Synthesis and Broad-Spectrum Antiviral Activity of Some Novel Benzo-Heterocyclic Amine Compounds

Abstract
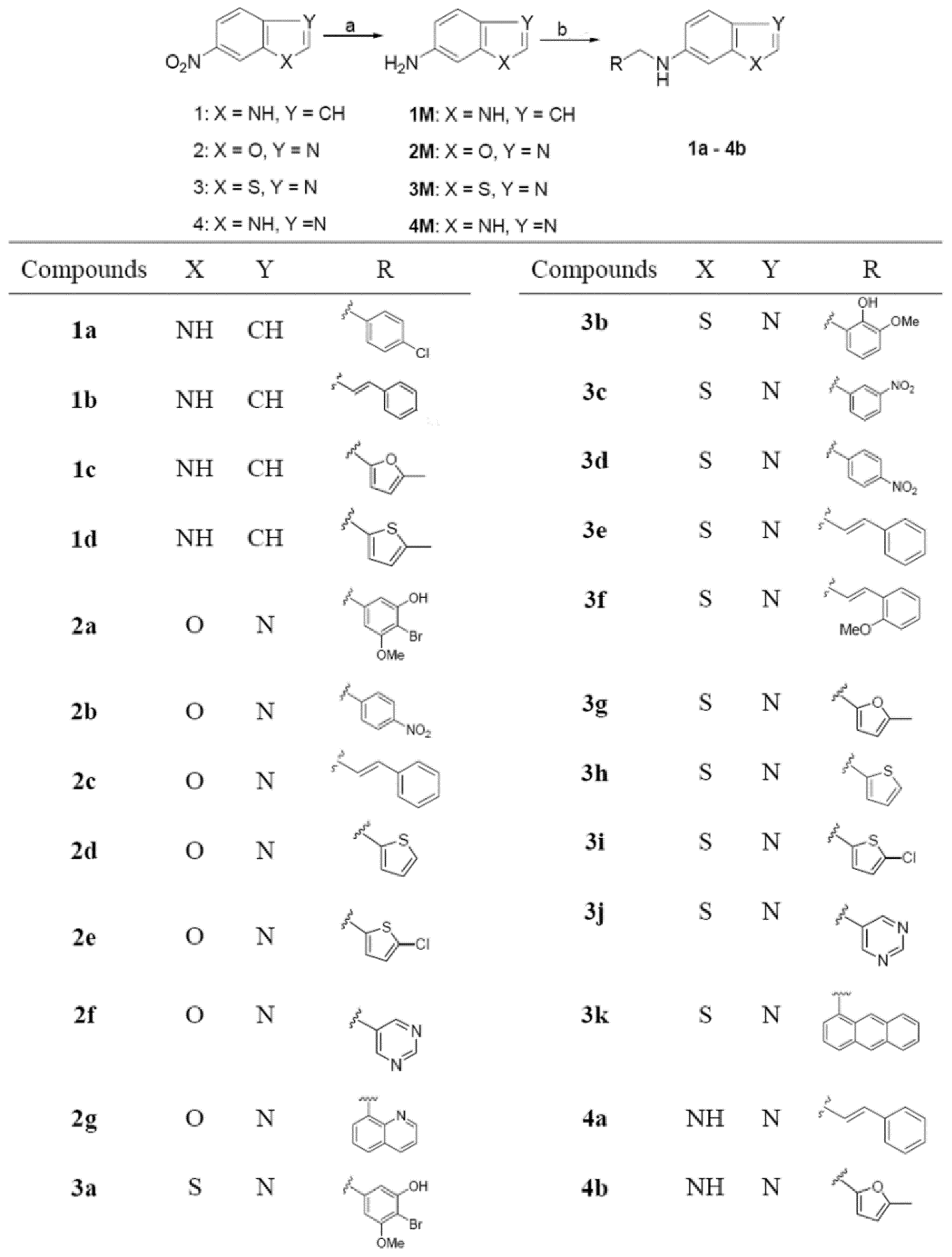
:1. Introduction


2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Anti-influenza Virus Activity

{kind=link}
{kind=link}
| NO. | Influenza A/hanfang/359/95 | Cox B3 | HCV | HBV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TC50 (μM) | IC50 (μM) | SI | TC50 (μM) | IC50 (μM) | SI | TC50 (μM) | IC50 (μM) | SI | TC50 (μM) | IC50 (μM) | SI | |
| 1a | 3.35 | >1.60 | - | 8.34 | 4.79 | 1.7 | 15.88 | 1.40 | 11.3 | 8.02 | 1.44 | 5.6 |
| 1b | 2.86 | >1.65 | - | 8.61 | 2.86 | 3.0 | NT | NT | NT | 3.21 | >1.85 | - |
| 1c | 4.52 | >0.66 | - | 25.15 | 1.93 | 13.0 | 46.78 | 2.77 | 16.8 | 16.67 | >5.56 | - |
| 1d | 26.50 | 11.84 | 2.2 | 66.10 | 1.69 | 39.1 | NT | NT | NT | 28.87 | >16.67 | - |
| 2a | 64.04 | >10.60 | - | 55.13 | 3.52 | 15.7 | 89.63 | 32.95 | 2.7 | 28.87 | >16.67 | - |
| 2b | 288.23 | 41.26 | 7.0 | 257.52 | 31.34 | 8.2 | >200 | >66.67 | <3.0 | 16.67 | >5.56 | - |
| 2c | 133.16 | 11.59 | 11.5 | 103.36 | 1.24 | 83.5 | 105.44 | 10.48 | 10.1 | 28.87 | >16.67 | - |
| 2d | 434.24 | 34.43 | 11.6 | 100.35 | 9.29 | 10.8 | 136.53 | >66.67 | <2.0 | NT | NT | NT |
| 2e | 87.30 | 10.95 | 8.0 | 29.08 | 3.55 | 8.2 | 45.78 | 17.72 | 2.6 | 19.25 | >11.11 | - |
| 2f | 425.44 | >98.32 | - | 295.10 | 56.73 | 5.2 | NT | NT | NT | NT | NT | NT |
| 2g | 56.04 | 9.04 | 6.2 | 80.80 | 8.98 | 9.0 | 27.57 | 14.18 | 1.9 | NT | NT | NT |
| 3a | 21.08 | >10.13 | - | 21.08 | 10.13 | 2.1 | 108.82 | 48.99 | 2.2 | 28.87 | >16.67 | - |
| 3b | 55.95 | 10.02 | 5.6 | 67.23 | 5.66 | 11.9 | 126.59 | 38.01 | 3.3 | 57.74 | >33.33 | - |
| 3c | 45.01 | 12.47 | 3.6 | 112.45 | 7.23 | 15.6 | NT | NT | NT | NT | NT | NT |
| 3d | 272.01 | 3.36 | 80.8 | 181.3 | 1.82 | 99.5 | 142.86 | 22.50 | 6.3 | 9.62 | >5.56 | - |
| 3e | 24.10 | 8.60 | 2.8 | 20.05 | 1.16 | 17.2 | 43.58 | 9.75 | 4.5 | 9.62 | >5.56 | - |
| 3f | 21.66 | 4.15 | 5.2 | 12.48 | 3.21 | 3.9 | 54.99 | 5.04 | 10.9 | 28.87 | 5.06 | 5.7 |
| 3g | 94.60 | 26.24 | 3.6 | 65.57 | 5.03 | 13.0 | 138.38 | 34.87 | 4.5 | 21.48 | 0.71 | 30.3 |
| 3h | 78.14 | 6.58 | 11.9 | 65.03 | 10.43 | 6.2 | 153.72 | 47.64 | 3.2 | NT | NT | NT |
| 3i | 27.42 | 3.42 | 8.0 | 13.18 | 2.53 | 5.2 | 38.24 | 9.81 | 3.9 | 9.62 | >5.56 | - |
| 3j | 826.44 | 213.80 | 3.9 | 826.44 | 52.98 | 15.6 | >200 | >66.67 | <3.0 | 28.87 | >16.67 | - |
| 3k | 1.73 | >0.41 | - | 2.09 | - | - | NT | NT | NT | NT | NT | NT |
| 4a | 14.84 | 4.93 | 3.0 | 25.75 | 6.38 | 4.0 | 79.04 | 5.51 | 14.3 | NT | NT | NT |
| 4b | 440.02 | 21.43 | 20.5 | 254.07 | 48.89 | 5.2 | >200 | 24.37 | >8.2 | >50 | >50 | - |
| RBV | 4765.44 | 8.76 | 544.0 | 8205.90 | 2120.4 | 3.9 | NT | NT | NT | NT | NT | NT |
| Oseltamivir | 4030.42 | 18.97 | 212.5 | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 3TC | NT | NT | NT | NT | NT | NT | NT | NT | NT | >100 | 1.85 | >54.1 |
| Telaprevir | NT | NT | NT | NT | NT | NT | 32.12 | 0.011 | 2920.0 | NT | NT | NT |
2.2.2. Anti Cox B3 Virus Activity
2.2.3. Anti HCV Activity
2.2.4. Anti-HBV Activity
3. Experimental
3.1. General Information
3.2. Chemistry
3.2.1. General procedure for the synthesis of 1M–4M
3.2.2. General procedure for the synthesis of 1a–1e, 2a–2g, 3a–3f
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Qi, X.; Zhang, Q.; Zeng, X.; Shi, Z.; Jin, Q.; Zhan, F.; Xu, Y.; Liu, Z.; Feng, Z.; et al. Human 4F5 single-chain Fv antibody recognizing a conserved HA1epitope has broad neutralizing potency against H5N1 influenza A viruses of different clades. Antivir. Res. 2013, 99, 91–99. [Google Scholar] [CrossRef]
- Metersky, M.L.; Masterton, R.G.; Lode, H.; File, T.M., Jr.; Babinchak, T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int. J. Infect. Dis. 2012, 16, 321–331. [Google Scholar]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-bornc non-A, non-B viral hepatitis genomc. Science 1989, 244, 359–362. [Google Scholar]
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar]
- Kesel, A.J. Broad-spectrum antiviral activity including human immunodeficiency and hepatitis C viruses mediated by a novel retinoid thiosemicarbazone derivative. Eur. J. Med. Chem. 2011, 46, 1656–1664. [Google Scholar] [CrossRef]
- Lawitz, E.; Sulkowski, M.; Jacobson, I.; Kraft, W.K.; Maliakkal, B.; Al-lbrahim, M.; Gordon, S.C.; Kwo, P.; Rockstroh, J.K.; Panorchan, P.; et al. Characterization of vaniprevir, a hepatitis C virus NS3/4A protease inhibitor, in patients with HCV genotype 1 infection: Safety, antiviral activity, resistance, and pharmacokinetics. Antivir. Res. 2013, 99, 214–220. [Google Scholar] [CrossRef]
- Huang, C.C.; Kuo, T.M.; Yeh, C.T.; Hu, C.P.; Chen, Y.L.; Tsai, Y.L.; Chen, M.L.; Chou, Y.C.; Chang, C. One single nucleotide difference alters the differential expression of spliced RNAs between HBV genotypes A and D. Virus Res. 2013, 174, 18–26. [Google Scholar] [CrossRef]
- Pinkert, S.; Klingel, K.; Lindig, V.; Dörner, A.; Zeichhardt, H.; Spiller, O.B.; Fechner, H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J. Virol. 2011, 85, 13409–13419. [Google Scholar] [CrossRef]
- Bedard, K.M.; Wang, M.L.; Proll, S.C.; Loo, Y.M.; Katze, M.G.; Gale, M., Jr.; Ladonato, S.P. Isoflavone agonists of IRF-3 dependent signaling have antiviral activity against RNA viruses. J. Virol. 2012, 86, 7334–7344. [Google Scholar] [CrossRef]
- Richman, D.D. Antiviral drug resistance. Antivir. Res. 2006, 71, 117–121. [Google Scholar] [CrossRef]
- Colman, P.M. New antivirals and drug resistance. Annu. Rev. Biochem. 2009, 78, 95–118. [Google Scholar] [CrossRef]
- Krepstakies, M.; Luciflra, J.; Naqel, C.H.; Zeisel, M.B.; Holstermann, B.; Hohenberq, H.; Kowalski, I.; Gutsmann, T.; Baumert, T.F.; Brandenburg, K.; et al. A new class of synthetic peptide inhibitors blocks attachment and entry of human pathogenic viruses. J. Infect. Dis. 2012, 205, 1654–1664. [Google Scholar] [CrossRef]
- ElSawy, K.M.; Twarock, R.; Verma, C.S.; Caves, L.S. Peptide inhibitors of viral assembly: A novel route to broad-spectrum antivirals. J. Chem. Inform. Model. 2012, 52, 770–776. [Google Scholar] [CrossRef]
- Zhong, Z.J.; Zhang, D.J.; Peng, Z.G.; Li, Y.H.; Shan, G.Z.; Zuo, L.M.; Wu, L.T.; Li, S.Y.; Gao, R.M.; Li, Z.R. Synthesis and antiviral activity of a novel class of (5-oxazolyl)phenyl amines. Eur. J. Med. Chem. 2013, 69, 32–43. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; de Clercq, E. Synthesis and antiviral/antiproliferative activity of some N-sulphonylbenzimidazoles. Bioorg. Med. Chem. Lett. 2002, 12, 2707–2710. [Google Scholar] [CrossRef]
- Cheng, J.; Xie, J.; Luo, X. Synthesis and antiviral activity against Coxsackie virus B3 of some novel benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 267–269. [Google Scholar] [CrossRef]
- De Sá Alves, F.R.; Barreiro, E.J.; Fraga, C.A. From nature to drug discovery: The indole scaffold as a ‘privileged structure’. Mini Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef]
- Yee, Y.K.; Bernstein, P.R.; Adams, E.J.; Brown, F.J.; Cronk, L.A.; Hebbel, K.C.; Vacek, E.P.; Krell, R.D.; Snyder, D.W. A novel series of selective leukotriene antagonists: Exploration and optimization of the acidic region in 1,6-disubstituted indoles and indazoles. J. Med. Chem. 1990, 33, 2437–2451. [Google Scholar]
- Jagadeesh, R.V.; Wienhöfer, G.; Westerhaus, F.A.; Surkus, A.E.; Pohl, M.M.; Junqe, H.; Junqe, K.; Beller, M. Efficient and highlyselectiveiron-catalyzedreduction of nitroarenes. Chem. Commun. 2011, 47, 10972–10974. [Google Scholar] [CrossRef]
- Hrobárik, P.; Hrobáriková, V.; Sigmundová, I.; Zahradník, P.; Fakis, M.; Polyzos, I.; Persephonis, P. Benzothiazoles with tunableelectron-withdrawingstrength and reverse polarity: A route to triphenylamine-based chromophores with enhanced two-photon absorption. J. Org. Chem. 2011, 76, 8726–8736. [Google Scholar] [CrossRef]
- Rahaim, R.J., Jr.; Maleczka, R.E., Jr. Pd-catalyzed silicon hydride reductions of aromatic and aliphatic nitro groups. Org. Lett. 2005, 7, 5087–5090. [Google Scholar] [CrossRef]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned Hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA. 1987, 84, 1005–1009. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the entire panel of target compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, D.-J.; Sun, W.-F.; Zhong, Z.-J.; Gao, R.-M.; Yi, H.; Li, Y.-H.; Peng, Z.-G.; Li, Z.-R. Synthesis and Broad-Spectrum Antiviral Activity of Some Novel Benzo-Heterocyclic Amine Compounds. Molecules 2014, 19, 925-939. https://doi.org/10.3390/molecules19010925
Zhang D-J, Sun W-F, Zhong Z-J, Gao R-M, Yi H, Li Y-H, Peng Z-G, Li Z-R. Synthesis and Broad-Spectrum Antiviral Activity of Some Novel Benzo-Heterocyclic Amine Compounds. Molecules. 2014; 19(1):925-939. https://doi.org/10.3390/molecules19010925
Chicago/Turabian StyleZhang, Da-Jun, Wen-Fang Sun, Zhao-Jin Zhong, Rong-Mei Gao, Hong Yi, Yu-Huan Li, Zong-Gen Peng, and Zhuo-Rong Li. 2014. "Synthesis and Broad-Spectrum Antiviral Activity of Some Novel Benzo-Heterocyclic Amine Compounds" Molecules 19, no. 1: 925-939. https://doi.org/10.3390/molecules19010925