An Efficient Synthesis of Enantiopure (R)-heteroarylpyrimidine Analogs

Abstract
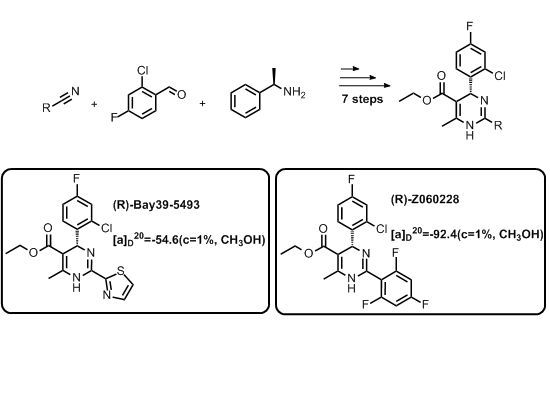
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
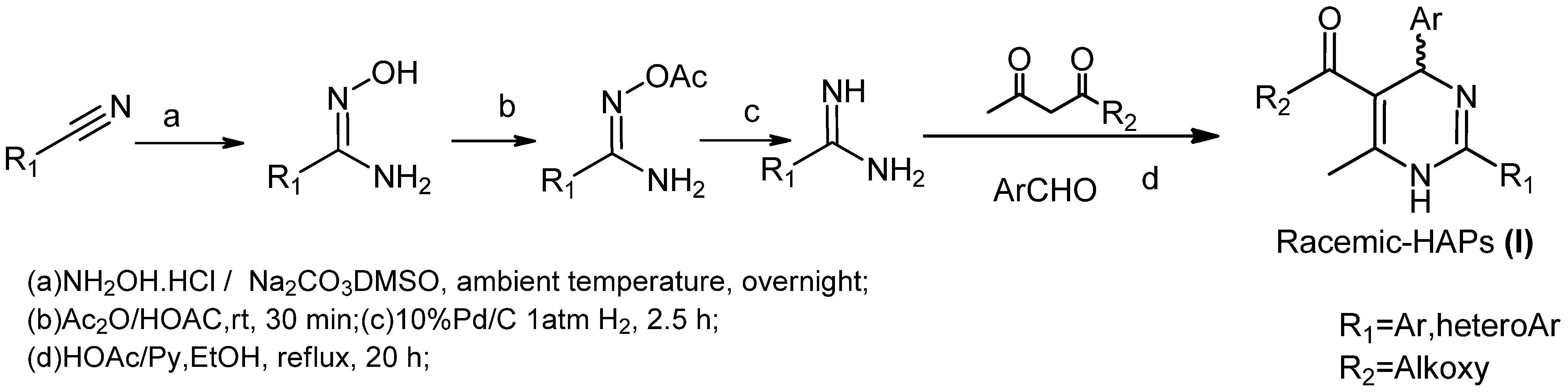
1. Introduction
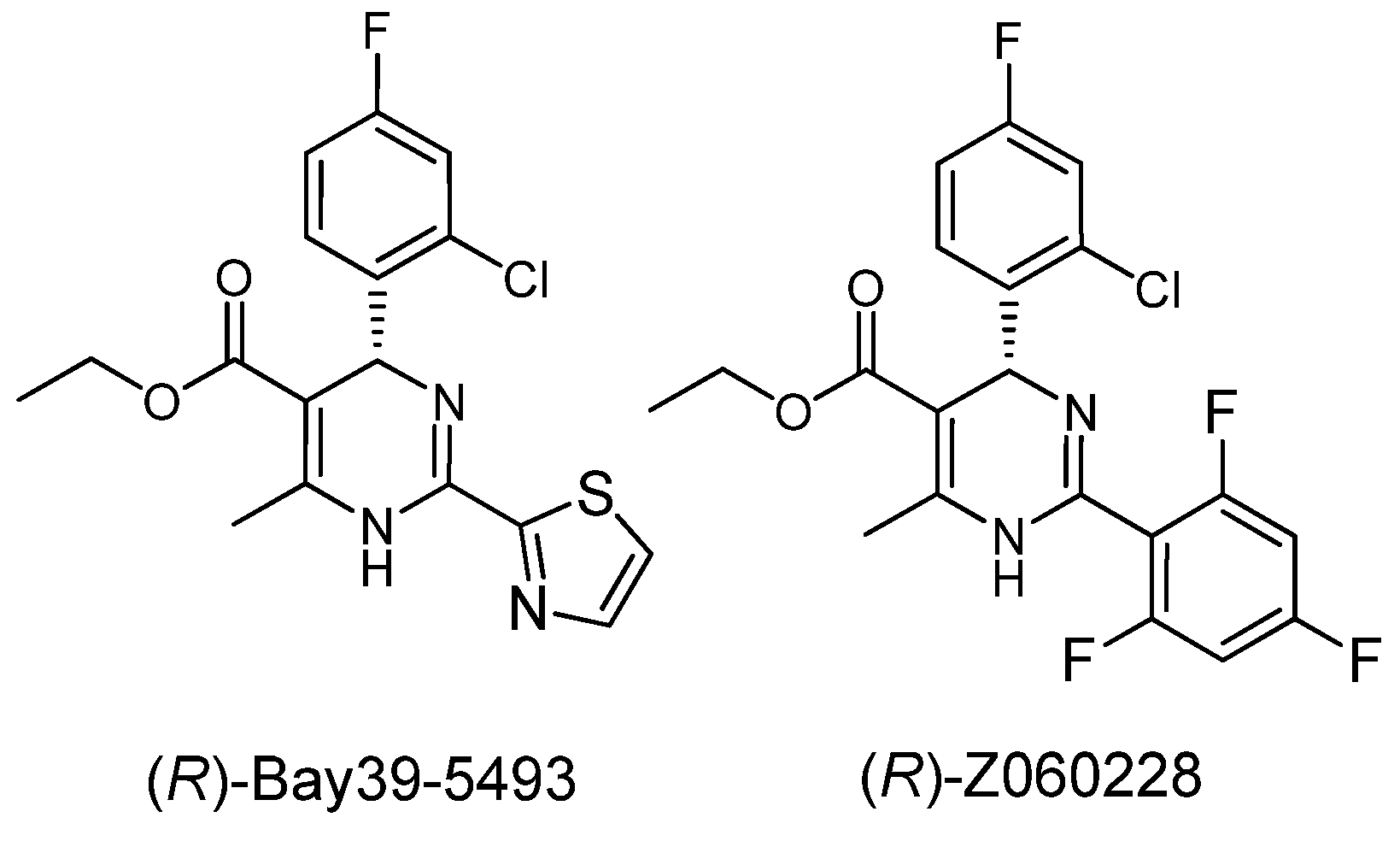
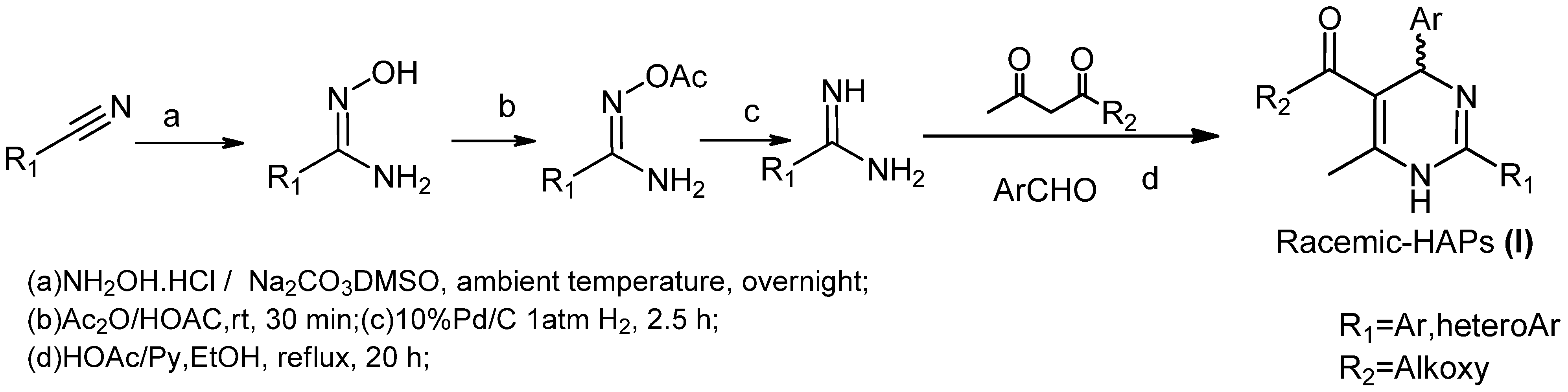
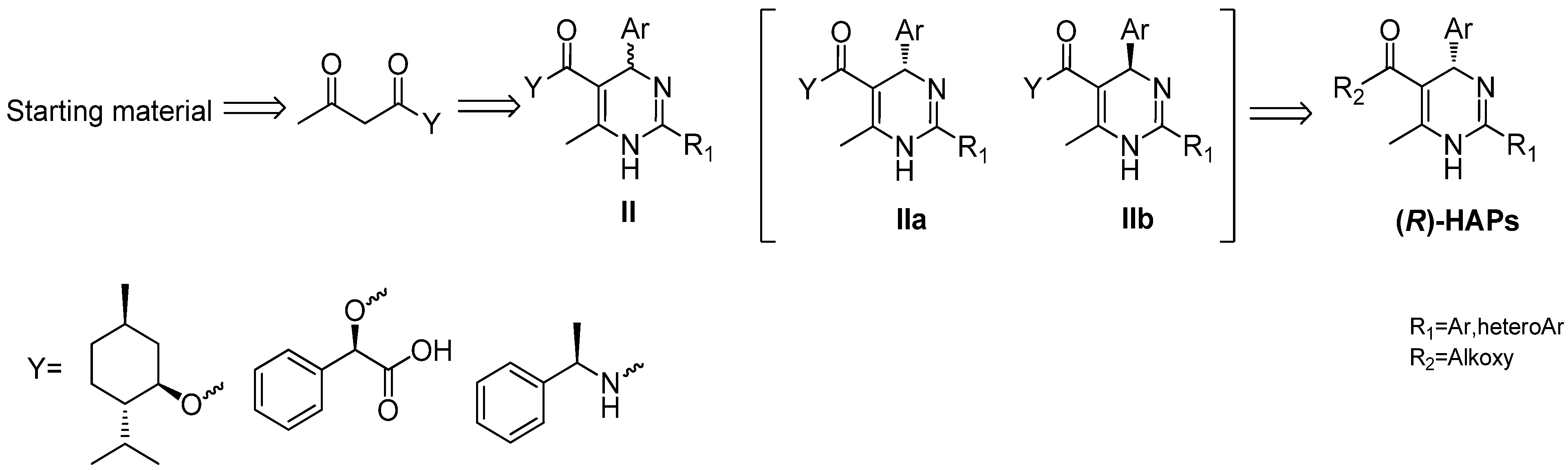
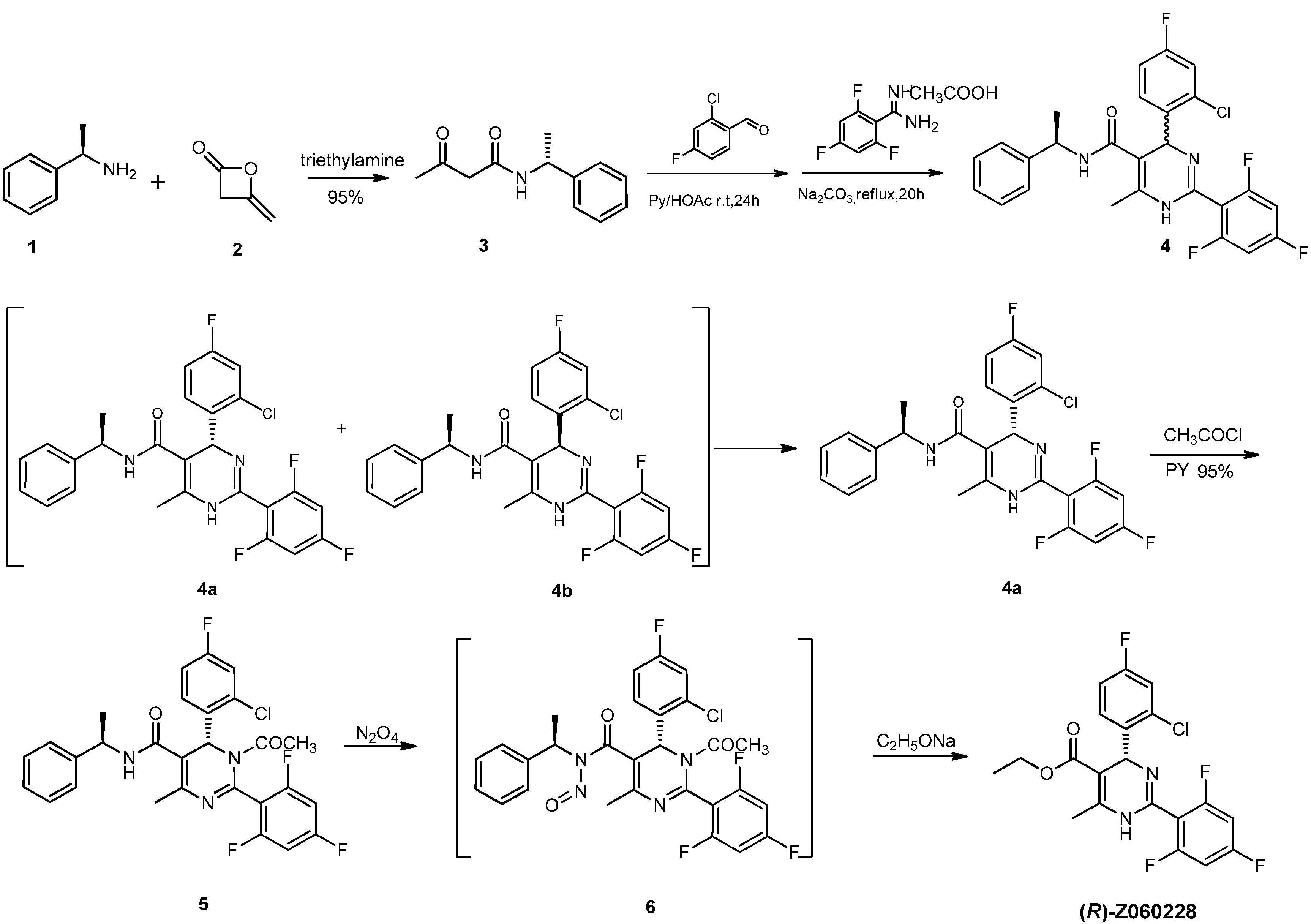
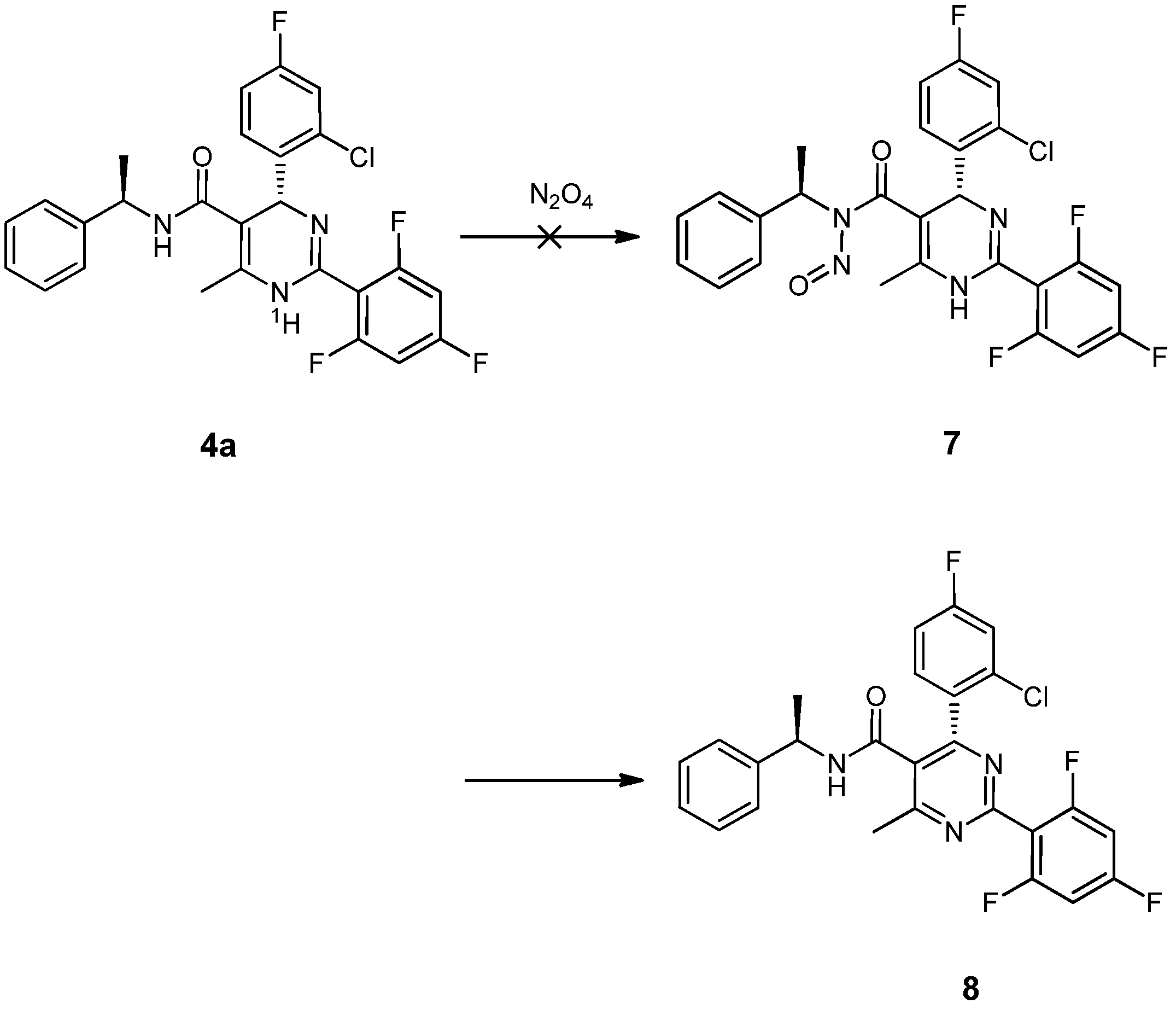
2. Results and Discussion

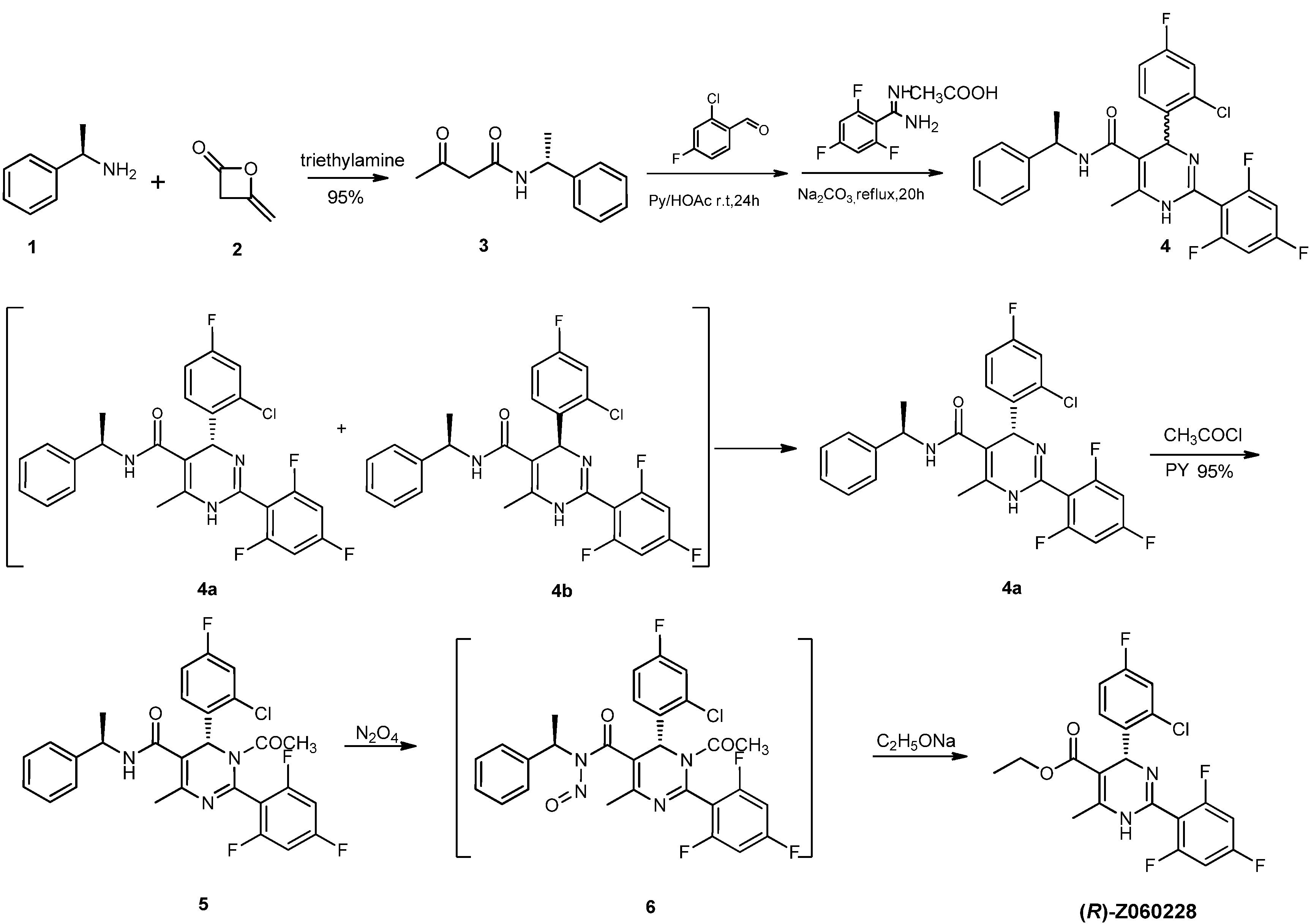
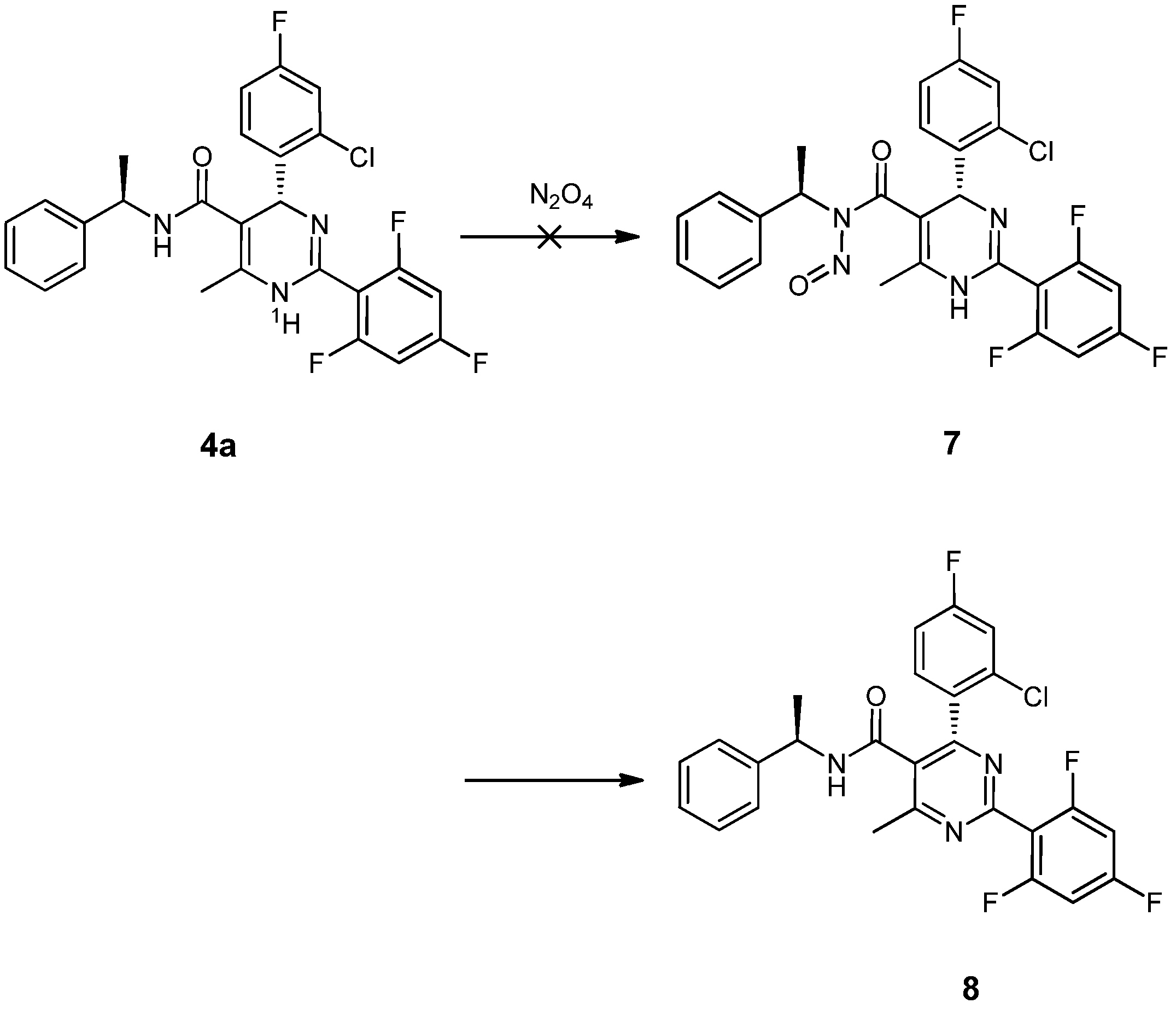
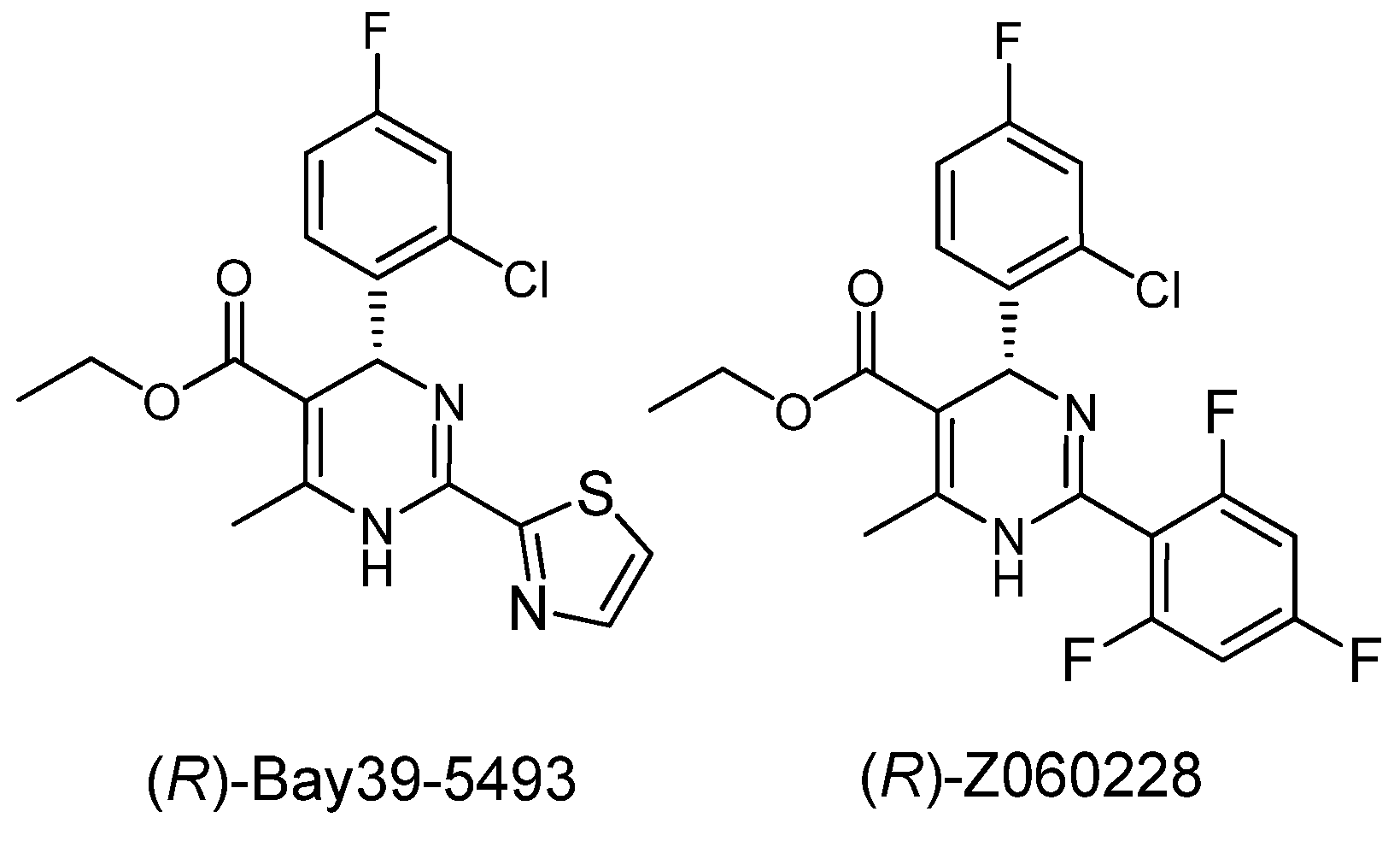
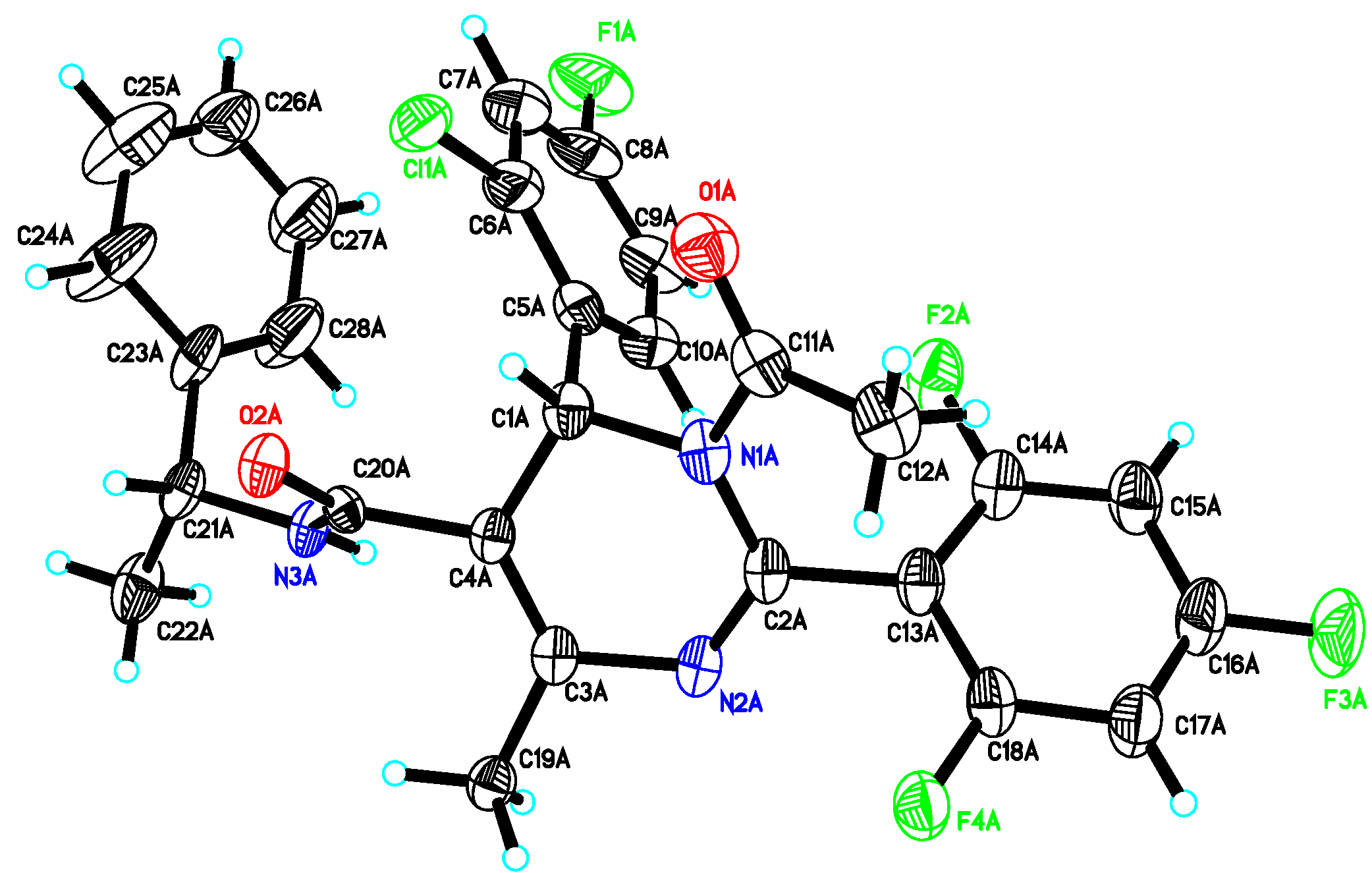
 = −92.4 (c = 1.0, CH3OH) and its absolute (R) configuration could be directly confirmed by determination of the absolute configuration of the intermediate (4R,2'R)-5 by X-ray crystallography (Figure 2) [26]. Using a similar reaction sequence, Bay39-5493 was prepared in 99% ee and its specific rotation closely matched the reported value (observed: = −54.6, c = 1.0, CH3OH; lit.: = −52, c = 1.0, CH3OH) [27].
= −92.4 (c = 1.0, CH3OH) and its absolute (R) configuration could be directly confirmed by determination of the absolute configuration of the intermediate (4R,2'R)-5 by X-ray crystallography (Figure 2) [26]. Using a similar reaction sequence, Bay39-5493 was prepared in 99% ee and its specific rotation closely matched the reported value (observed: = −54.6, c = 1.0, CH3OH; lit.: = −52, c = 1.0, CH3OH) [27].
3. Experimental
3.1. General
3.2. Synthesis
= −92.4 (c = 1.0, CH3OH), 1H-NMR (DMSO-d6) δ: 1.02–1.05 (3H, t); 2.32 (3H, s); 3.92–3.96 (2H, m); 5.97 (1H, s); 7.21–7.45 (5H, m); 9.86 (1H, s). 13C-NMR (DMSO-d6) δ: 14.4, 17.9, 56.0, 59.6, 96.2, 101.3, 101.5, 101.8, 115.2, 116.6, 116.8, 131.2, 131.2, 132.6, 139.8, 142.0, 147.6, 160.0, 162.4, 166.1. HRMS (ESI) m/z: Calcd for C20H15N2O2F4Cl, 427.0831 [M+H]+; Found: 427.0827. = −54.6 (c = 1.0, CH3OH). 1H-NMR (DMSO-d6) δ: 1.01–1.04 (3H, m); 2.50 (3H, s); 3.90–3.94 (2H, m); 5.99 (1H, s); 7.36–7.43 (3H, m); 7.90–7.91 (1H, d, J = 4 Hz); 7.97–7.98 (1H, d, J = 4 Hz); 9.96 (1H, s); HRMS (ESI) m/z: Calcd for C17H15N3O2FSCl, 380.0630 [M+H]+; Found: 380.0628.4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Janssen, H.L.; Van-Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Zoulim, F.; Poynard, T.; Degos, F.; Slama, A.; El Hasnaoui, A.; Blin, P.; Mercier, F.; Deny, P.; Landais, P.; Parvaz, P.; et al. A prospective study of the evolution of lamivudine resistance mutations in patients with chronic hepatitis B treated with lamivudine. J. Viral. Hepat. 2006, 13, 278–288. [Google Scholar] [CrossRef]
- Hacker, H.J.; Deres, K.; Mildenberger, M.; Schröder, C.H. Antivirals interacting with hepatitis B virus core protein and core mutations may misdirect capsid assembly in a similar fashion. Biochem. Pharmacol. 2003, 66, 2273–2279. [Google Scholar] [CrossRef]
- Stray, S.J.; Bourne, C.R.; Punna, S.; Lewis, W.G.; Finn, M.G.; Zlotnick, A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 8138–8143. [Google Scholar]
- Bourne, C.R.; Finn, M.G.; Zlotnick, A. Global Structural Changes in Hepatitis B Virus Capsids Inducedby the Assembly Effector HAP1. J. Virol. 2006, 80, 11055–11061. [Google Scholar] [CrossRef]
- Li, S.; Zhao, G.M; Xia, G.Q. Dihydropyrimidine compounds and their uses in preparation of medicaments for treating and preventing antiviral diseases. U.S. Patent 8168642 B2, 1 May 2012. [Google Scholar]
- Zhao, G.M.; Wang, X.K.; Ju, X.J.; Guan, H.; Zhong, W.; Wang, L.L.; Xiao, J.H.; Yang, X.H.; Li, S. Synthesis and Anti-HBV Activity of Novel Dihydropyrimidine Analogs. Scientia Sinica Vitae 2011, 41, 971–977. [Google Scholar] [CrossRef]
- Deres, K.; Schroder, C.H.; Paessens, A.; Goldmann, S.; Hacker, H.J.; Weber, O.; Krämer, T.; Niewöhner, U.; Pleiss, U.; Stoltefuss, J.; et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [Google Scholar] [CrossRef]
- Goldmann, S.; Stoltefuss, J.; Niewohner, U.; Schelemmer, K.H. Dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis B. DE 10012824A1, 20 September 2001. [Google Scholar]
- Goldmann, S.; Stoltefuss, J.; Paessens, A.; Graef, E.; Lottmann, S. 2-heterocyclically substituted dihydropyrimidines. U.S. Patent 6503913B1, 7 January 2003. [Google Scholar]
- Evans, P.A.; Qin, J.; Robinson, J.E.; Bazin, B. Enantioselective total synthesis of the polycyclic guanidine-containing marine alkaloid (−)-batzelladine D. Angew. Chem. Int. Ed. 2007, 46, 7417–7419. [Google Scholar]
- Kamaljit, S.; Divya, A.; Danielle, F.; Liu, Q.X.; Moreland, R.S. An Efficacious Protocol for 4-Substituted 3,4-Dihydropyrimidinones: Synthesis and Calcium Channel Binding Studies. Eur. J. Org. Chem. 2009, 19, 3258–3264. [Google Scholar]
- Siegfried, G.; Jiirgen, S. 1,4-Dihydropyridines: Effects of Chirality and Conformation on the Calcium Antagonist and Calcium Agonist Activities. Angew. Chem. Inr. Ed. Engl. 1991, 30, 1559–1578. [Google Scholar]
- Hudlicky, T.; Gillman, G.; Andersen, Catherine. Homochiral amine synthesis by baker's yeast resolution of a β-keto amide: 1-Phenylethylamine. Tetrahedron Asymmetry 1992, 3, 281–286. [Google Scholar]
- Slee, H.D.; Chen, Y.; Zhang, X. 2-Amino-N-pyrimidin-4-ylacetamides as A2A receptor antagonists: 1. Structure-activity relationships and optimization of heterocyclic substituents. J. Med. Chem. 2008, 51, 1719–1729. [Google Scholar]
- Judkins, B.D.; Allen, D.G.; Cook, T.A. A Versatile Synthesis of Amidines from Nitriles Via Amidoximes. Synth. Commun. 1996, 26, 4351–4367. [Google Scholar]
- Zhu, X.J.; Zhao, G.M.; Li, S. 2,4-Diaryl-4,6,7,8-tetrahydroquinazolin-5(1 H)-one derivatives as anti-HBV agents targeting at capsid assembly. Med. Chem. Lett. 2010, 20, 299–301. [Google Scholar]
- Evans, D.A.; Carter, P.H.; Dinsmore, C.J.; James, C.B.; Jeffrey, L.K.; Daniel, W.K. Mild nitrosation and hydrolysis of polyfunctional amides. Tetrahedron Lett. 1997, 38, 4535–4538. [Google Scholar]
- Julio, C.; Albino, C.; Ramon Leis, J.; Manuel, M.; Pena, M.E. Kinetic Studies on the Formation of N-Nitroso Compounds X. The Nitrosation of N-Methylacetamide and Its Differences with Respect to the Nitrosation of Amines. Monatshefte für Chemie 1984, 115, 1047–1057. [Google Scholar]
- Nasser, I.; Habib, F.; AliReza, P. Dinitrogen Tetroxide – ImpregnatedCharcoal (N2O4/Charcoal): Selective Nitrosation of Amines, Amides,Ureas, and Thiols. Synth. Commun. 2005, 35, 1517–1526. [Google Scholar]
- Hoang-V, L.; Li, J.F.; Bruce, G. A practical and inexpensive ‘convertible’ isonitrilefor use in multicomponent reactions. Tetrahedron Lett. 2011, 52, 2209–2211. [Google Scholar]
- Fujita, D.; Suzuki, K.; Sato, S.; Maho, Y.U.; Kurimoto, E.; Yamaguchi, Y.; Kato, K.; Fujita, M. Synthesis of a Bridging Ligand with a Non-denatured Protein Pendant: Toward Protein Encapsulation in a Coordination Cage. Chem. Lett. 2012, 3, 313–315. [Google Scholar]
- Monika, C.; Alexis, B.; Caroline, W.; Martial, T.; Jérôme, B.; Caroline, W.; Krzystof, L.; Isabelle, G. P–C Cross-Coupling Onto Enamides: Versatile Synthesis of α-Enamido Phosphane Derivatives. Eur. J. Org. Chem. 2012, 6, 1101–1106. [Google Scholar]
- Daniel-S, L.; Anupong, T.; Ramajeyam, S.; Taylor, M.T.; Fox, J.M.; Ting, A.Y. Diels-Alder cycloaddition for fluorophore targeting to specific proteins inside living cells. J. Am. Chem. Soc. 2012, 134, 792–795. [Google Scholar]
- Bisset, A.; Desmond, L.; Clarkson, J.; Dishington, A.; Jones, T.; Clarkson, G.J.; Ikariya, T.; Wills, M. Synthesis and asymmetric hydrogenation of (3E)-1-benzyl-3-[(2-oxopyridin-1(2H)-yl)methylidene]piperidine-2,6-dione. Chem. Comm. 2012, 48, 11978–11980. [Google Scholar]
- Yang, X.Y.; Zhao, G.M.; Yang, S.Y. Synthesis and Crystal Structure of (R)-1-acetyl-6-(2-chloro-4-fluorophenyl)-4-methyl-N-((R)-1-phenylethyl)-2-(2,4,6-trifluorophenyl)-1,6-dihydropyr-imidine-5-carboxamide. Chin. J. Struct. Chem. 2013, 32, 989–992. [Google Scholar]
- Weber, O.; Schlemmer, K.-H.; Hartmann, E.; Hagelschuer, I.; Paessens, A.; Graef, E.; Deres, K.; Goldmann, S.; Niewoehner, U.; Stoltefuss, J.; et al. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antiviral Res. 2002, 54, 69–78. [Google Scholar]
- CCDC 908381 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- Sample Availability: Samples of the compound (R)-Z060228 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, X.-Y.; Xia, G.-Q.; Wang, X.-K.; Zheng, Z.-B.; Zhao, D.-M.; Zhao, G.-M.; Li, S. An Efficient Synthesis of Enantiopure (R)-heteroarylpyrimidine Analogs. Molecules 2013, 18, 11144-11152. https://doi.org/10.3390/molecules180911144
Yang X-Y, Xia G-Q, Wang X-K, Zheng Z-B, Zhao D-M, Zhao G-M, Li S. An Efficient Synthesis of Enantiopure (R)-heteroarylpyrimidine Analogs. Molecules. 2013; 18(9):11144-11152. https://doi.org/10.3390/molecules180911144
Chicago/Turabian StyleYang, Xiu-Yan, Guang-Qiang Xia, Xiao-Kui Wang, Zhi-Bing Zheng, Dong-Mei Zhao, Guo-Ming Zhao, and Song Li. 2013. "An Efficient Synthesis of Enantiopure (R)-heteroarylpyrimidine Analogs" Molecules 18, no. 9: 11144-11152. https://doi.org/10.3390/molecules180911144