Comprehensive Theoretical Studies on the Reaction of 1-Bromo-3,3,3-trifluoropropene with OH Free Radicals

Abstract
:
1. Introduction
2. Results and Discussion
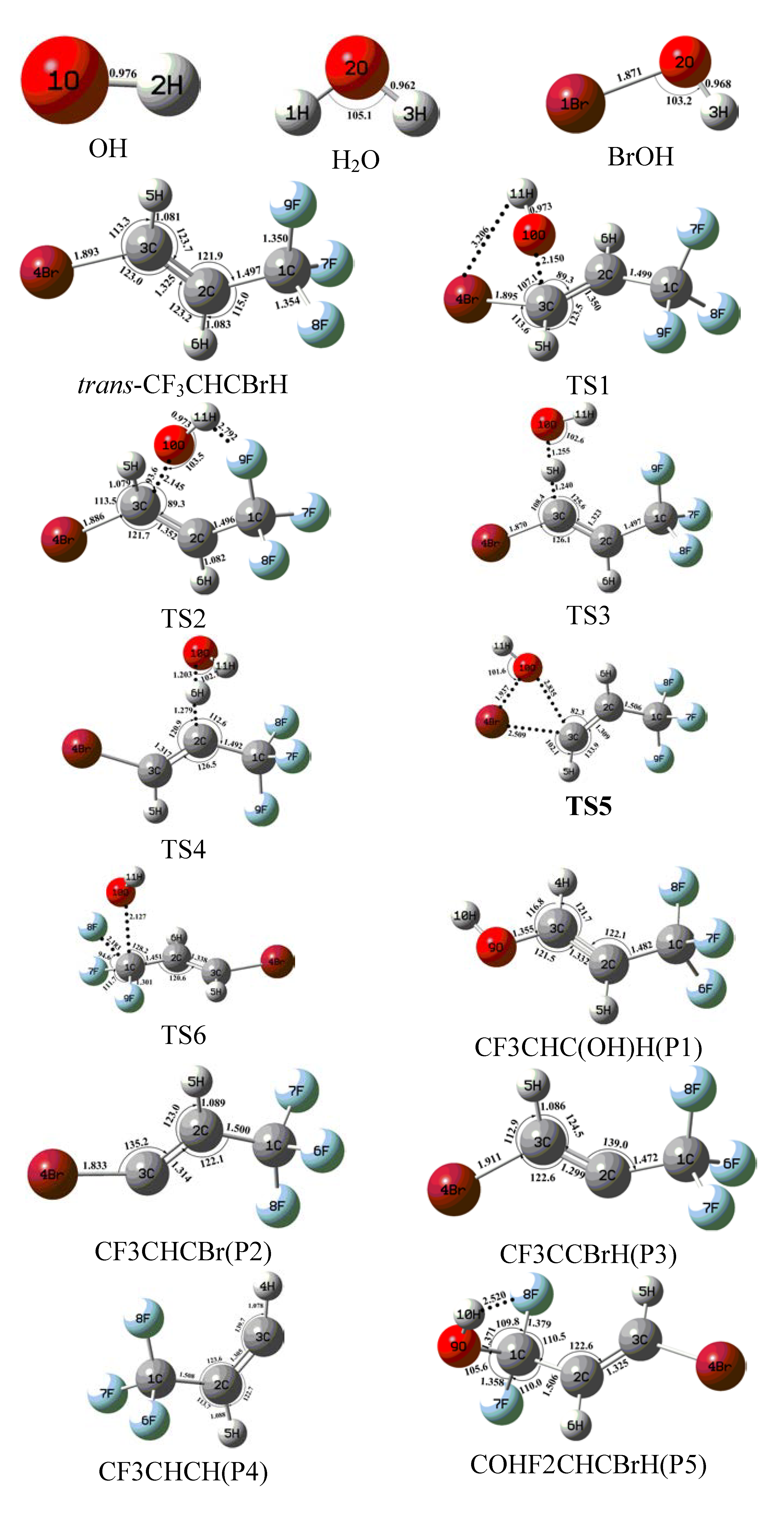

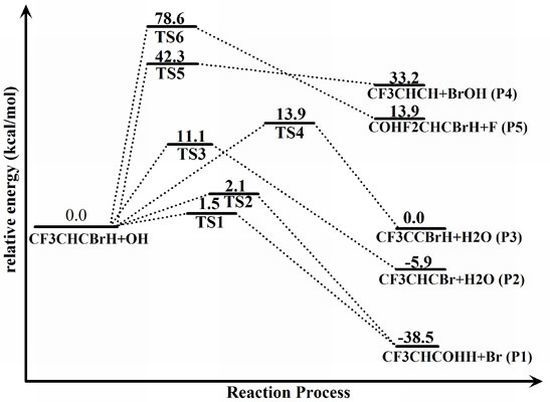
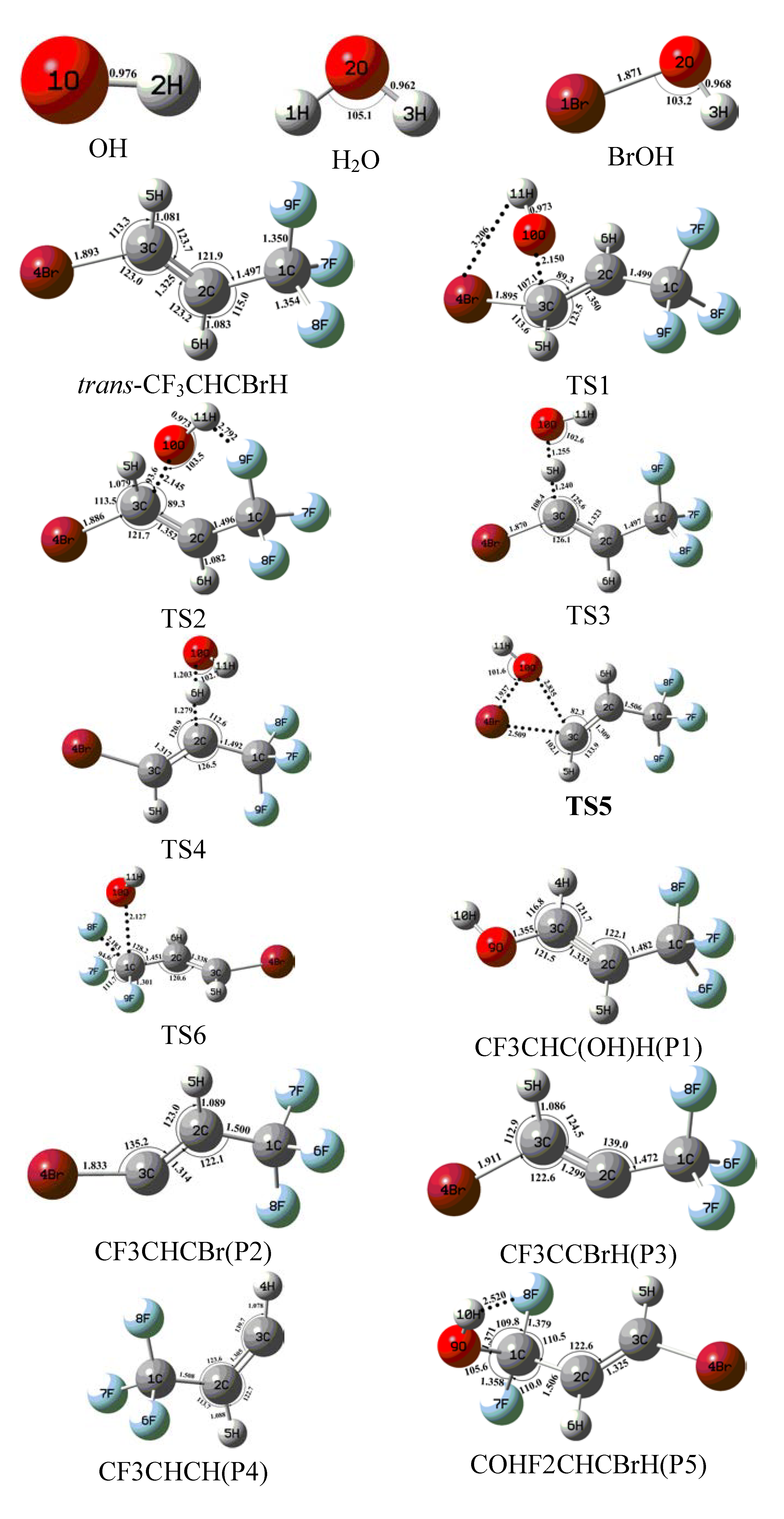
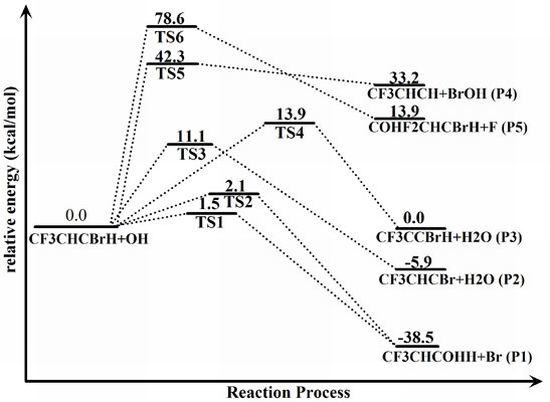{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Column heading | Species | ΔE(CCSD) | ΔE(DFT) | ZPE | ΔH298(CCSD) |
|---|---|---|---|---|---|
| Reactants | trans-CF3CHCBrH + OH | 0.0 | 0.0 | 34.9 | 0.0 |
| Transition states | TS1 | 1.5 | −1.4 | 36.5 | 2.3 |
| TS2 | 2.1 | −1.0 | 36.4 | 2.8 | |
| TS3 | 11.1 | 3.5 | 32.8 | 8.4 | |
| TS4 | 13.9 | 6.7 | 32.5 | 11.1 | |
| TS5 | 42.3 | 36.5 | 36.5 | 42.6 | |
| TS6 | 78.6 | 59.8 | 36.3 | 79.2 | |
| Products | CF3CHC(OH)H +Br (P1) | −38.5 | −38.8 | 38.2 | −35.3 |
| CF3CHCBr+H2O (P2) | −5.9 | −8.3 | 34.9 | −5.6 | |
| CF3CCBrH+H2O (P3) | 0.0 | −3.3 | 34.5 | 0.1 | |
| CF3CHCH+BrOH (P4) | 33.2 | 31.2 | 34.9 | 32.8 | |
| COHF2CHCBrH+F (P5) | 13.9 | 16.0 | 37.1 | 15.9 |
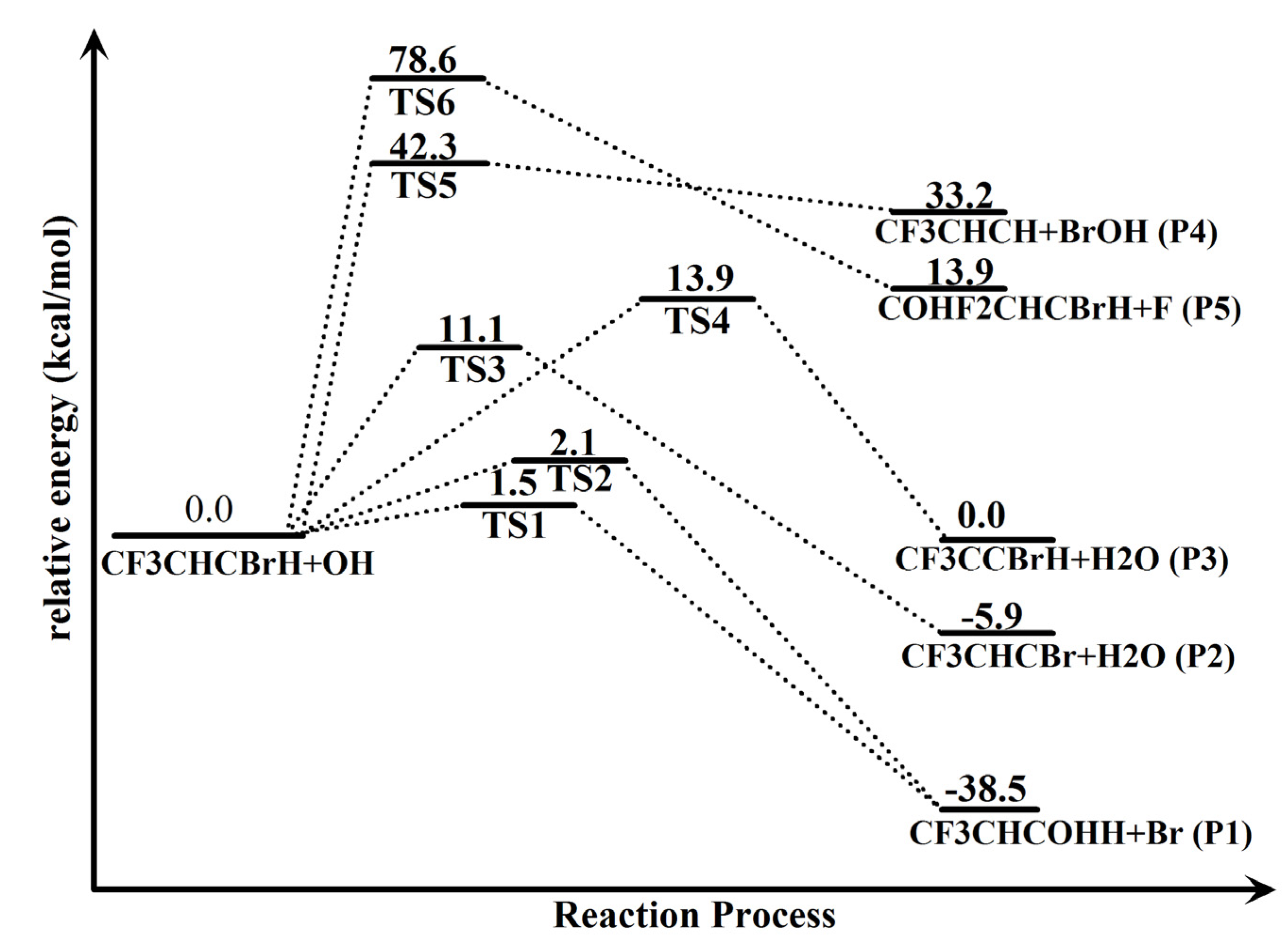
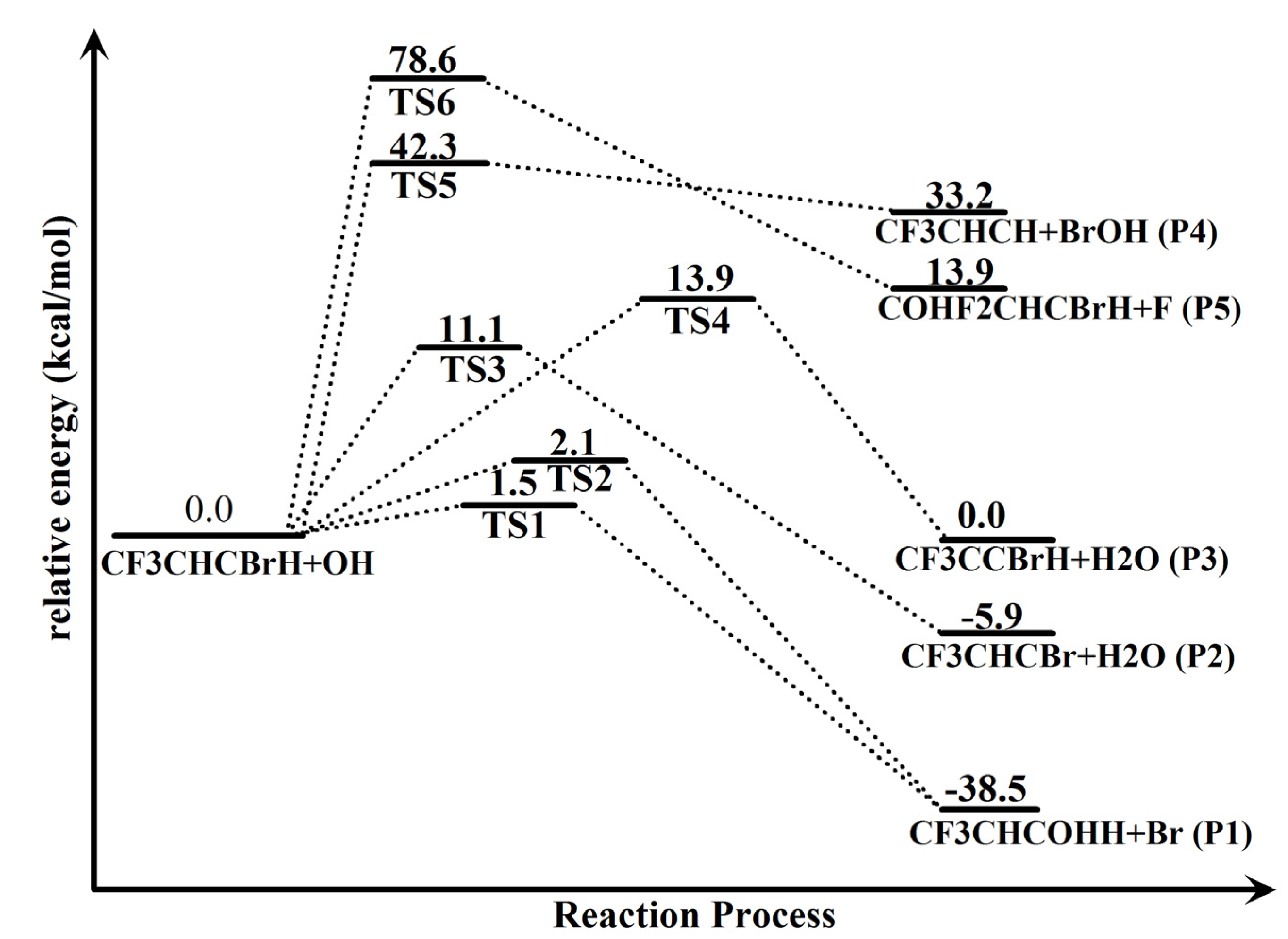
2.1. Conformational Surface
2.2. Reaction Mechanism of Trans-CF3CHCBrH + OH






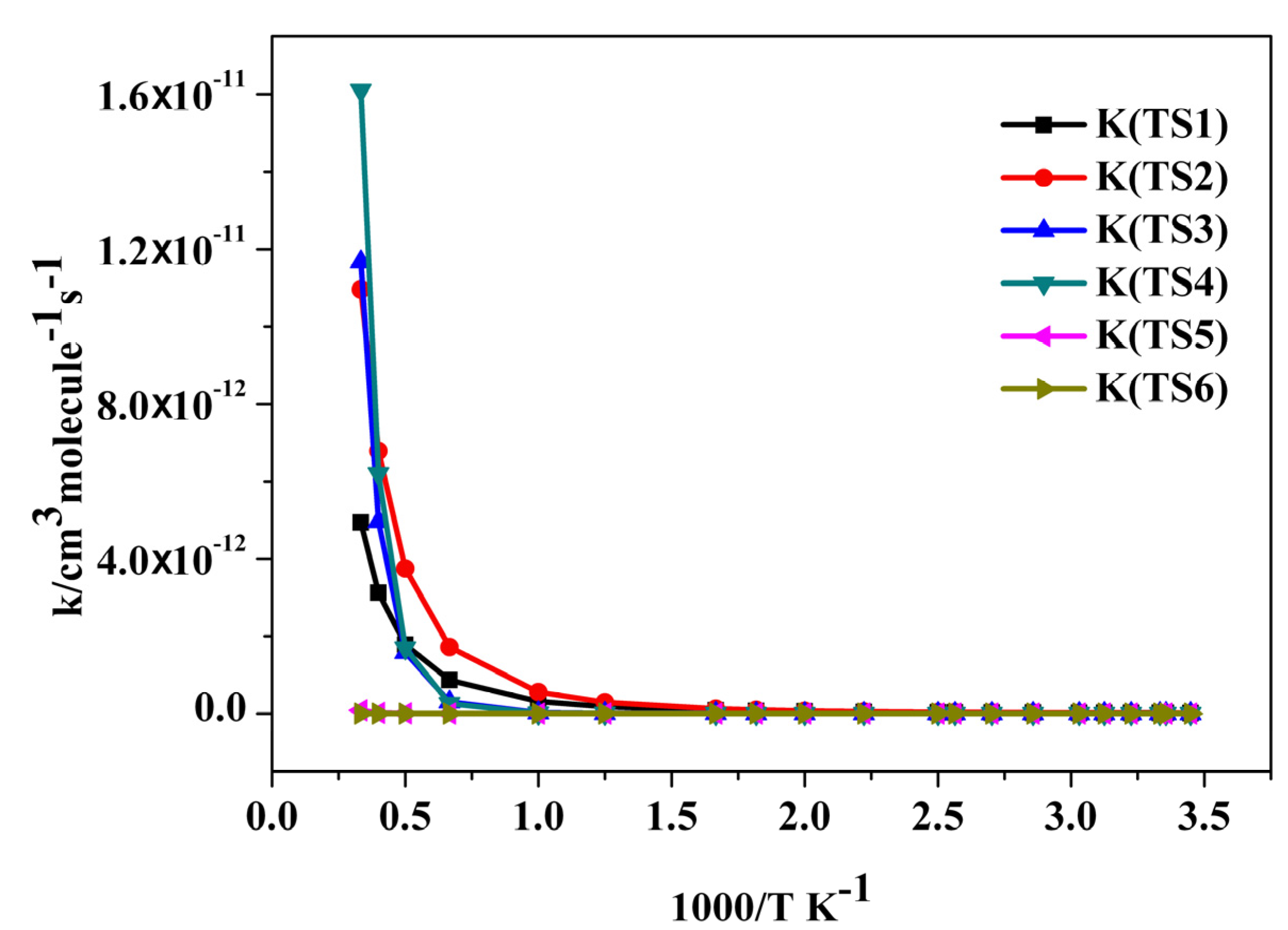
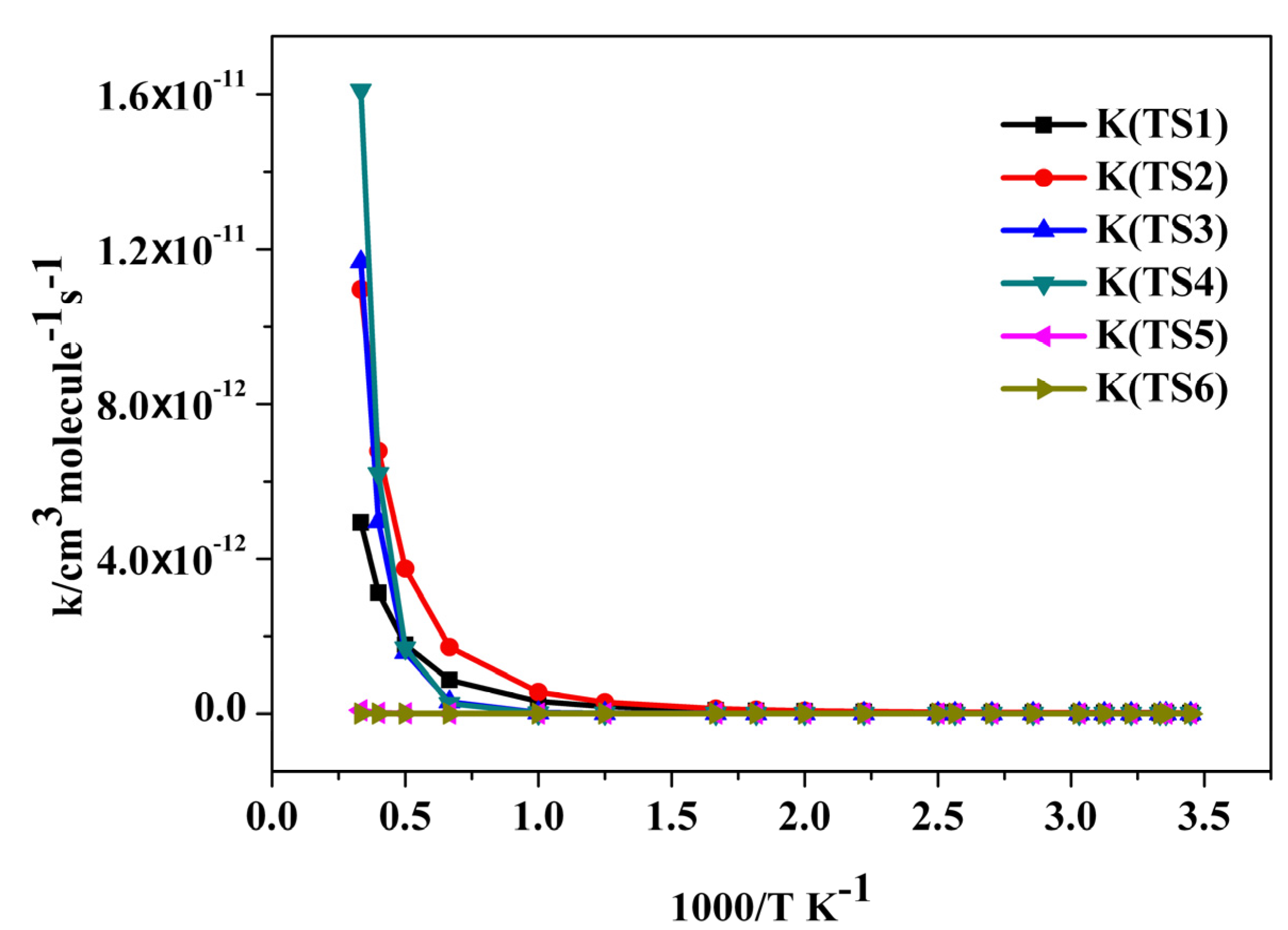
2.3. Reaction Rate Results




3. Computational Methods

 are the continued product of the reactions partition function, ƒ≠′ are the partition function of the transition state, kb is the Boltzman’s constant, and h is the Planck’s constant.
are the continued product of the reactions partition function, ƒ≠′ are the partition function of the transition state, kb is the Boltzman’s constant, and h is the Planck’s constant.
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Proceedings of Halon Options Technical Working Conference, University of New Mexico, Albuquerque, NM, USA, 2–4 May 2000.
- Saso, Y.; Ogawa, Y.; Saito, N. Binary CF3Br- and CHF3-inert flame suppressants: effect of temperature on the flame inhibition effectiveness of CF3Br and CHF3. Combust Flame. 1999, 118, 489–499. [Google Scholar] [CrossRef]
- Mather, J.D.; Robert, E. Troprodegradable bromocarbon extinguishants-compound selection and testing issues. Available online: http://fire.nist.gov/bfrlpubs/fire02/PDF/f02120.pdf (accessed on 7 April 2005).
- Lifke, J.; Martinez, A.; Tapscott, R.E.; Mather, J.D. Tropodegradable bromocarbon extinguishants. Available online: http://www.bfrl.nist.gov/866/NGP/publications/Tropo_Final_Rpt.pdf (accessed on 6 April 2005).
- Mather, J.D.; Tapscott, R.E. Tropodegradable bromocarbon extinguishants—progress overview. In Proceedings of the Halon Options Technical Working Conference, Sheraton Old Town, Albuquerque, NM, USA, May 2000; pp. 2–4, 154–163.
- Gann, R.G. Next-generation Fire Suppression Technology Program (NGP): Technical Highlights. In Proceedings of the Halon Options Technical Working Conference, Sheraton Old Town, Albuquerque, NM, USA, April 1999; pp. 27–29, 87–94.
- Tapscott, R.E.; Mather, J.D.; Moore, T.A. Clean, Tropodegradable Agents with Low Ozone Depletion and Global Warming Potentials to Protect Against Fires and Explosions. U.S. Patent 1998. [Google Scholar]
- Nyden, M.R.; Yang, J.C.; Mather, J.D. Screening of candidate fire suppressants. In Proceedings of the Halon Options Technical Working Conference, Sheraton Old Town, Albuquerque, NM, USA, May 2000; pp. 2–4, 104–114.
- Tapscott, R.E.; Heinonen, E.W.; Lac, J.L.; Mather, J.D.; Moore, T.A. Tropodegradable Bromocarbons as Halon Replacements, Proceedings of Halon Options Technical Working Conference, Sheraton Old Town, Albuquerque, NM, USA, 6–8 May 1991.
- Grzyll, L.R.; Back, D.D. Development of Quantitative Structure-property Relationships for Tropodegradable Halocarbon Fire Suppression Agents, Final Report, SSG Subtask 3.20, Subcontract S-5000.48, ARA, Inc., Tyndall Air Force Base, Florida, Mainstream Engineering Corporation, Rockledge. Florida. Mar. 1997. [Google Scholar]
- Zhang, M.L.; Lin, Z.J. Ab initio studies of the thermal decomposition pathways of 1-bromo-3,3,3-trifluoropropene. J. Mol. Struc.-Theochem. 2009, 899, 98–110. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Jin, X.; Liao, G.X. Experimental study of the fire-extinguishing effectiveness of 1-bromo-3,3,3-trifluoropropene/nitrogen mixtures. J. Fire Sci. 2007, 25, 177–187. [Google Scholar] [CrossRef]
- Johnston, H.S. Gas-Phase Reaction Rate Theory; the Roland Press Co.: New York, NY, USA, 1966. [Google Scholar]
- Laidler, K.J. Theories of Chemical Reaction Rates; McGraw-Hill: New York, NY, USA, 1969. [Google Scholar]
- Weston, R.E.; Schwartz, H.A. Chemical Kinetics; Prentice Hall: New York, NY, USA, 1972. [Google Scholar]
- Rapp, D. Statistical Mechanics; Holt, Reinhard, and Winston: New York, NY, USA, 1972. [Google Scholar]
- Nikitin, E.E. Theory of Elementary Atomic and Molecular Processes in Gases; Claredon Press: Oxford, UK, 1974. [Google Scholar]
- Smith, I.W.M. Kinetics and Dynamics of Elementary Gas Reactions; Butterworth: London, UK, 1980. [Google Scholar]
- Steinfeld, J.I.; Francisco, J.S.; Hase, W.L. Chemical Kinetics and Dynamics; Prentice Hall: Englewood Cliffs, NJ, USA, 1989. [Google Scholar]
- Louis, F.; Gonzalez, C.A.; Sawerysyn, J.P. Ab Initio Study of the Oxidation Reaction of CO by ClO Radicals. J. Phys. Chem. A 2003, 107, 9931–9936. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G.; Bowman, J.M.; Wagner, A.F.; Robie, D.; Arepalli, S.; Presser, N.; Gordon, R.J. Ab initio predictions and experimental confirmation of large tunneling contributions to rate constants and kinetic isotope effects for hydrogen atom transfer reactions. J. Am. Chem. Soc. 1986, 108, 3515–3516. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. II. The effect of the Perdew-Wang generalized-gradient correlation correction. J. Chem. Phys. 1992, 97, 9173–9177. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265. [Google Scholar] [CrossRef]
- Tran, K.M.; McAnoy, A.M.; Bowie, J.H. Do the interstellar molecules CCCO and CCCS rearrange when energised? Org. Biomol. Chem. 2004, 2, 999–1006. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions - the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Page, M.; Mclver, J.W. On evaluating the reaction path Hamiltonian. J. Chem. Phys. 1988, 88, 922–935. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction Path Following in Mass-Weighted Internal Coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Wigner, E.P. Über das Überschreiten von Potentialschwellen bei chemischen Reaktionen. Z. Phys. Chem. B 1932, 19, 203–216. [Google Scholar]
- Garret, B.C.; Truhlar, D.G. Semiclassical tunneling calculations. J. Phys. Chem. 1979, 83, 2921–2926. [Google Scholar] [CrossRef]
- Garret, B.C.; Truhlar, D.G. WKB approximation for the reaction-path Hamiltonian: Application to variational transition state theory, vibrationally adiabatic excited-state barrier heights, and resonance calculation. J. Chem. Phys. 1984, 81, 309–317. [Google Scholar] [CrossRef]
- Skodke, R.T.; Garret, B.C.; Truhlar, D.G. A General Small-Curvature Approximation for Transition-State-Theory Transmission Coefficients. J. Phys. Chem. 1981, 85, 3019–3023. [Google Scholar] [CrossRef]
- Skodje, R.T.; Garret, B.C.; Truhlar, D.G. Vibrationally Adiabatic Models for Reactive Tunneling. J. Chem. Phys. 1982, 77, 5955–5976. [Google Scholar] [CrossRef]
- Garret, B.C.; Truhlar, D.G.; Grev, R.S.; Magnuson, A.W. Improved Treatment of Threshold Contributions in Variational Transition State Theory. J. Chem. Phys. 1980, 84, 1730–1748, Erratum: 1983, 87, 4554. [Google Scholar] [CrossRef]
- Miller, W.H.; Shi, S.H. Unified semiclassical perturbation and infinite order sudden approximation, with application to the reaction path hamiltonian model. J. Chem. Phys. 1981, 75, 2258–2264. [Google Scholar] [CrossRef]
- Miller, W.H.; Smith, F.T. Semiclassical Perturbation Theory of Electron-Molecule Collisions. Phys. Rev. A 1978, 17, 939–953. [Google Scholar] [CrossRef]
- Bell, R.P. The Tunnel Effect in Chemistry; Chapman and Hall: New York, NY, USA, 1980. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Amos, R.D.; Bernhardsson, A.; Berning, A.; Celani, P.; Cooper, D.L.; Deegan, M.J.O.; Dobbyn, A.J.; Eckert, F.; et al. MOLPRO package, A package of ab initio programs; Universitat Stuttgart: Stuttgart, Germany; University of Birmingham: Birmingham, UK, 2002. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, M.; Song, C.; Tian, Y. Comprehensive Theoretical Studies on the Reaction of 1-Bromo-3,3,3-trifluoropropene with OH Free Radicals. Molecules 2013, 18, 7873-7885. https://doi.org/10.3390/molecules18077873
Zhang M, Song C, Tian Y. Comprehensive Theoretical Studies on the Reaction of 1-Bromo-3,3,3-trifluoropropene with OH Free Radicals. Molecules. 2013; 18(7):7873-7885. https://doi.org/10.3390/molecules18077873
Chicago/Turabian StyleZhang, Meiling, Ce Song, and Yan Tian. 2013. "Comprehensive Theoretical Studies on the Reaction of 1-Bromo-3,3,3-trifluoropropene with OH Free Radicals" Molecules 18, no. 7: 7873-7885. https://doi.org/10.3390/molecules18077873