Decomposition Mechanisms and Kinetics of Novel Energetic Molecules BNFF-1 and ANFF-1: Quantum-Chemical Modeling

Abstract
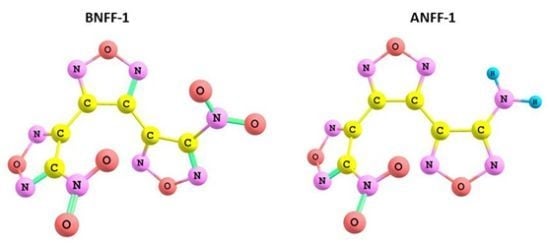
:
1. Introduction
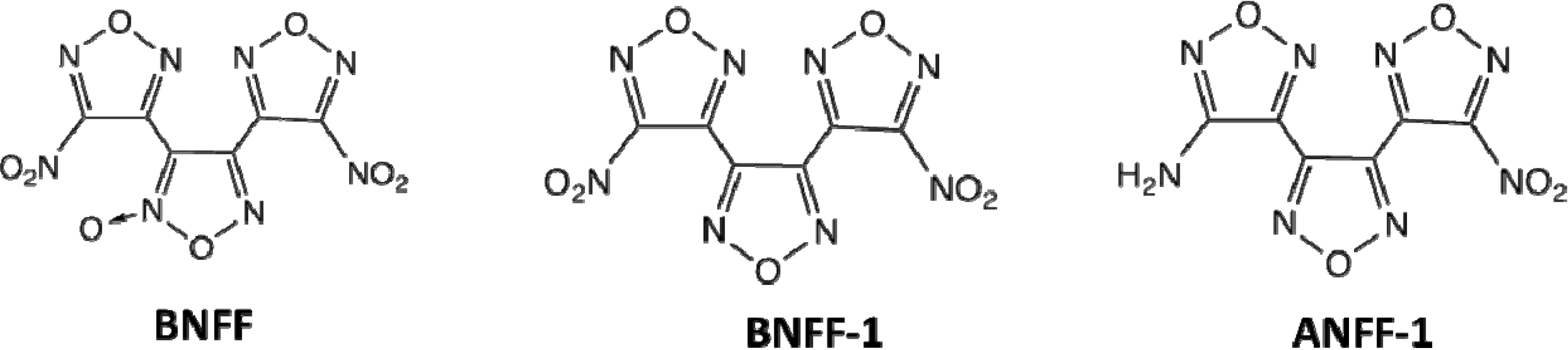
2. Computational Details
3. Simulating Chemical Decomposition Reactions
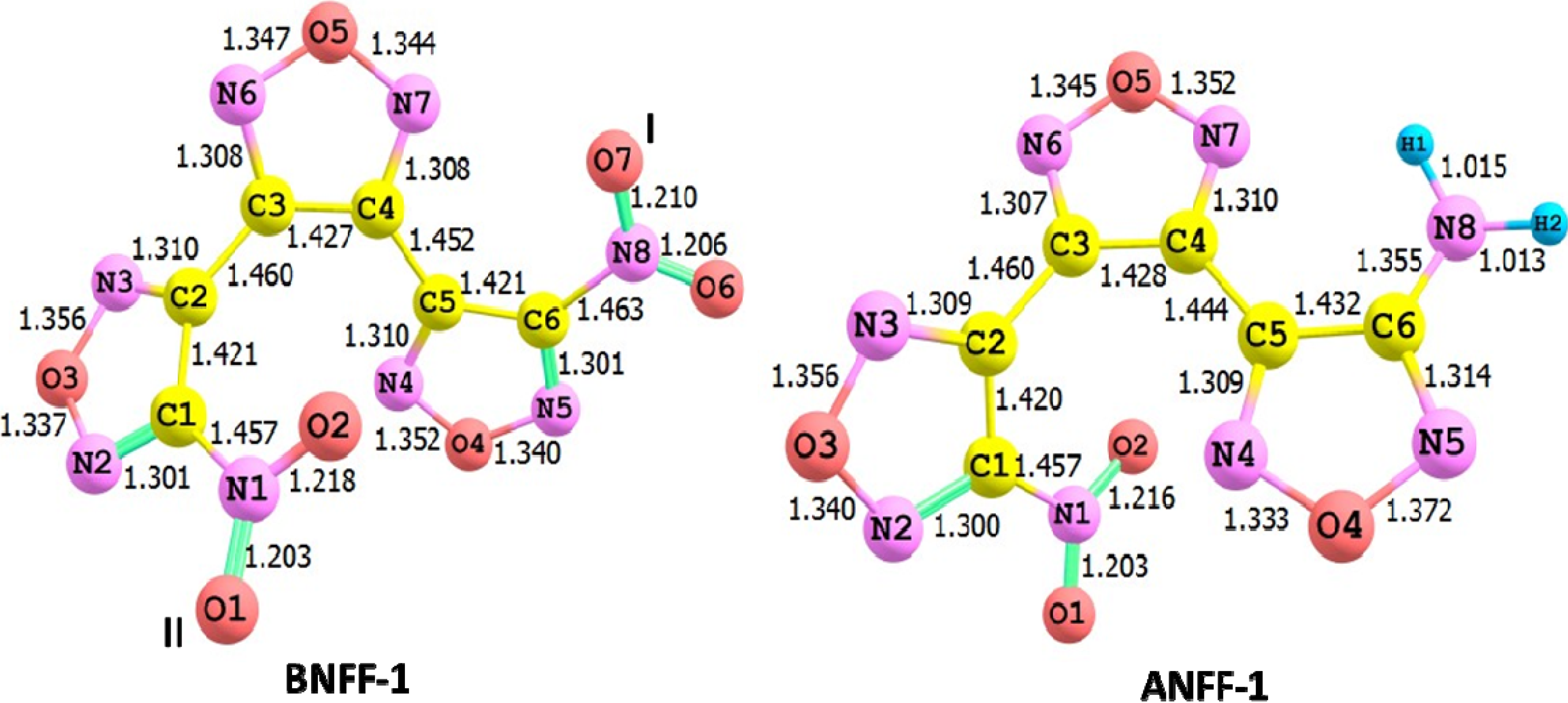

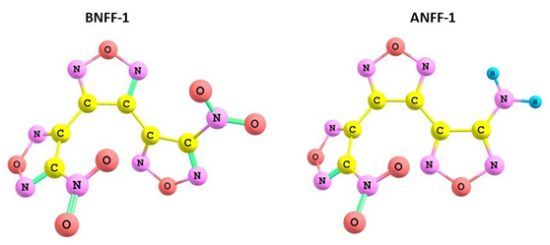{kind=link}
{kind=link}
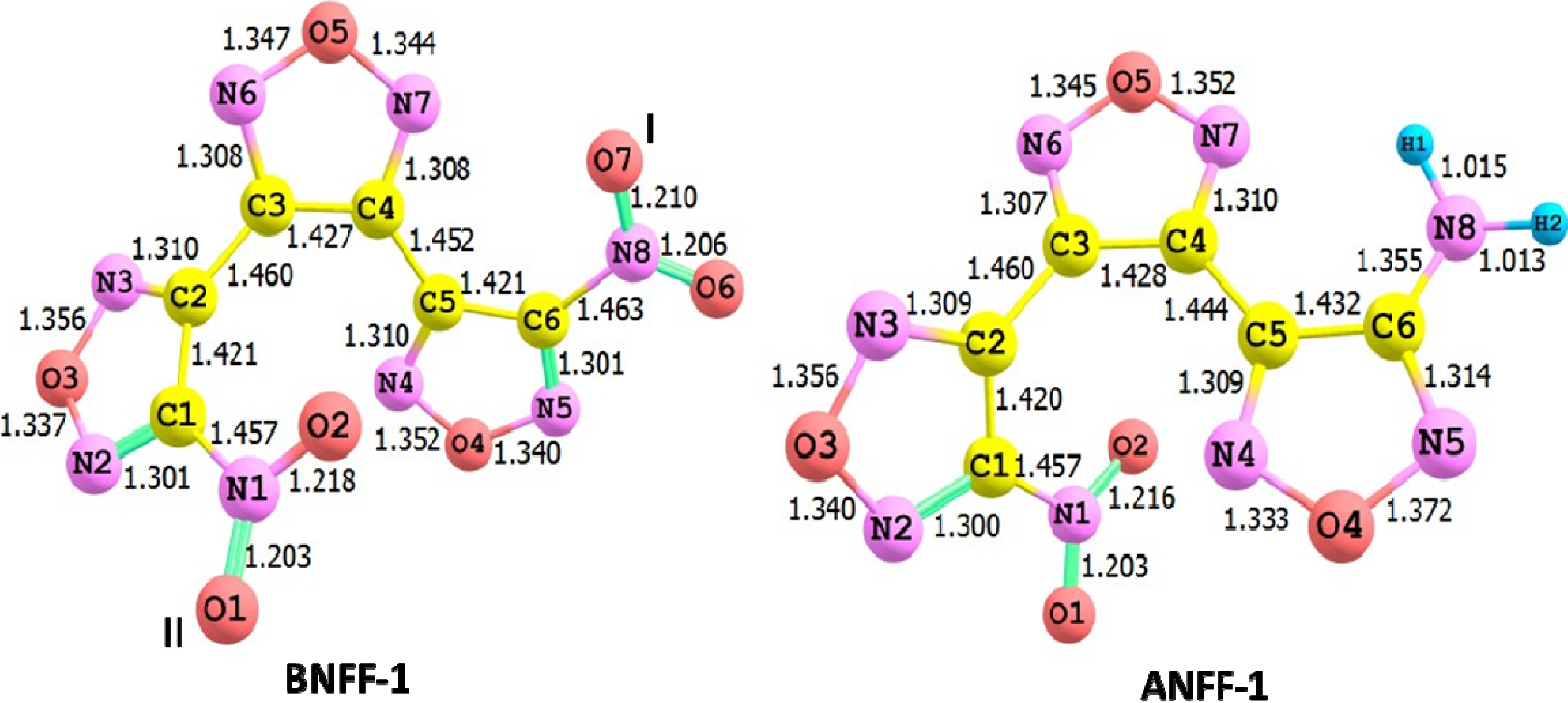{kind=link}
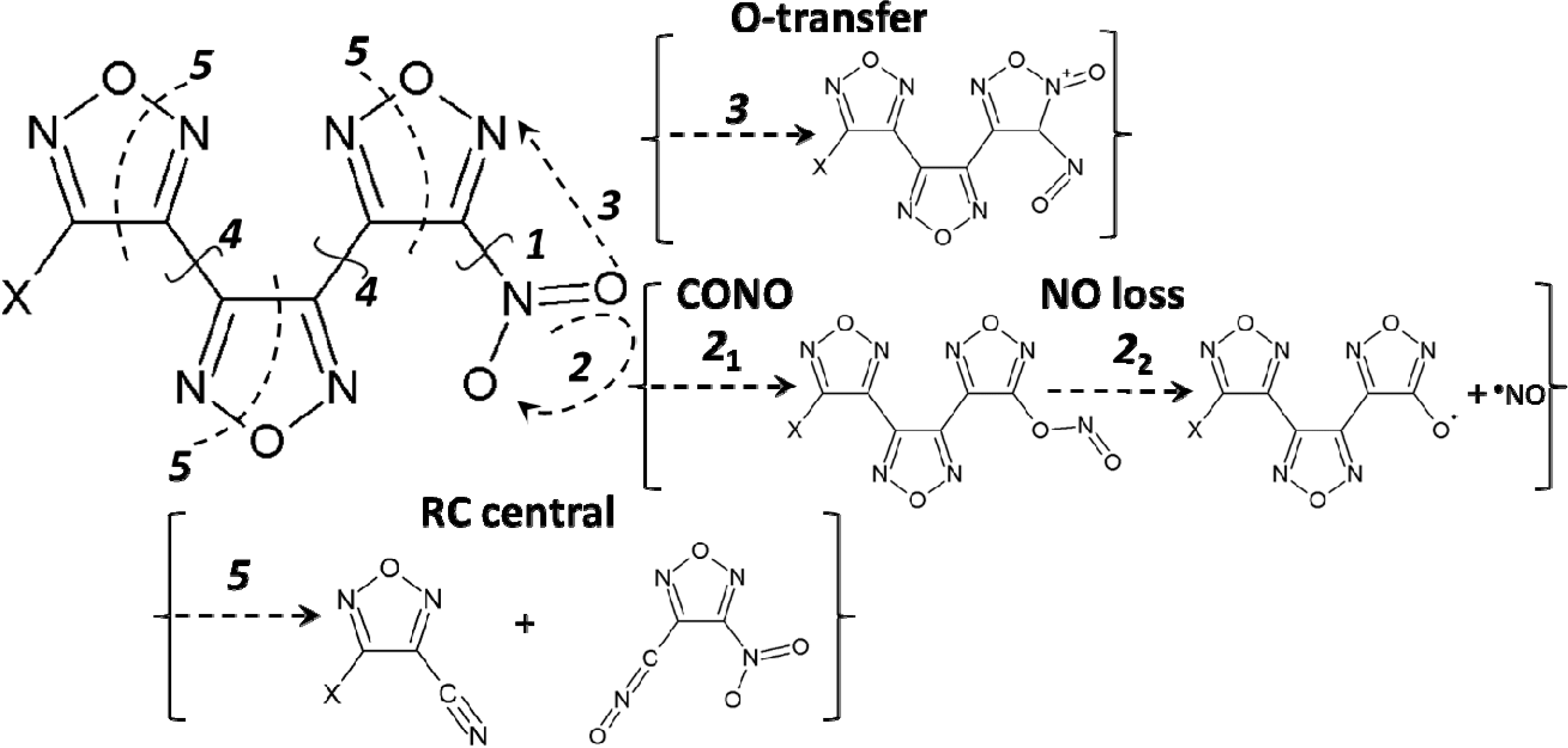{kind=link}
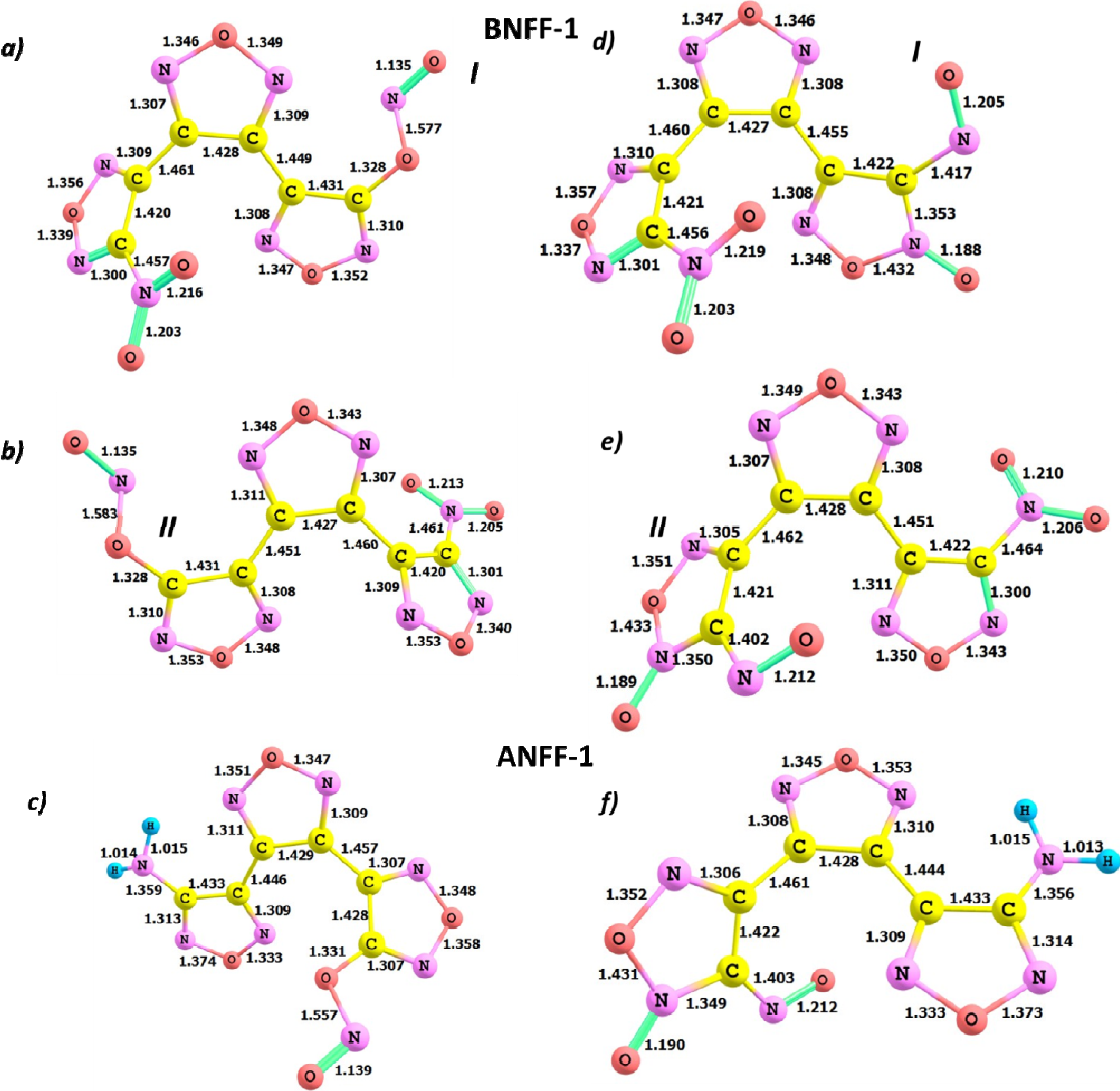{kind=link}
{kind=link}
{kind=link}
{kind=link}
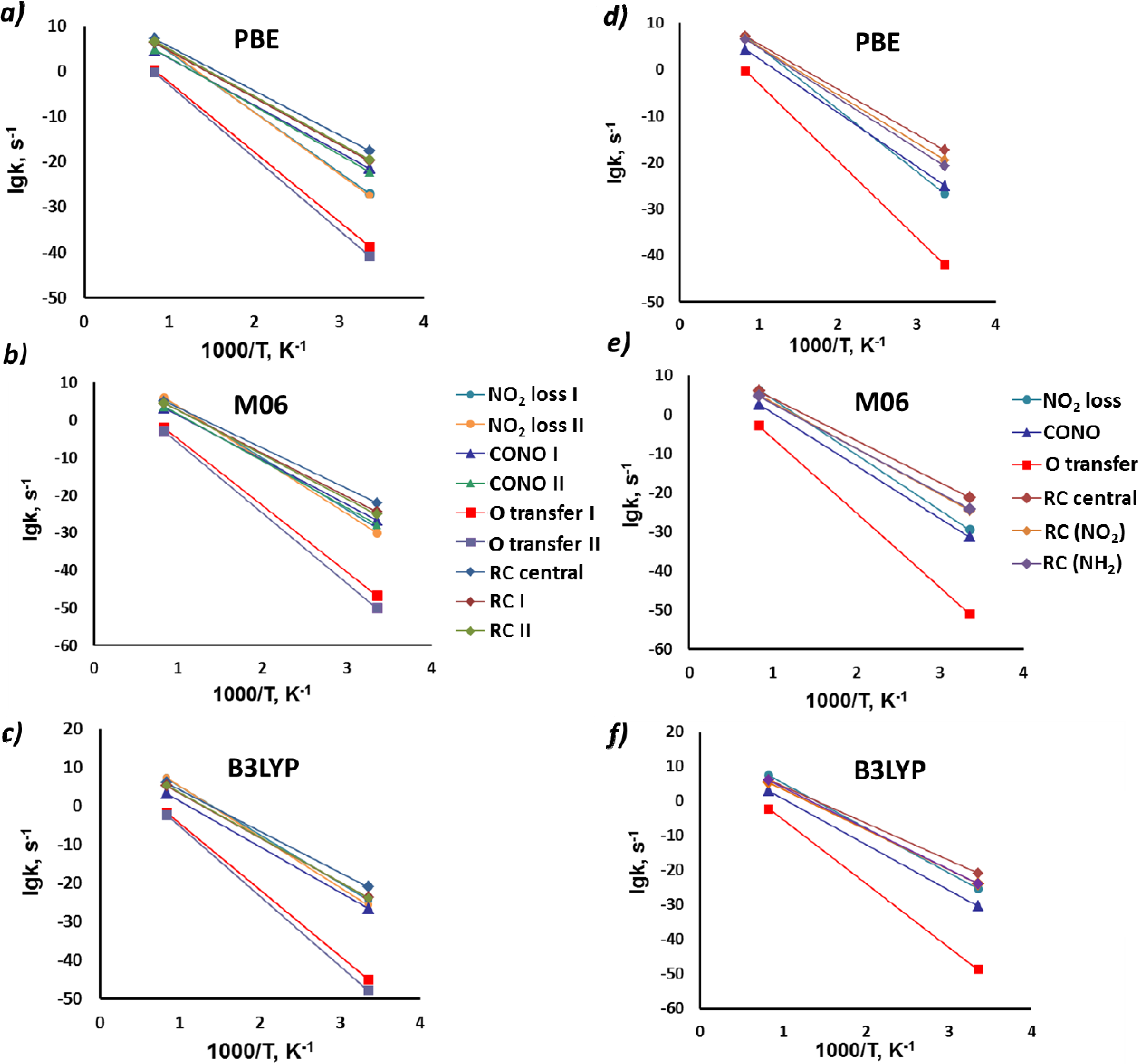
| Reaction | PBE | M06 | B3LYP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E | EZPE | logA | E | EZPE | logA | E | EZPE | logA | ||
| BNFF-1 (LLM-172) | ||||||||||
| 1 | NO2 loss (I) a | 64.9 | 61.1 | 17.6 | 67.0 | 63.1 | 17.2 | 61.6 | 57.6 | 17.6 |
| NO2 loss (II) a | 66.3 | 62.2 | 18.1 | 69.7 | 65.6 | 17.9 | 64.3 | 60.0 | 18.2 | |
| 2 | CONO (I) | 48.3 | 46.1 (−13.3) b | 13.2 | 55.4 | 53.2 (−12.8) | 13.2 | 55.4 | 53.2 (−14.6) | 13.2 |
| CONO (II) | 50.2 | 47.9 (−11.6) | 13.8 | 58.2 | 55.9 (−10.0) | 14.0 | - | - | - | |
| NO loss (I)c | 34.1 | 30.9 | - | 27.9 | 25.0 | - | 22.8 | 19.8 | - | |
| NO loss (II) | 33.9 | 30.8 | - | 27.9 | 24.9 | - | - | - | - | |
| 3 | O-transfer(I) | 71.6 | 69.4 (17.9) | 12.9 | 82.3 | 80.3 (24.3) | 12.7 | 80.1 | 77.9 (22.7) | 12.6 |
| O-transfer(II) | 74.8 | 72.4 (13.0) | 13.0 | 86.8 | 84.6 (25.1) | 12.5 | 84.3 | 81.9 (23.1) | 12.7 | |
| 4 | C-C (I) | 127.1 | 122.6 | - | 129.5 | 125.4 | - | 125.3 | 120.9 | - |
| C-C (II) | 129.3 | 124.8 | - | 131.2 | 127.1 | - | 126.6 | 122.2 | - | |
| 5 | RC central | 45.7 | 42.9 (27.6) | 15.3 | 50.5 | 47.6 (27.3) | 14.0 | 50.4 | 47.5 (23.3) | 15.1 |
| RC (I) | 48.3 | 45.4 (36.6) | 14.9 | 53.7 | 50.7 (37.5) | 14.0 | 54.0 | 50.8 (34.1) | 14.8 | |
| RC (II) | 48.3 | 45.3 (36.4) | 15.2 | 55.2 | 52.1 (38.0) | 14.3 | 54.3 | 51.1 | 15.0 | |
| ANFF-1 (LLM-175) | ||||||||||
| 1 | NO2 loss | 65.4 | 61.3 | 18.1 | 65.2 | 61.5 | 18.0 | 63.8 | 59.5 | 18.3 |
| 2 | CONO | 54.3 | 51.9 (−9.8) | 13.8 | 62.6 | 60.3 (−8.2) | 13.7 | 61.6 | 59.1 (−10.7) | 13.9 |
| NO lossc | 32.9 | 29.8 | - | 26.8 | 23.8 | - | 21.6 | 18.6 | - | |
| 3 | O-transfer | 77.0 | 74.6 (18.4) | 13.4 | 88.6 | 88.5 (25.0) | 12.9 | 85.8 | 83.5 (23.1) | 13.1 |
| 4 | C-C (NO2) | 130.3 | 125.8 | - | 132.1 | 128.1 | - | 127.5 | 123.2 | - |
| C-C (NH2) | 134.3 | 129.8 | - | 136.4 | 132.4 | - | 132.2 | 127.7 | - | |
| 5 | RC central | 45.5 | 42.7 (27.5) | 15.2 | 50.6 | 47.6 (24.6) | 14.9 | 50.0 | 47.1 (23.8) | 15.0 |
| RC (NO2) | 48.5 | 45.5 (32.4) | 15.2 | 54.9 | 51.8 (30.3) | 14.2 | 54.4 | 51.3 (31.1) | 15.1 | |
| RC (NH2) | 50.6 | 47.3 (40.6) | 15.4 | 53.7 | 50.8 (39.7) | 14.6 | 55.6 | 52.2 (36.3) | 15.8 | |
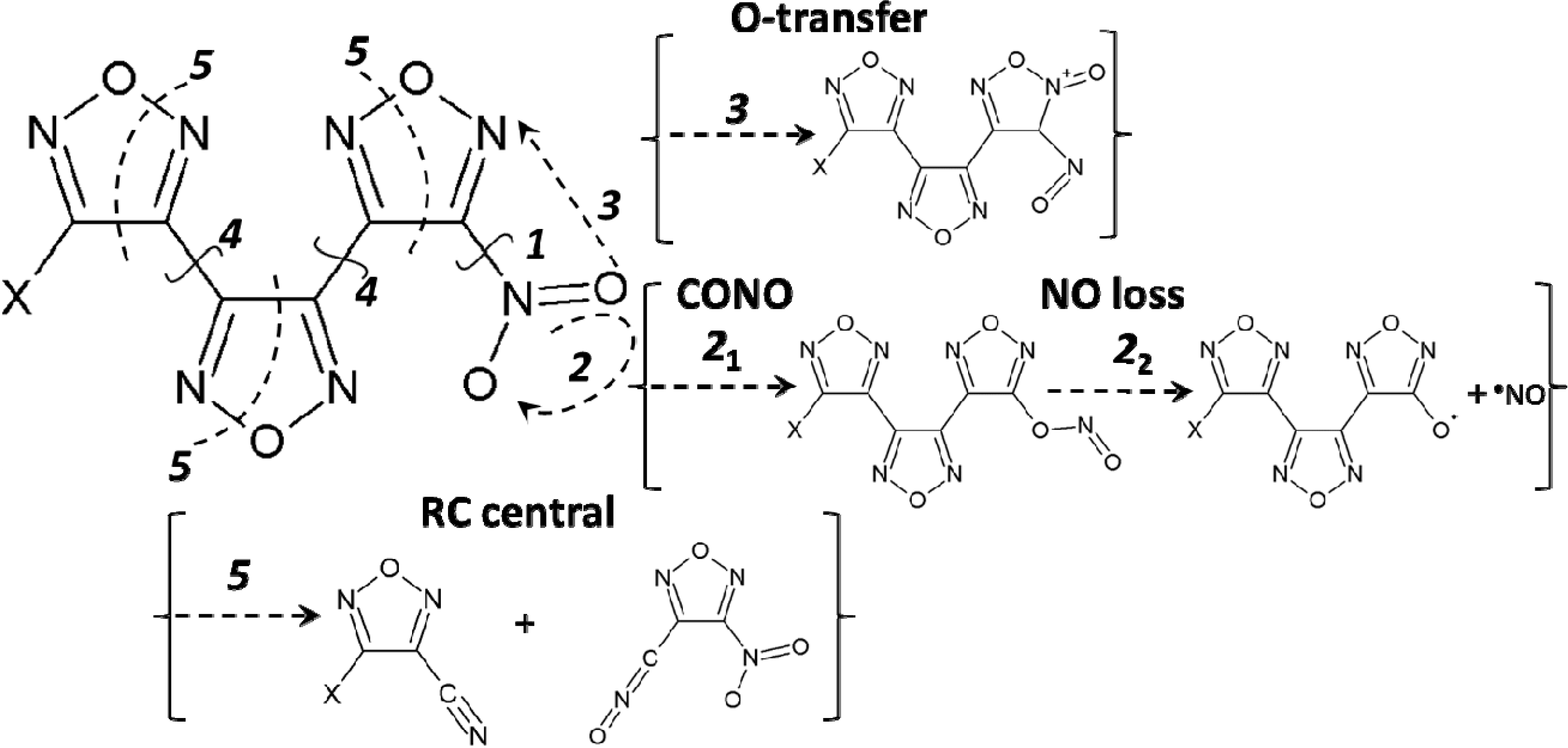
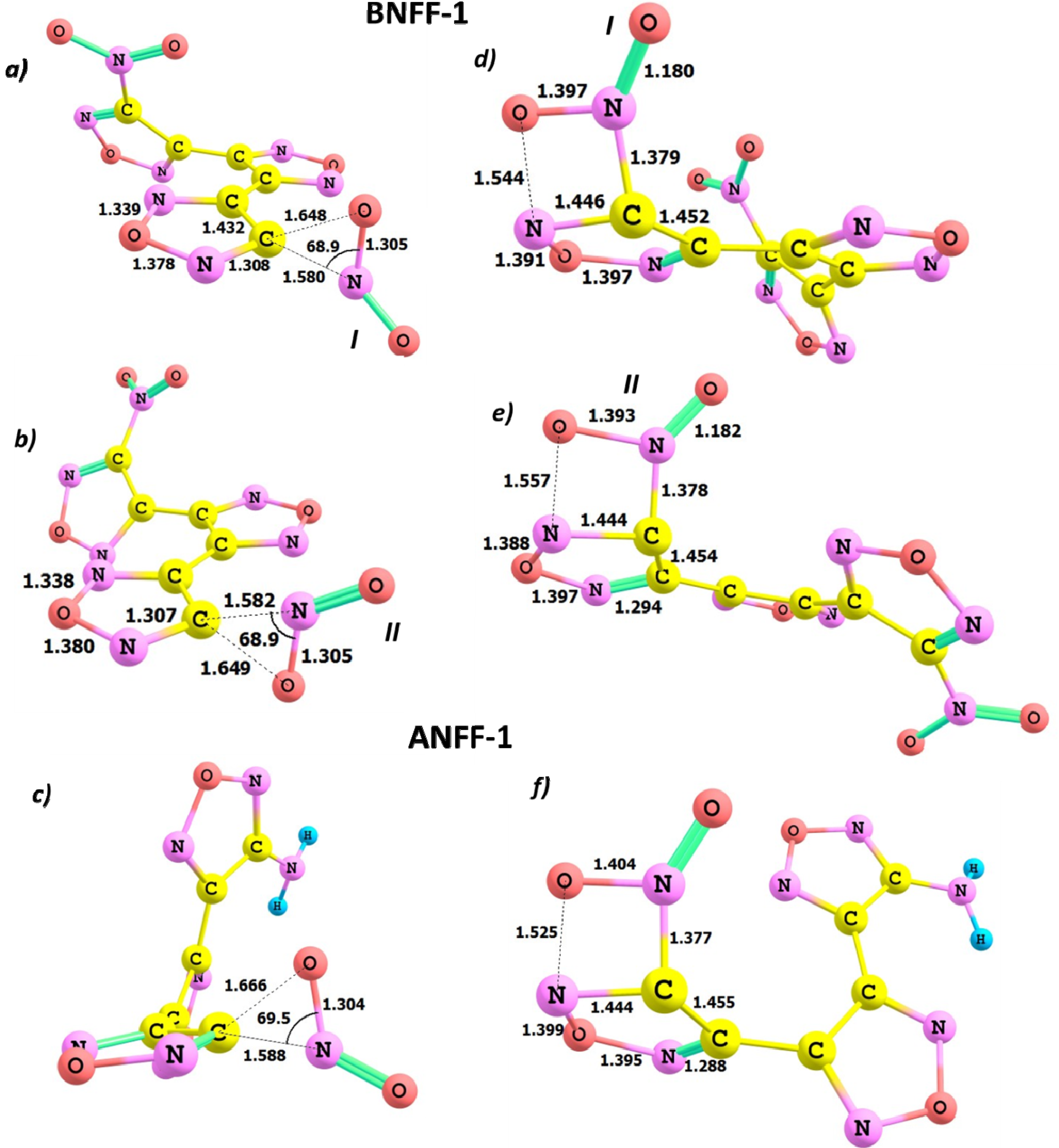
3.1. The Homolytic C-NO2 Break
3.2. The CONO-Isomerization Precursor of the NO Loss


3.3. Oxygen Transfer

3.4. The Homolytic C-C Break
3.5. A Heterocyclic Ring Cleavage (RC)

4. Comparing BNFF-1 and ANFF-1
5. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- See, for example, Fried, L.E.; Manaa, M.R.; Pagoria, P.F.; Simpson, R.L. Design and synthesis of energetic materials. Annu. Rev. Mater. Res. 2001, 31, 291–321. [Google Scholar] [CrossRef]
- See, for example, Kotomin, E.A.; Merkle, R.; Mastrikov, Yu.A.; Kuklja, M.M.; Maier, J. Energy Conversion—Solid Oxide Fuel Cells: First-Principles Modeling of Elementary Processes. In Computational Approaches to Energy Materials; Catlow, R., Sokol, A., Walsh, A., Eds.; John Wiley & Sons: The Atrium, Southern Gate, Chichester, West Sussex, UK, 2013; pp. 149–186. [Google Scholar]
- See, for example, Carter, J.A.; Wang, Z.; Dlott, D.D. Ultrafast nonlinear coherent vibrational sum-frequency spectroscopy methods to study thermal conductance of molecules at interfaces. Acc. Chem. Res. 2009, 42, 1343–1351. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Self-accelerated mechanochemistry in nitroarenes. J. Phys. Chem. Lett. 2010, 1, 363–367. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Molecular mechanisms of Shear-strain sensitivity of energetic crystals DADNE and TATB. J. Energetic Mater. 2010, 28, 66–77. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Interplay of decomposition mechanisms at shear-strain interface. J. Phys. Chem. C 2009, 113, 17–20. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Shear-strain induced chemical reactivity of layered molecular crystals. Appl. Phys. Lett. 2007, 90, 151913. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Shear-strain induced structural and electronic modifications of the molecular crystal 1,1-diamino-2,2-dinitroethylene: Slip-plane flow and band gap relaxation. Phys. Rev. B 2007, 75, 104111. [Google Scholar] [CrossRef]
- Bolton, O.; Simke, L.R.; Pagoria, P.F.; Matzger, A.J. High Power Explosive with Good Sensitivity: A 2: 1 Cocrystal of CL-20: HMX. Cryst. Growth Des. 2012, 12, 4311–4314. [Google Scholar] [CrossRef]
- Millar, D.I.A.; Maynard-Casely, H.E.; Allan, D.R.; Cumming, A.S.; Lennie, A.R.; Mackay, A.J.; Oswald, I.D.H.; Tang, C.C.; Pulham, C.R. Crystal engineering of energetic materials: Co-crystals of CL-20. CrystEngComm 2012, 14, 3742–3749. [Google Scholar] [CrossRef]
- Göbel, M.; Klapötke, T.M. Development and testing of energetic materials: the concept of high densities based on the trinitroethyl functionality. Adv. Funct. Mater. 2009, 19, 347–365. [Google Scholar] [CrossRef]
- Kimmel, A.V.; Sushko, P.V.; Kuklja, M.M. The structure and decomposition chemistry of isomer defects in a crystalline DADNE. J. Energetic Mater. 2010, 28, 128–140. [Google Scholar] [CrossRef]
- Sharia, O.; Kuklja, M.M. Rapid materials degradation induced by surfaces and voids: ab initio modeling of β-octatetramethylene tetranitramine. J. Am. Chem. Soc. 2012, 134, 11815–11820. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Modeling of defect induced phenomena in energetic materials. In Static Compression of Energetic Materials; Peiris, S., Piermarini, G., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2008; pp. 322–361. [Google Scholar]
- Kuklja, M.M. On the Initiation of chemical reactions by electronic excitations in molecular solids. Appl. Phys. A 2003, 76, 359–366. [Google Scholar] [CrossRef]
- Loebbecke, S.; Schuppler, H.; Schweikert, W. Thermal Properties of Different Substituted Energetic Furoxans. In Energetic Materials: Synthesis, Production and Application, 33rd ed., Proceedings of International Annual Conference of ICT, Karlsruhe, Germany, 25–28 June 2002.
- Zheng, W.; Wang, J.; Ren, X.; Zhang, L.; Zhou, Y. An investigation on thermal decomposition of BNFF-CMDB propellants. Propel. Explos. Pyrotech. 2007, 32, 520–524. [Google Scholar] [CrossRef]
- Kotomin, A.A.; Kozlov, A.S.; Dushenok, S.A. Detonatability of high-energy-density heterocyclic compounds. Russ. J. Phys. Chem. 2007, 1, 573–575. [Google Scholar]
- Wang, J.; Dong, H.S.; Huang, Y.G.; Li, J.S. Properties of 3,4-Bis(nitrofurazano)furoxan. In Energetic Materials: Insensitivity, Ageing, Monitoring, Proceedings of the 37th International Annual Conference of ICT, Karlsruhe, Germany, 27–30 June 2006.
- Stepanov, A.I.; Dashko, D.V.; Astrat’ev, A.A. 3,4-Bis(4′-nitrofurazan-3′-yl)furoxan: a melt cast powerful explosive and a valuable building block in 1,2,5-Oxadiazole chemistry. Cent. Eur. J. Energetic Mater. 2012, 9, 329–342. [Google Scholar]
- Feng-qi, Z.; Pei, C.; Rong-zu, Hu; Yang, L.; Zhi-zhong, Z.; Yan-shui, Z.; Xu-wu, Y.; Yin, G.; Sheng-li, G.; Qi-zhen, S. Thermochemical properties and non-isothermal decomposition reaction kinetics of 3,4-dinitrofurazanfuroxan (DNTF). J. Haz. Mater. 2004, 113, 67–71. [Google Scholar]
- Wei, T.; Wu, J.; Zhu, W.; Zhang, C.; Xiao, H. Characterization of nitrogen-bridged 1,2,4,5-tetrazine-,furazan-, and 1H-tetrazole-based polyheterocyclic compounds: heats of formation, thermal stability, and detonation properties. J. Mol. Model. 2012, 18, 3467–3479. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, W.; Xiao, H. Comparative theoretical studies of energetic substituted carbon- and nitrogen-bridged difurazans. J. Phys. Chem. A 2010, 114, 603–612. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, W.; Xiao, H. Theoretical studies on heats of formation, detonation properties, and bond dissociation energies of monofurazan derivatives. Int. J. Quant. Chem. 2010, 110, 1549–1558. [Google Scholar]
- Wang, L.; Yi, C.; Zou, H.; Liu, Y.; Li, S. Theoretical study on the thermal decomposition mechanism of 3,3’-dinitro-4,4’-azoxyfurazan. Comput. Theoret. Chem. 2011, 963, 135–140. [Google Scholar] [CrossRef]
- Guo, Y.Q.; Bhattacharya, A.; Bernstein, E.R. Excited electronic state decomposition of furazan based energetic materials: 3,3'-diamino-4,4'-azoxyfurazan and its model systems, diaminofurazan and furazan. J. Chem. Phys. 2008, 128, 34303. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Guo, Y.Q.; Bernstein, E.R. Nonadiabatic reaction of energetic molecules. Acc. Chem. Res. 2010, 43, 1476–1485. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, Noncovalent interactions, Excited states, And transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef]
- Hanggi, P; Talkner, P; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C. Variational transition state theory. Ann. Rev. Phys. Chem. 1984, 35, 159–189. [Google Scholar] [CrossRef]
- Sharia, O.; Kuklja, M.M. Ab Initio kinetics of gas phase decomposition reactions. J. Phys. Chem. A 2010, 114, 12656–12661. [Google Scholar] [CrossRef]
- Sharia, O.; Kuklja, M.M. Modeling Thermal decomposition mechanisms in Gaseous and Crystalline molecular materials: Application to β-HMX. J. Phys. Chem. B 2011, 115, 12677–12686. [Google Scholar] [CrossRef]
- Sharia, O.; Kuklja, M.M. Surface-enhanced decomposition kinetics of molecular materials illustrated with cyclotetramethylene-tetranitramine. J. Phys. Chem. C 2012, 116, 11077–11081. [Google Scholar] [CrossRef]
- Brill, T.B. Energetic Materials: Detonation, Combustion; Politzer, P.A., Murray, J.S., Eds.; Elsevier: Amsterdam, The Netherland and New York, NY, USA, 2003. [Google Scholar]
- Brill, T.B.; James, K.J. Kinetics and mechanisms of thermal decomposition of nitroaromatlc explosives. Chem. Rev. 1993, 93, 2667–2692. [Google Scholar] [CrossRef]
- Brill, T.B.; James, K.J. Thermal decomposition of energetic materials. 61. Perfidy in the amino-2,4,6-trinitrobenzene series of explosives. J. Phys. Chem. 1993, 97, 8752. [Google Scholar] [CrossRef]
- Zhang, C. Computational investigation of the detonation properties of furazans and furoxans. J. Mol. Struct. THEOCHEM 2006, 765, 77–83. [Google Scholar] [CrossRef]
- Korolev, V.L.; Petukhova, T.V.; Pivina, T.S.; Porollo, A.A.; Sheremetev, A.B.; Suponitskii, K.Yu.; Ivshin, V.P. Thermal decomposition mechanisms of nitro-1,2,4-triazoles: a theoretical study. Russ. Chem. Bull. 2006, 55, 1388–1410. [Google Scholar] [CrossRef]
- Wu, C.J.; Fried, L.E. Ring closure mediated by intramolecular hydrogen transfer in the decomposition of a push−pull nitroaromatic: TATB. J. Phys. Chem. A 2000, 104, 6447–6452. [Google Scholar] [CrossRef]
- Rice, B.M.; Sahu, S.; Owens, F.J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J. Mol. Struct. THEOCHEM. 2002, 583, 69–72. [Google Scholar] [CrossRef]
- Shao, J.; Cheng, X.; Yang, X. The C-NO2 bond dissociation energies of some nitroaromatic compounds: DFT study. Struct. Chem. 2006, 17, 547–550. [Google Scholar] [CrossRef]
- Kimmel, A.V.; Sushko, P.V.; Shluger, A.L.; Kuklja, M.M. Effect of charged and excited states on the decomposition of 1,1-diamino-2,2-dinitroethylene molecules. J. Chem. Phys. 2007, 126, 234711–234721. [Google Scholar] [CrossRef]
- Politzer, P.; Concha, M.C.; Grice, M.E.; Murray, J.S.; Lane, P.; Habibollazadeh, D. Computational investigation of the structures and relative stabilities of amino/nitro derivatives of ethylene. J. Mol. Struct. THEOCHEM 1998, 452, 75–83. [Google Scholar] [CrossRef]
- This heat can be expended to facilitate the further decomposition, which is especially important for the condensed phase processes.
- Gindulyté, A.; Masaa, L.; Huang, L.; Karle, J. Proposed mechanism of 1,1-Diamino-dinitroethylene decomposition: A density functional theory study. J. Phys. Chem. A 1999, 103, 11045–11051. [Google Scholar]
- Wodtke, A.M.; Hintsa, E.J.; Lee, Y.T. Infrared multiphoton dissociation of three nltroalkanes. J. Phys. Chem. 1986, 90, 3549–3558. [Google Scholar] [CrossRef]
- Saxon, R.P.; Yoshimine, M. Theoretical study of nitro-nitrite rearrangement of CH3NO2. Can. J. Chem. 1992, 70, 572–579. [Google Scholar] [CrossRef]
- Gindulyté, A.; Masaa, L.; Huang, L.; Karle, J. Ab Initio study of unimolecular decomposition of Nitroethylene. J. Phys. Chem. A 1999, 103, 11040–11044. [Google Scholar] [CrossRef]
- Xu, S.; Lin, M.C. Computational study on the kinetics and mechanism for the unimolecular decomposition of C6H5NO2 and the related C6H5 + NO2 and C6H5O + NO reactions. J. Phys. Chem. B 2005, 109, 8367–8373. [Google Scholar] [CrossRef]
- Fayet, G.; Joubert, L.; Rotureau, P.; Adamo, C. Theoretical Study of the Decomposition Reactions in Substituted Nitrobenzenes. J. Phys. Chem. A 2008, 112, 4054–4059. [Google Scholar] [CrossRef]
- Cohen, R.; Zeiri, Y.; Wurzberg, E.; Kosloff, R. Mechanism of Thermal Unimolecular Decomposition of TNT (2,4,6-Trinitrotoluene): A DFT Study. J. Phys. Chem. A 2007, 111, 11074–11083. [Google Scholar] [CrossRef]
- Kimmel, A.V.; Sushko, P.V.; Shluger, A.L.; Kuklja, M.M. Effect of Molecular and Lattice Structure on Hydrogen Transfer in Molecular Crystals of Diamino-Dinitroethylene and Triamino-Trinitrobenzene. J. Phys. Chem. A 2008, 112, 4496–4500. [Google Scholar] [CrossRef]
- Sharia, O.; Tsyshesvky, R.; Kuklja, M.M. Surface-accelerated decomposition chemistry of δ-HMX. J. Phys. Chem. Lett. 2013, 4, 730–734. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsyshevsky, R.V.; Kuklja, M.M. Decomposition Mechanisms and Kinetics of Novel Energetic Molecules BNFF-1 and ANFF-1: Quantum-Chemical Modeling. Molecules 2013, 18, 8500-8517. https://doi.org/10.3390/molecules18078500
Tsyshevsky RV, Kuklja MM. Decomposition Mechanisms and Kinetics of Novel Energetic Molecules BNFF-1 and ANFF-1: Quantum-Chemical Modeling. Molecules. 2013; 18(7):8500-8517. https://doi.org/10.3390/molecules18078500
Chicago/Turabian StyleTsyshevsky, Roman V., and Maija M. Kuklja. 2013. "Decomposition Mechanisms and Kinetics of Novel Energetic Molecules BNFF-1 and ANFF-1: Quantum-Chemical Modeling" Molecules 18, no. 7: 8500-8517. https://doi.org/10.3390/molecules18078500
APA StyleTsyshevsky, R. V., & Kuklja, M. M. (2013). Decomposition Mechanisms and Kinetics of Novel Energetic Molecules BNFF-1 and ANFF-1: Quantum-Chemical Modeling. Molecules, 18(7), 8500-8517. https://doi.org/10.3390/molecules18078500