A Fully Automated Radiosynthesis of [18F]Fluoroethyl-Diprenorphine on a Single Module by Use of SPE Cartridges for Preparation of High Quality 2-[18F]Fluoroethyl Tosylate

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
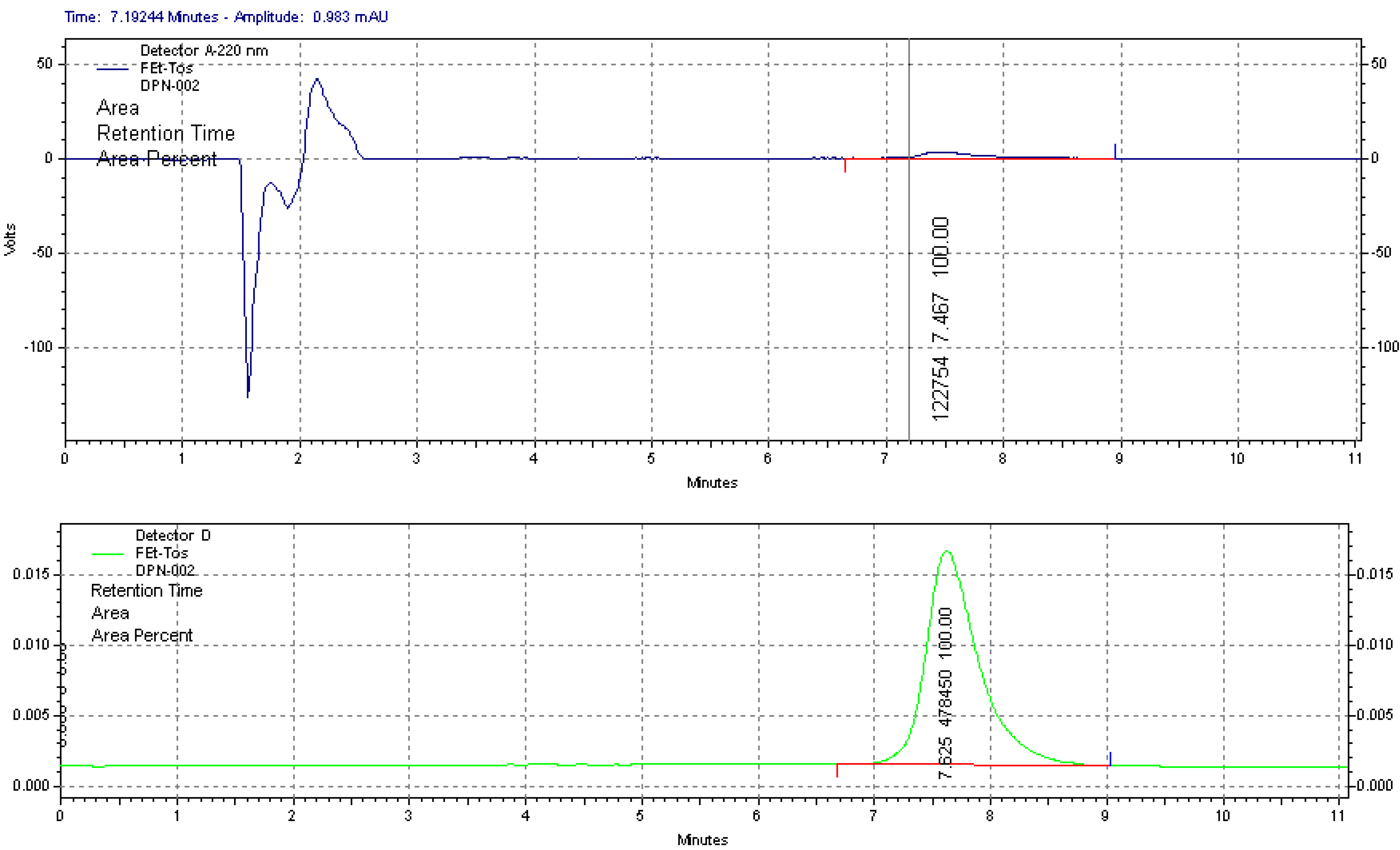
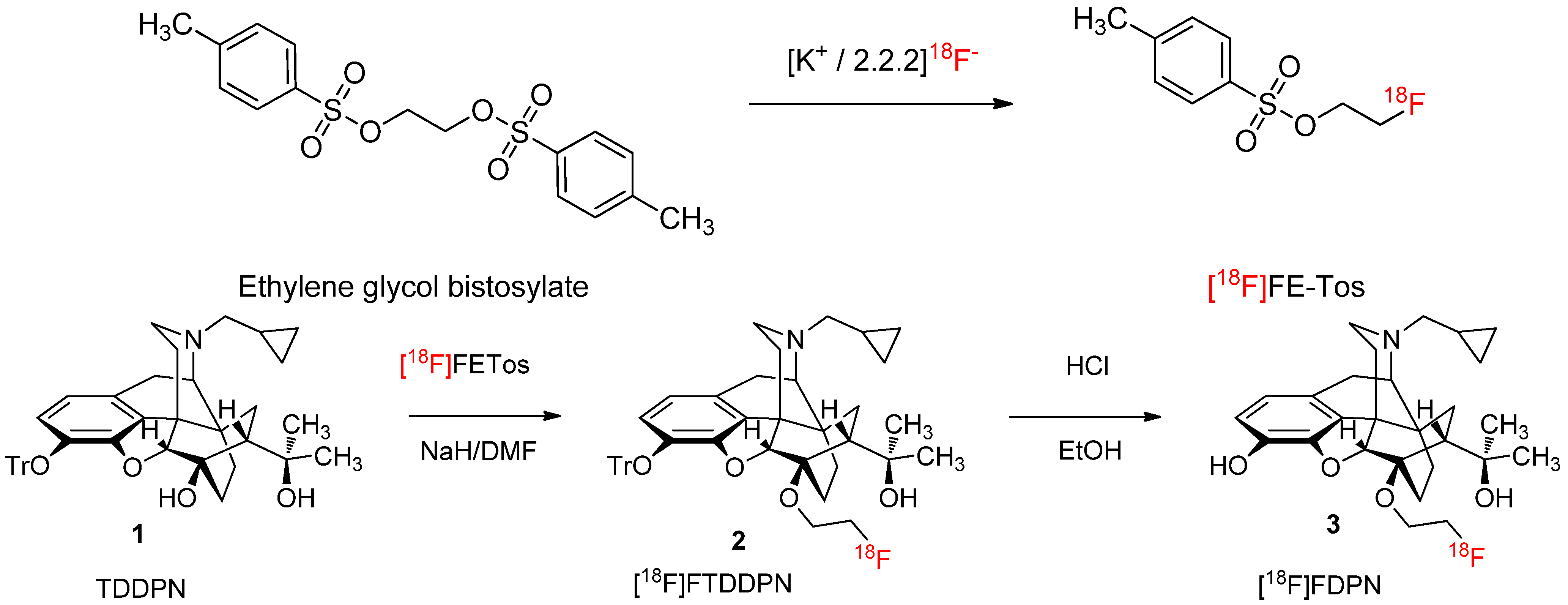
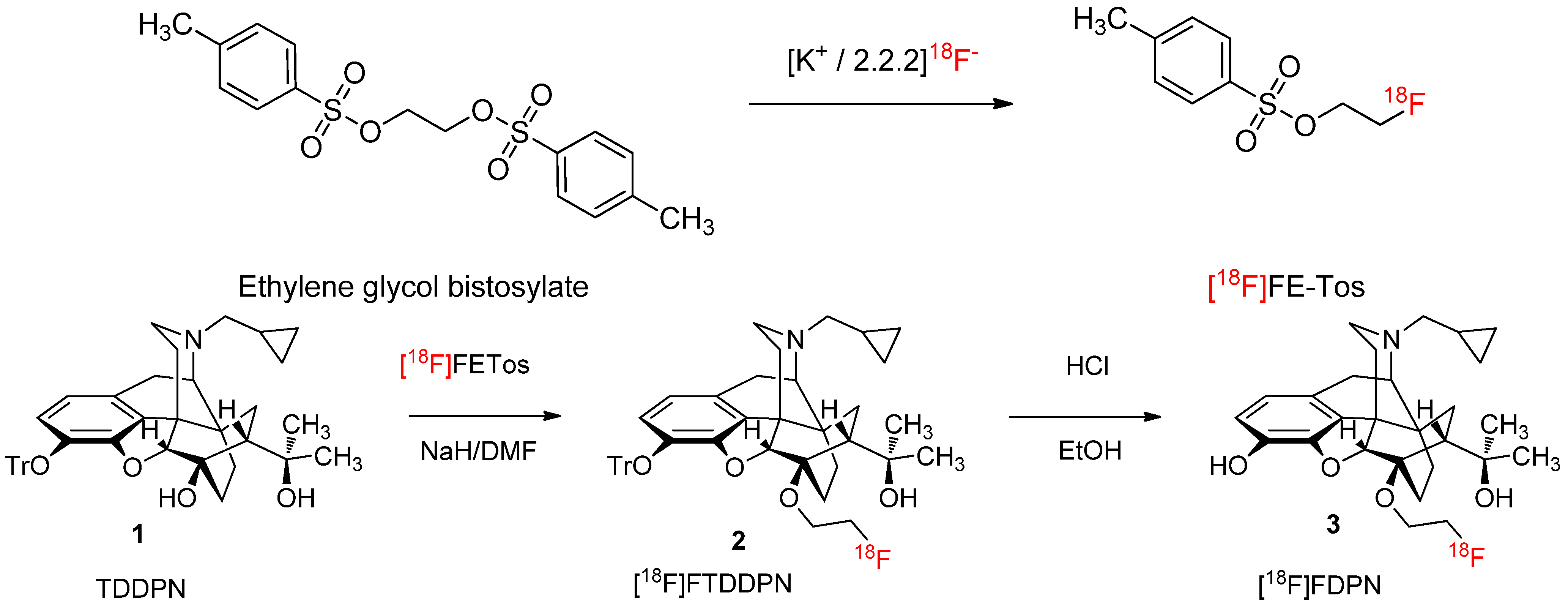
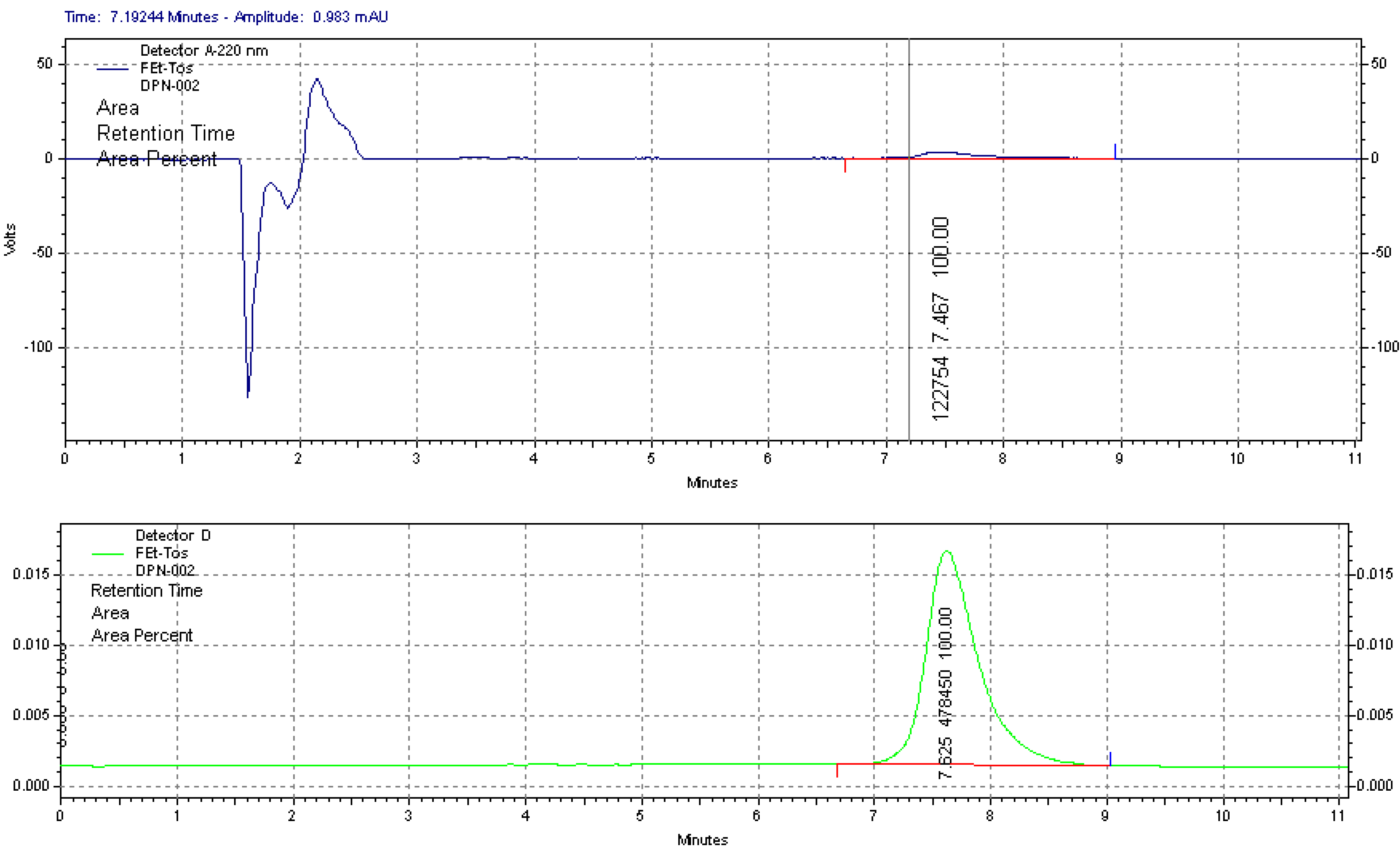
2.1.1. Radiolabelling and Purification of [18F]FETos
2.1.2. Production Runs

2.1.3. 18F-Fluoroalkylation and Purification of [18F]Fluoroethyldiprenorphine
3. Experimental
3.1. General Methods
3.2. Radiolabelling
3.3. 18F-Fluoroalkylation of 3-O-Trityl-6-O-desmethyl-Diprenorphine
3.4. Quality Control
4. Conclusions
Abbreviations
| PET | positron emission tomography |
| SPE | solid phase extraction |
| HPLC | high performance liquid chromatography |
| [18F]FETos | 2-[18F]fluoroethyl tosylate |
| [18F]FDPN | [18F]fluoro-ethyldiprenorphine |
| TDDPN | 3-O-trityl-6-O-desmethyldiprenorphine |
| HBIII | Hot Box III, synthesis module |
| TLC | thin layer chromatography |
| PBS | phosphate buffered saline |
Acknowledgments
Conflicts of Interest
References
- Schirrmacher, R.; Wängler, C.; Schirrmacher, E. Recent developments and trends in 18F-radiochemsitry: Syntheses and applications. Minirev. Org. Chem. 2007, 4, 317–329. [Google Scholar] [CrossRef]
- Wadsak, W.; Mien, L.-K.; Ettlinger, D.E.; Eidherr, H.; Haeusler, D.; Sindelar, K.-M.; Keppler, B.K.; Dudczak, R.; Kletter, K.; Mitterhauser, M. 18F-fluoroethylations: Different strategies for the rapid translation of 11C-methylated radiotracers. Nucl. Med. Biol. 2007, 34, 1019–1028. [Google Scholar] [CrossRef]
- Musachio, J.L.; Shah, J.; Pike, V.W. Radiosynthesis and reactivities of novel [18F]2-fluoro-ethyl arylsulfonates. J. Labelled Compd. Radiopharm. 2005, 48, 735–747. [Google Scholar] [CrossRef]
- Bauman, A.; Piel, M.; Schirrmacher, R.; Rösch, F. Efficient alkali iodide promoted 18F-fluoroethylations with 2-[18F]fluoroethyltosylate and 1-bromo-2-[18F]fluoroethane. Tetrahedron Lett. 2003, 44, 9165–9167. [Google Scholar] [CrossRef]
- Block, D.; Coenen, H.H.; Stöcklin, G. The n.c.a. nucleophilic 18F-fluorination of 1,N-disubstituted alkanes as fluoralkylation agents. J. Labelled Compd. Radiopharm. 1987, 24, 1029–1042. [Google Scholar] [CrossRef]
- Wester, H.J.; Herz, M.; Weber, W.; Heiss, P.; Senekowitch-Schmidtke, R.; Schwaiger, M.; Stöcklin, G. Synthesis and Radiopharmacology of O-(2-[18F]fluoroethyl-l-Tyrosine for tumor imaging. J. Nucl. Med. 1999, 40, 205–212. [Google Scholar]
- Wester, H.J.; Willoch, F.; Tölle, T.R.; Munz, F.; Herz, M.; Øye, I.; Schadrack, J.; Schwaiger, M.; Bartenstein, P. 6-O-(2-[18F]fluoroethyl)-6-O-desmethyldiprenorphine ([18F]DPN): Synthesis, biological evaluation, and comparison with [11C]DPN in humans. J. Nucl. Med. 2000, 41, 1279–1286. [Google Scholar]
- Schreckenberger, M.; Klega, A.; Gründer, G.; Buchholz, H.-G.; Scheurich, A.; Schirrmacher, R.; Schirrmacher, E.; Müller, C.; Henriksen, G.; Bartenstein, P. Opioid receptor PET reveals the psychobiologic correlates of reward processing. J. Nucl. Med. 2008, 49, 1257–1261. [Google Scholar]
- Mueller, C.; Klega, A.; Buchholz, H.G.; Rolke, R.; Magerl, W.; Schirrmacher, R.; Schirrmacher, E.; Birklein, F.; Treede, R.-D.; Schreckenberger, M. Basal opioid receptor binding is associated with differences in sensory perception in healthy human subjects: A [18F]diprenorphine PET study. Neuroimage 2010, 49, 731–737. [Google Scholar] [CrossRef]
- Klega, A.; Eberle, T.; Buchholz, H.-G.; Maus, S.; Maihöfner, C.; Schreckenberger, M.; Birklein, F. Central opioidergic neurotransmission in complex regional pain syndrome. Neurology 2010, 75, 129–136. [Google Scholar] [CrossRef]
- Marton, J.; Schoultz, B.W.; Hjørnevik, T.; Drzezga, A.; Yousefi, B.; Wester, H.-J.; Willoch, F.; Henriksen, G. Synthesis and Evaluation of a full-agonist orvinol for PET-imaging of opioid receptors: [11C]PEO. J. Med. Chem. 2009, 52, 5586–5589. [Google Scholar] [CrossRef]
- Sample Availability: Samples of fluoroethyl-Diprenorphine, protected precursors of Diprenorphine, flouroethyltosylate and ethylene glycol bistosylate are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schoultz, B.W.; Reed, B.J.; Marton, J.; Willoch, F.; Henriksen, G. A Fully Automated Radiosynthesis of [18F]Fluoroethyl-Diprenorphine on a Single Module by Use of SPE Cartridges for Preparation of High Quality 2-[18F]Fluoroethyl Tosylate. Molecules 2013, 18, 7271-7278. https://doi.org/10.3390/molecules18067271
Schoultz BW, Reed BJ, Marton J, Willoch F, Henriksen G. A Fully Automated Radiosynthesis of [18F]Fluoroethyl-Diprenorphine on a Single Module by Use of SPE Cartridges for Preparation of High Quality 2-[18F]Fluoroethyl Tosylate. Molecules. 2013; 18(6):7271-7278. https://doi.org/10.3390/molecules18067271
Chicago/Turabian StyleSchoultz, Bent W., Brian J. Reed, János Marton, Frode Willoch, and Gjermund Henriksen. 2013. "A Fully Automated Radiosynthesis of [18F]Fluoroethyl-Diprenorphine on a Single Module by Use of SPE Cartridges for Preparation of High Quality 2-[18F]Fluoroethyl Tosylate" Molecules 18, no. 6: 7271-7278. https://doi.org/10.3390/molecules18067271