | 0.16 (0.03) |
| “Naive” 2D Model | 0.12 (0.03) |
| 3D Model (Hidden z) | 0.46 (0.04) |
Figure 6.
Same as
Figure 5, except that effects of dimensionality of the underlying state model are investigated (see description in text).
Figure 6.
Same as
Figure 5, except that effects of dimensionality of the underlying state model are investigated (see description in text).
4. Conclusions
We demonstrated how the HDP-SLDS method can model simulations mimicking 3D single-molecule data. The technique was demonstrated by analyzing a large collection of long control simulation trajectories containing a varying mix of classical SPT “modes of motion” [
2,
5,
44] as well as more difficult to detect changes (e.g., abrupt change in the spatial location of a “harmonic well-minimum” where statistical time correlation in the relaxation to the new harmonic well-minimum is non-negligible). Parameters selected were motivated by studies of transmembrane protein kinetics in the primary cilium [
42]. It was shown that the HDP-SLDS framework can systematically account for spatially varying forces, the statistical effects of measurement noise, and an
a priori unknown number of underlying latent states where other methods encountered problems due to neglecting key statistical features or making unnecessary approximations. The HDP-SLDS can obtain state-of-the-art segmentation results using only a single “long” trajectory (
i.e., one containing many time samples). For situations where there is benefit to pooling information from multiple long trajectories, alternative approaches similar in spirit to the HDP-SLDS show promise in single-molecule analysis [
45]. The HDP-SLDS and other nonparametric Bayesian approaches extracting information from long time ordered sets of measurements [
45] are nice complements to the technique of Persson
et al. [
17], which aims at pooling kinetic information from multiple short trajectories to identify the number of states. However, it should be noted that in the analysis of single-molecule data, a small finite set of discrete states describing a trajectory (or groups of trajectories) may not always be an appropriate representation of data measured in complex heterogeneous environments [
29]. In cases where a small set of discrete SLDS states (driven by standard diffusive noise) can be informative about the underlying single-molecule system and one has “long” trajectories, the HDP-SLDS approach is useful because it is capable of producing accurate state estimation and temporal segmentation when compared with other state segmentation routines used in SPT data analysis. The HDP-SLDS approach also provides a systematic framework for the “time window” selection problem mentioned in [
29]. Note that the HDP-SLDS method has been successfully applied to experimental SPT trajectories containing as few as 150 observations uniformly sampled at 22 frames per second [
33].
Despite the fact that the HDP-SLDS technique is labeled as a nonparametric Bayesian method, we demonstrated that the parameters characterizing the base measure can still heavily influence state estimation and segmentation results (we also presented results confirming that sensitivity to the concentration parameters and hyperparameters is minimal [
36] in the situations studied). The “nonparametric Bayesian” monicker attached to the HDP-SLDS is slightly misleading since the base measure depends heavily on an SDE model with an SLDS parametric structure; the model also has priors depending on a parametric structure. Prior parameter sensitivity is not unique to the HDP-SLDS approach; priors and hyperparameters affecting algorithm performance is typically common amongst Bayesian approaches [
15,
17]. Other approaches that are closer to a “nonparametric” spirit are potential alternatives (e.g., anomalous and standard diffusion driven models can be considered as in [
18]), but such methods can encounter technical difficulties when faced with trajectories where velocity or forces are spatially dependent and the measured signal contains inherent “thermal noise” as well as measurement noise.
If accurate quantitative information about single-molecule trajectories are not available
a priori (a common situation in single-molecule analysis), techniques for extracting data-driven base measure and priors parameters in a “single-molecule fashion” can be considered (see a companion manuscript [
33]). Note also that goodness-of-fit testing can be leveraged to assess the fundamental HDP-SLDS assumptions against data without “ground truth ” available [
29,
33]; this feature is useful since in the analysis of live cell experimental data, one does not typically have the luxury of “ground truth”. In such situations, it becomes important to determine if there is adequate statistical evidence in the data to justify one segmentation over another. After a good segmentation is believed to be in hand, one can then attempt to refine parameters estimates characterizing the motion of the single-molecule trajectory [
33]. Hence using nonparametric Bayesian ideas (such as the HDP-SLDS) along with frequentist ideas (such as those in [
29]) shows great promise in reliably extracting new quantitative information from single-molecule data [
33].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}