New Tacrine Analogs as Acetylcholinesterase Inhibitors — Theoretical Study with Chemometric Analysis

 ,
,
Abstract
:
1. Introduction
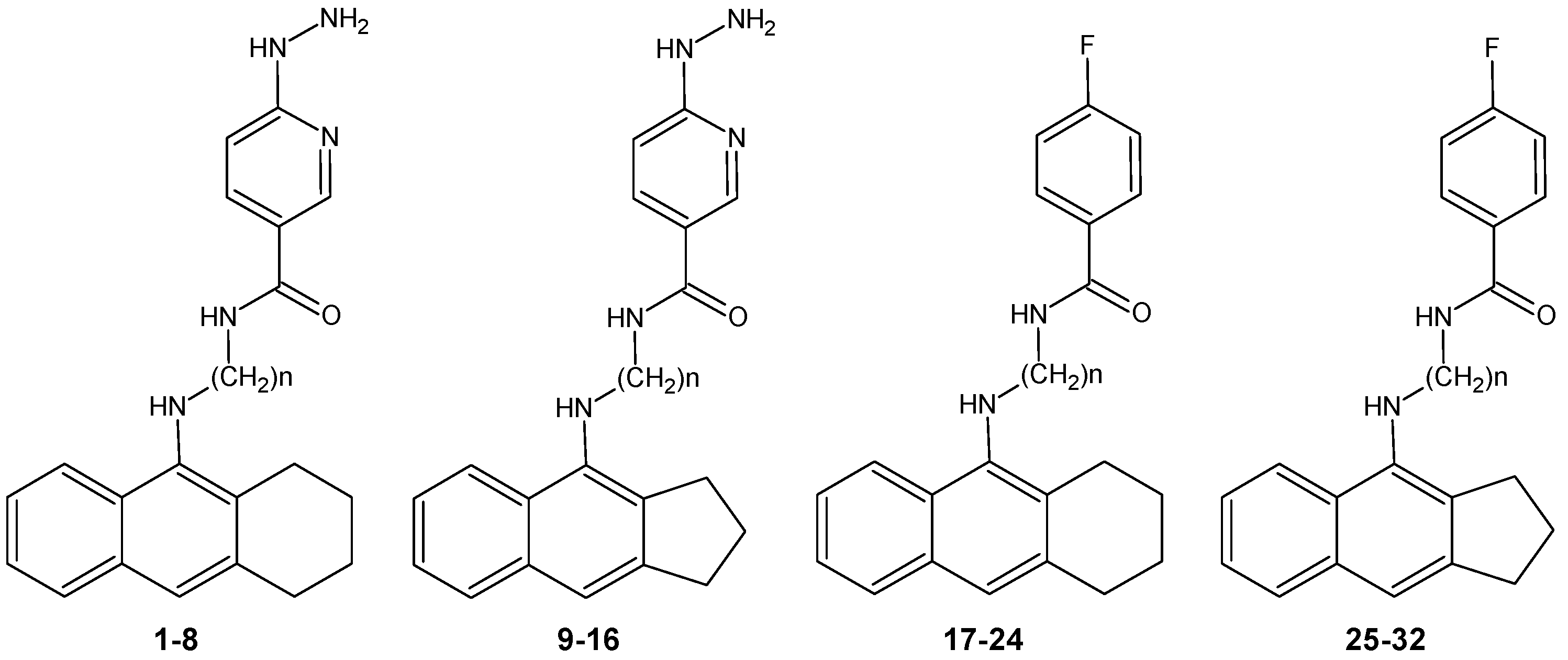
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Results and Discussion
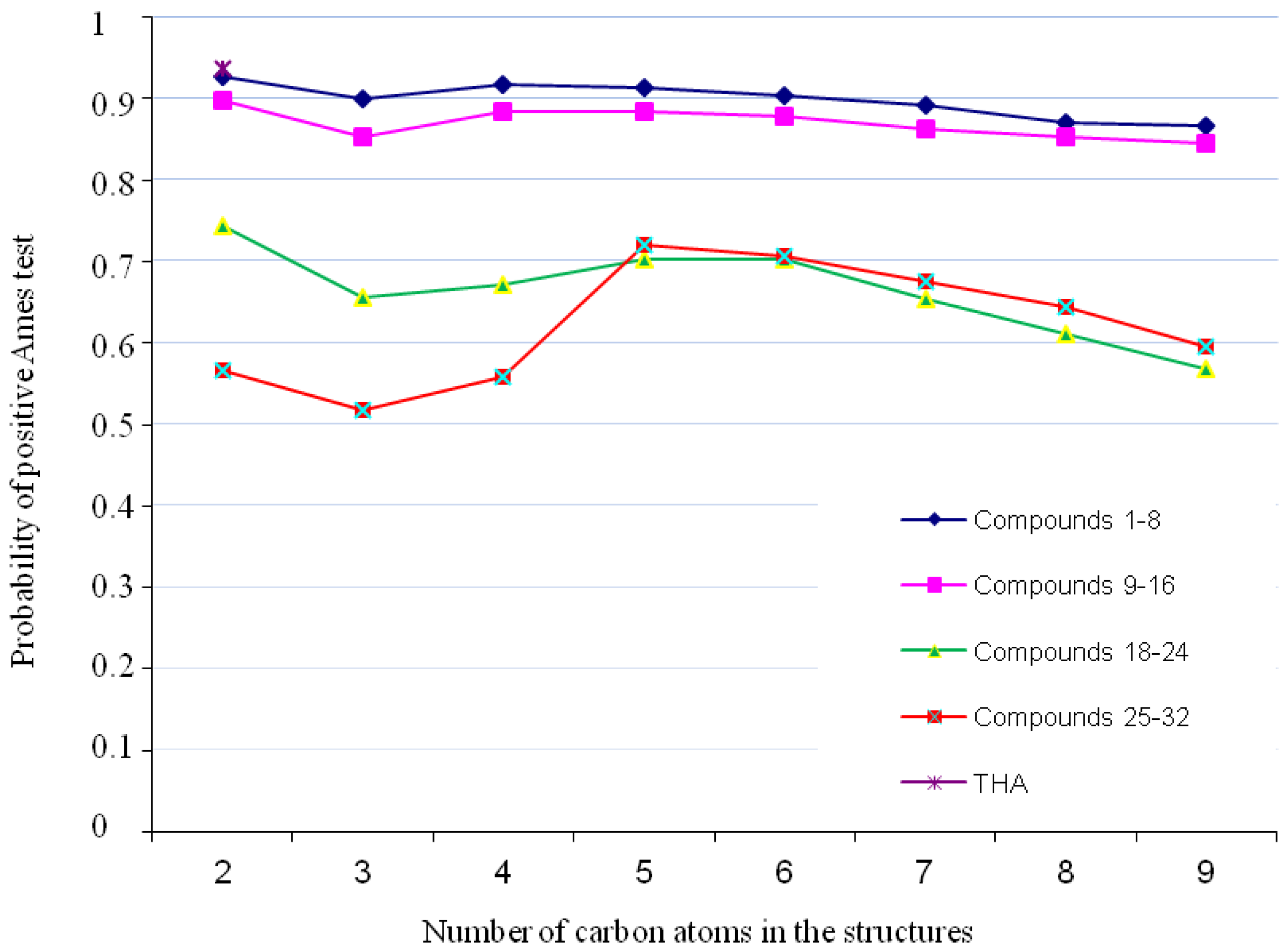
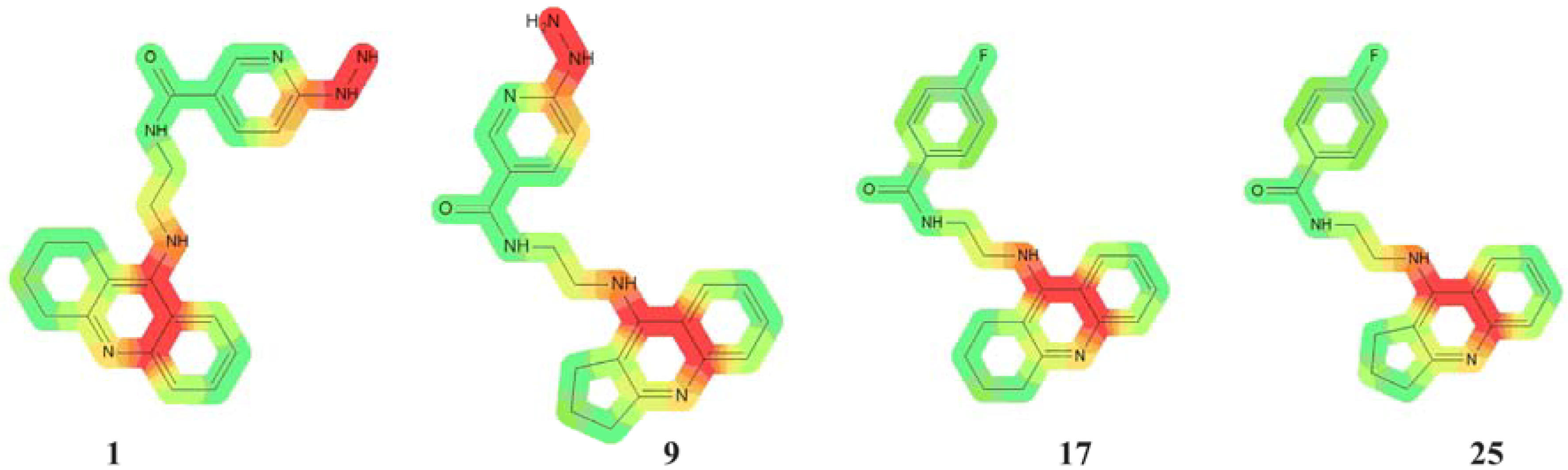
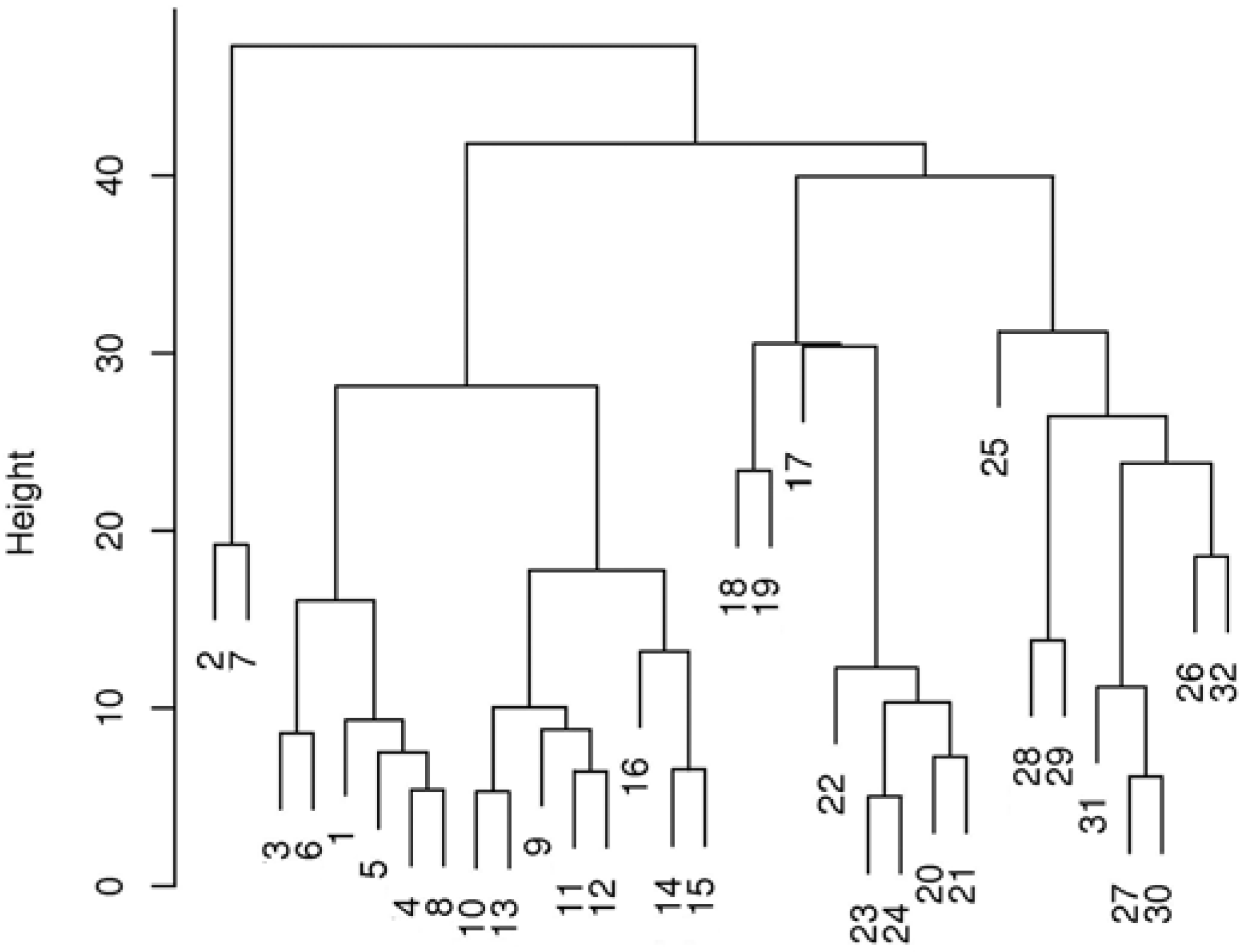
2.1. Genotoxicity


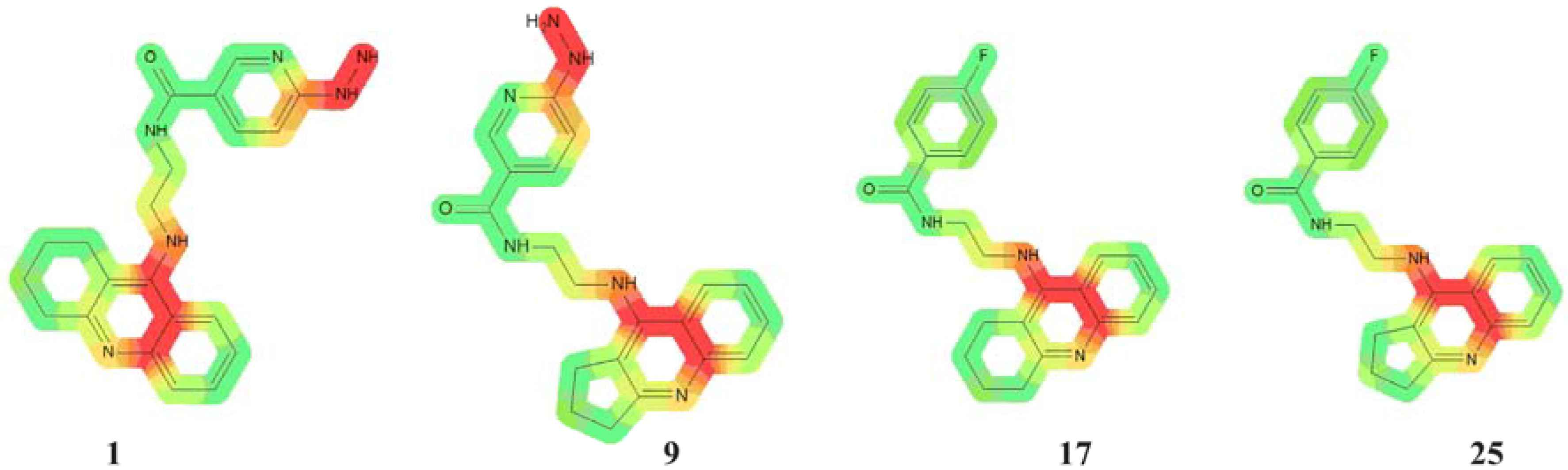
2.2. Cardiotoxicity
2.3. P-Glycoprotein Specificity
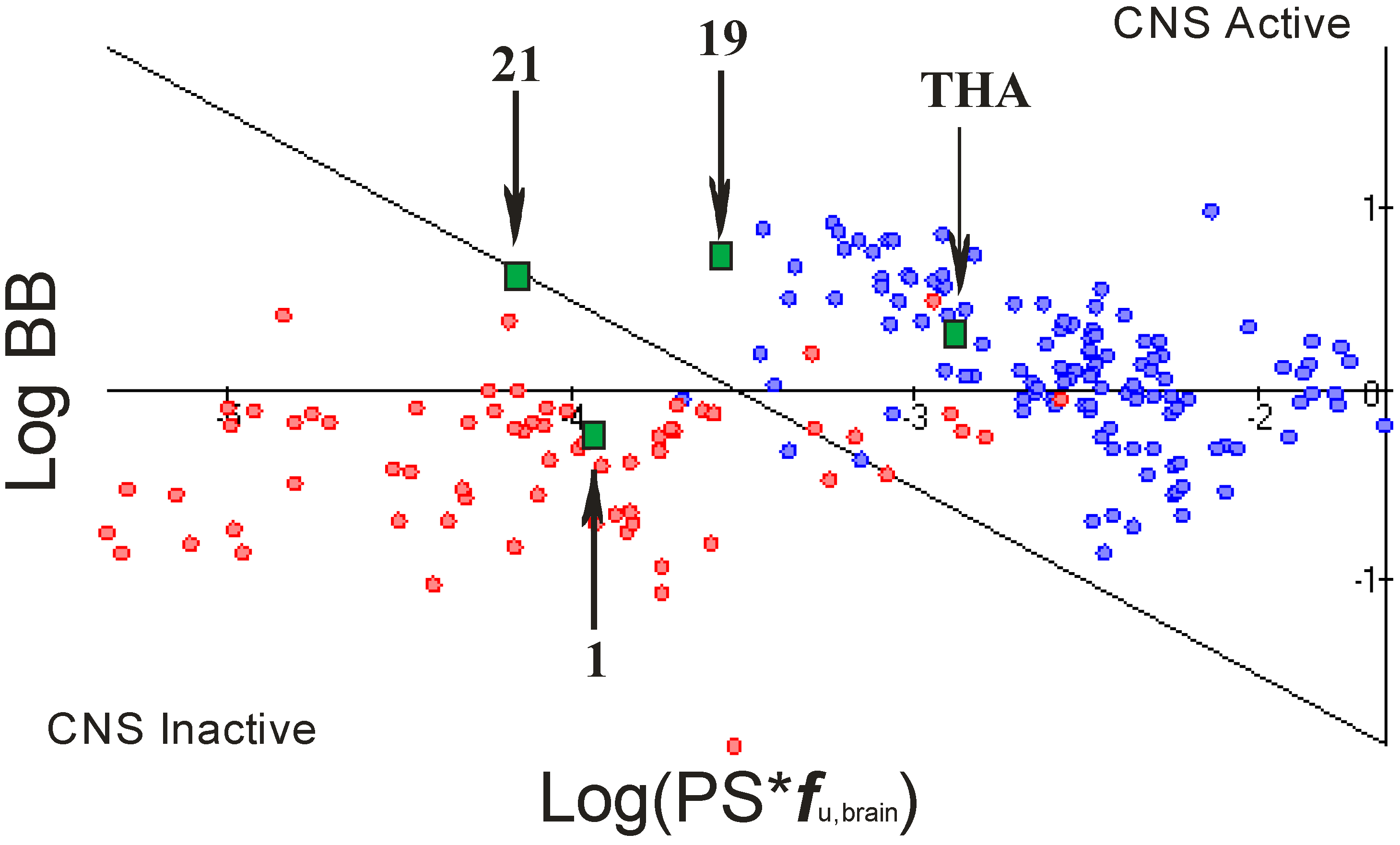
2.4. Blood Brain Barier (BBB) Penetration
- -
- rate of brain penetration (LogPS); this parameter depends on essential physicochemical properties such as lipophilicity, ionization constants, hydrogen bonding parameters, and molecular size;
- -
- extent of brain penetration (LogBB); cumulative effect of drug binding to brain and plasma (LogBB is assumed to correspond to the passive transport across blood-brain barrier);
- -
- brain/plasma equilibration rate (Log(PS*fu,brain)); fu,brain – fraction unbound in plasma, calculated automatically [23].
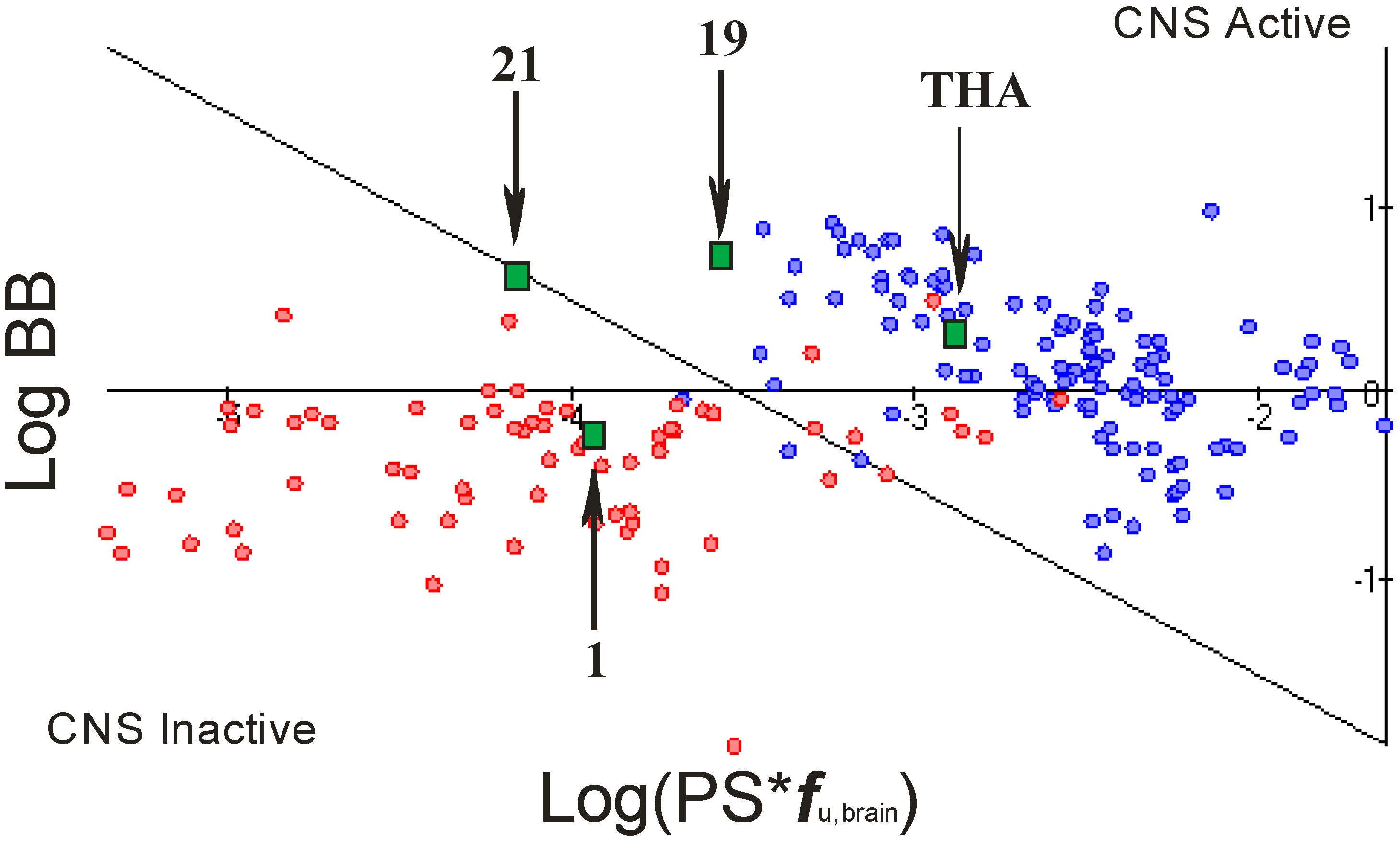
2.5. Distribution
2.6. EC50 and LC50
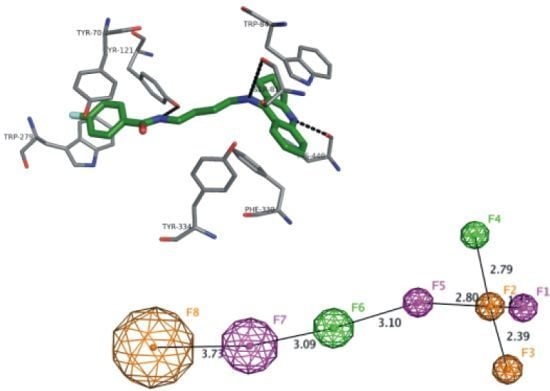
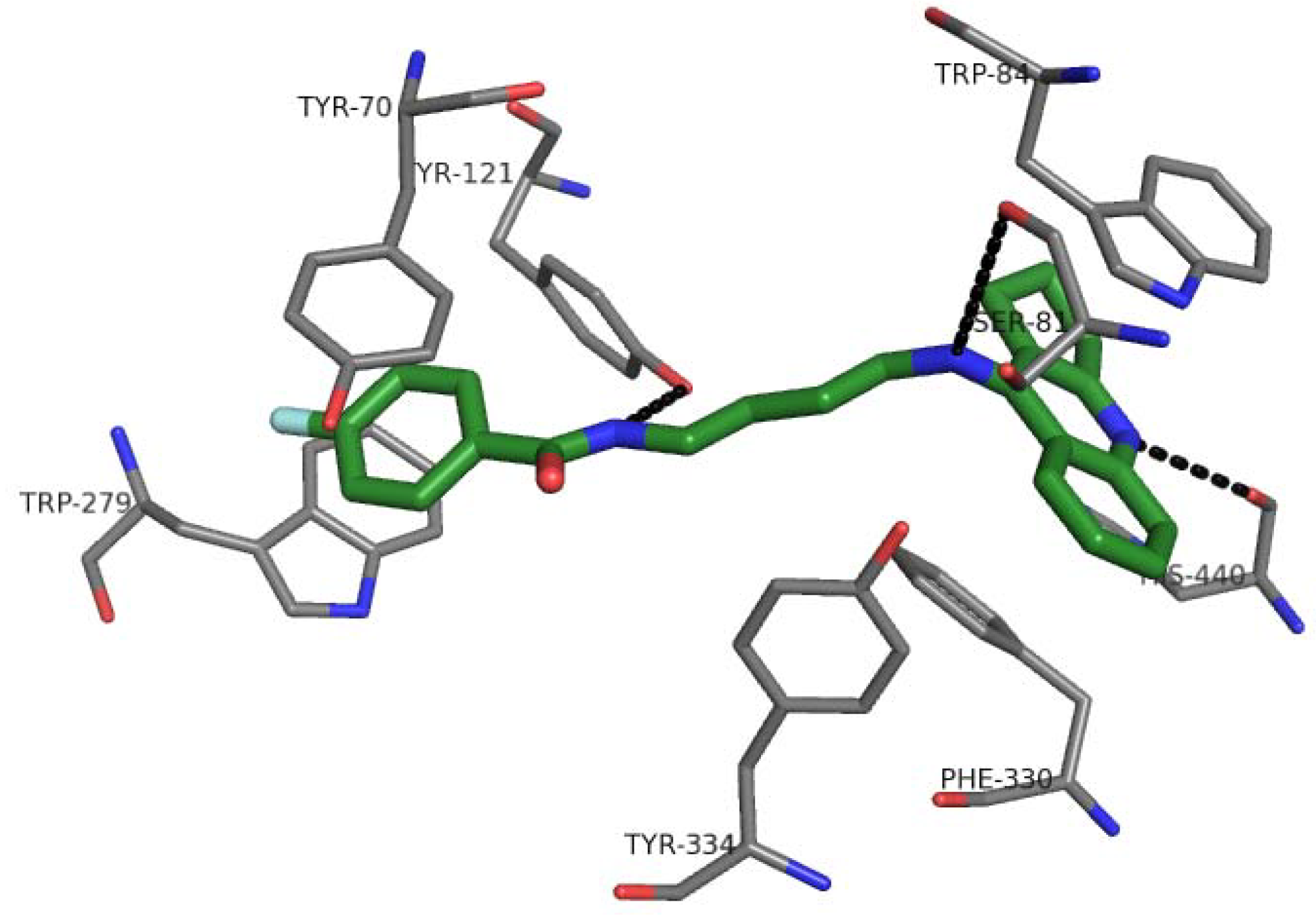
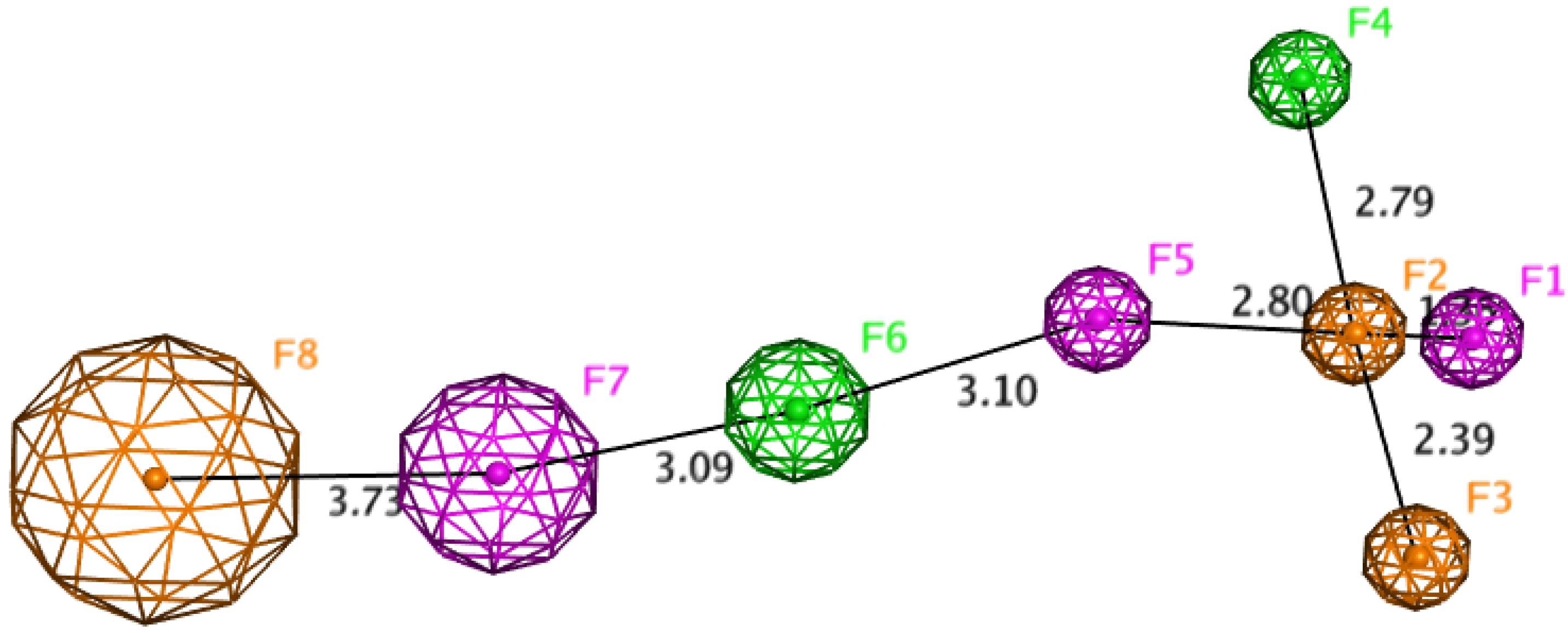
2.7. Docking Studies and Pharmacophore Search
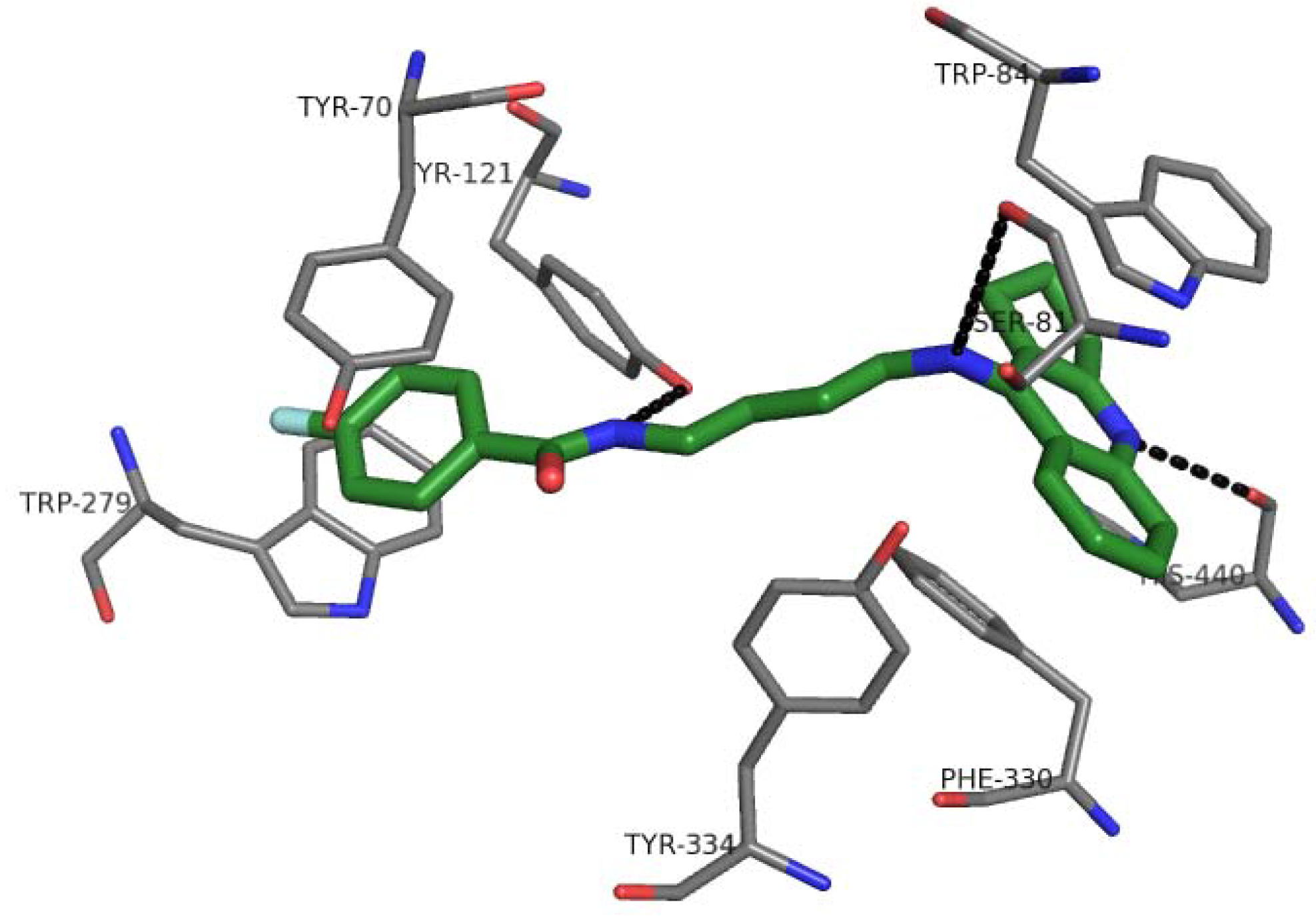
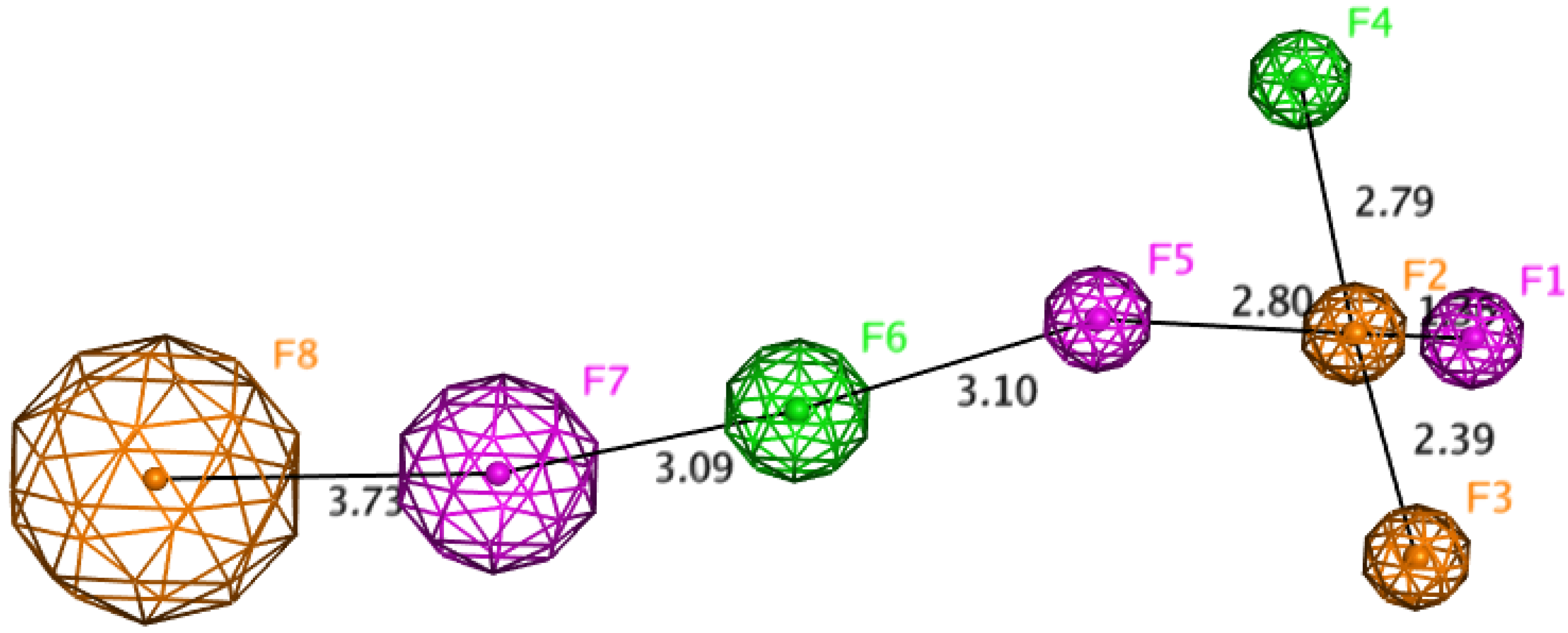
2.8. Chemometric Analysis


3. Experimental
3.1. Genotoxicity
- -
- the Ames test to provide the mutagenic potential of new compounds
- -
- a knowledge-based system in identifying and visualizing hazardous fragments of structures.
3.2. Cardiotoxicity
3.3. P-Glycoprotein Specificity
3.4. Blood Brain Barier (BBB) Penetration
3.5. Distribution
3.6. EC50 and LC50
3.7. Molecular Modeling
3.8. ATR-FT-IR and Multivariate Chemometric Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Albert, M.S.; Drachman, D.A. Alzheimer’s disease. What is it, how many people have it, and why do we need to known? Neurology 2000, 55, 166–168. [Google Scholar] [CrossRef]
- Mikiciuk-Olasik, E.; Szymański, P.; Żurek, E. Diagnostics and Therapy of Alzheimer’s Disease. Ind. J. Exp. Biol. 2007, 45, 315–325. [Google Scholar]
- Pakaski, M.; Kalman, J. Interaction between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 2008, 53, 103–111. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of generic memory dysfunction. Science 1982, 217, 408–417. [Google Scholar]
- Kozurkova, M.; Hamulakova, S.; Gazova, Z.; Paulikova, H.; Kristian, P. Neuroactive multifunctional tacrine congeners with cholinesterase, anti-amyloid aggregation and neuroprotective properties. Pharmaceuticals 2011, 4, 382–418. [Google Scholar]
- Sarter, M.; Nelson, C.L.; Bruno, J.B. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophrenia Bull 2005, 31, 117–138. [Google Scholar] [CrossRef]
- Korabecny, J.; Musilek, K.; Holas, O.; Nepovimova, E.; Jun, D.; Zemek, F.; Opletalova, V.; Patocka, J.; Dohnal, V.; Nachon, F.; et al. Synthesis and in vitro evaluation of N-(bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a cholinesterase inhibitor with regard to Alzheimer’s disease treatment. Molecules 2010, 15, 8804–8812. [Google Scholar] [CrossRef]
- McKenna, M.T.; Proctor, G.R.; Young, L.C.; Harvey, A.L. Novel tacrine analogues for potential use against alzheimer’s disease: Potent and selective acetylcholinesterase inhibitors and 5-HT uptake inhibitors. J. Med. Chem. 1997, 40, 3516–3523. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Gonzalez-Munoz, G.C.; Conde, S.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; Rodriguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef]
- Hu, M.K.; Wu, L.J.; Hsiao, G.; Yen, M.H. Homodimeric tacrine congeners as acetylcholinesterase inhibitors. J. Med. Chem. 2002, 45, 2277–2282. [Google Scholar] [CrossRef]
- Szymański, P.; Karpiński, A.; Mikiciuk-Olasik, E. Synthesis, biological activity and HPLC validation of 1,2,3,4-tetrahydroacridine. Eur. J. Med. Chem. 2011, 46, 3250–3257. [Google Scholar] [CrossRef]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Synthesis and biological activity of derivatives of tetrahydroacridine as acetylcholinesterase inhibitors. Bioorg. Chem. 2011, 39, 138–142. [Google Scholar] [CrossRef]
- Szymański, P.; Markowicz, M.; Bajda, M.; Malawska, B.; Mikiciuk-Olasik, E. Synthesis and biological activity of new 2,3-dihydro-1H-cyclopenta[b]-quinoline derivatives as acetylcholinesterase inhibitors. Lett. Drug Des. Discov. 2012, 9, 645–654. [Google Scholar] [CrossRef]
- Szymański, P.; Laznickova, A.; Laznicek, M.; Bajda, M.; Malawska, B.; Markowicz, M.; Mikiciuk-Olasik, E. 2,3-Dihydro-1H-cyclopenta[b]quinoline derivatives as acetylcholinesterase inhibitors—synthesis, radiolabelling and biodistribution. Int. J. Mol. Sci. 2012, 13, 10067–10090. [Google Scholar] [CrossRef]
- Szymański, P.; Markowicz, M.; Bajda, M.; Malawska, B.; Mikiciuk-Olasik, E. Synthesis, biological activity and molecular modeling of 4-fluoro-N-[ω-(1,2,3,4-tetrahydroacridin-9-ylamino)-alkyl]-benzamide derivatives as cholinesterase inhibitors. Arzneimittelforschung 2012, 62, 655–660. [Google Scholar] [CrossRef]
- Szymański, P.; Żurek, E.; Mikiciuk-Olasik, E. New tacrine-hydrazinonicotinamide hybrids as acetylcholinesterase inhibitors of potential interest for the early diagnostics of Alzheimer’s disease. Pharmazie 2006, 61, 269–273. [Google Scholar]
- Didziapetris, R.; Lanevskij, K.; Japertas, P. Trainable QSAR model of ames genotoxicity. Toxicol. Lett. 2008, 180, S152–S153. [Google Scholar] [CrossRef]
- Tiemersma, E.W.; Voskuil, D.W.; Bunschoten, A.; Hogendoorn, E.A.; Witteman, B.J.M.; Nagengast, F.M.; Glatt, H.; Kok, F.J.; Kampman, E. Risk of colorectal adenomas in relation to meat consumption, meat preparation, and genetic susceptibility in a dutch population. Cancer Causes Control 2004, 15, 225–236. [Google Scholar] [CrossRef]
- Westwood, I.M.; Holton, S.J.; Rodrigues-Lima, F.; Dupret, J.M.; Bhakta, S.; Noble, M.E.; Sim, E. Expression, purification, characterization and structure of Pseudomonas aeruginosa arylamine N-acetyltransferase. Biochem. J. 2005, 385, 605–612. [Google Scholar] [CrossRef]
- Shet, M.S.; McPhaul, M.; Fisher, C.W.; Stallings, N.R.; Estabrook, R.W. Metabolism of the antiandrogenic drug (Flutamide) by human CYP1A2. Drug Metab. Dispos. 1997, 25, 1234–1241. [Google Scholar]
- Kazius, J.; McGuire, R.; Bursi, R. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 2005, 48, 312–320. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Ionization-specific QSAR models of blood–brain penetration of drugs. Chem. Biodivers. 2009, 6, 2050–2054. [Google Scholar]
- Abraham, M.H.; Chadha, H.S.; Martins, F.; Mitchell, R.C.; Bradbury, M.W.; Gratton, J.A. Hydrogen bonding part 46: A review of the correlation and prediction of transport properties by an LFER method: Physicochemical properties brain penetration and skin permeability. Pestic. Sci. 1999, 55, 78–88. [Google Scholar]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Camenish, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding characteristics. J. Drug Target 1998, 6, 151–165. [Google Scholar] [CrossRef]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981, 33, 17–22. [Google Scholar]
- Chen, Y.; Fang, L.; Peng, S.; Liao, H.; Lehmann, J.; Zhang, Y. Discovery of a novel acetylcholinesterase inhibitor by structure-based virtual screening techniques. Bioorg. Med. Chem. Lett. 2012, 22, 3181–3187. [Google Scholar] [CrossRef]
- Sazonovas, A.; Japertas, P.; Didziapetris, R. Estimation of reliability of predictions and model applicability domain evaluation in the analysis of acute toxicity (LD50). SAR QSAR Environ Res. 2010, 21, 127–148. [Google Scholar] [CrossRef]
- Zhu, B.Y.; Jia, Z.J.; Zhang, P.; Su, T.; Huang, W.; Goldman, E.; Tumas, D.; Kadambi, V.; Eddy, P.; Sinha, U.; Scarborough, R.M.; Song, Y. Inhibitory effect of carboxylic acid group on hERG binding. Bioorg. Med. Chem. Lett. 2006, 16, 5507–5512. [Google Scholar] [CrossRef]
- Ramakrishnan, P. The role of P-glycoprotein in the blood-brain barrier. Einstein Quart. J. Biol. Med. 2003, 19, 160–165. [Google Scholar]
- Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Drug discovery interface lonization-specifie prediction of blood-brain permeability. J. Pharm. Sci. 2009, 98, 122–134. [Google Scholar] [CrossRef]
- Lanevskij, K.; Dapkunas, J.; Juska, L.; Japertas, P.; Didziapetris, R. QSAR analysis of blood-brain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci. 2011, 100, 2147–2160. [Google Scholar] [CrossRef]
- Liu, X.; Smith, B.J.; Chen, C.; Callegari, E.; Becker, S.L.; Chen, X.; Cianfrogna, J.; Doran, A.C.; Doran, S.D.; Gibbs, J.P.; et al. Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: An experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J. Pharmacol. Exp. Ther. 2005, 313, 1254–1262. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Hall, L.M.; Hall, L.H.; Kier, L.B. QSAR modeling of β-lactam binding to human serum proteins. J. Comput. Aided Mol. Des. 2003, 17, 103–118. [Google Scholar] [CrossRef]
- Johnson, D.J.; Sanderson, H.; Brain, R.A.; Wilson, C.J.; Solomon, K.R. Toxicity and hazard of selective serotonin reuptake inhibitor antidepressants fluoxetine, fluvoxamine, and sertraline to algae. Ecotoxicol. Environ. Saf. 2007, 67, 128–139. [Google Scholar] [CrossRef]
- Berg, L.; Andersson, C.D.; Artursson, E.; Hörnberg, A.; Tunemalm, A.-K.; Linusson, A.; Ekström, F. Targeting acetylcholinesterase: Identification of chemical leads by high throughput screening, structure determination and molecular modeling. PLoS One 2011, 6, e26039. [Google Scholar] [CrossRef]
- Yan, A.; Wang, K. Quantitative structure and bioactivity relationship study on human acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3336–3342. [Google Scholar] [CrossRef]
- Chaudhaery, S.S.; Roy, K.K.; Shakya, N.; Saxena, G.; Sammi, S.R.; Nazir, A.; Nath, C.; Saxena, A.K. Novel carbamates as orally active acetylcholinesterase inhibitors found to improve Scopolamine-induced cognition impairment: Pharmacophore-based virtual screening, synthesis, and pharmacology. J. Med. Chem. 2010, 53, 6490–6505. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–33 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szymański, P.; Skibiński, R.; Inglot, T.; Bajda, M.; Jończyk, J.; Malawska, B.; Mikiciuk-Olasik, E. New Tacrine Analogs as Acetylcholinesterase Inhibitors — Theoretical Study with Chemometric Analysis. Molecules 2013, 18, 2878-2894. https://doi.org/10.3390/molecules18032878
Szymański P, Skibiński R, Inglot T, Bajda M, Jończyk J, Malawska B, Mikiciuk-Olasik E. New Tacrine Analogs as Acetylcholinesterase Inhibitors — Theoretical Study with Chemometric Analysis. Molecules. 2013; 18(3):2878-2894. https://doi.org/10.3390/molecules18032878
Chicago/Turabian StyleSzymański, Paweł, Robert Skibiński, Tadeusz Inglot, Marek Bajda, Jakub Jończyk, Barbara Malawska, and Elżbieta Mikiciuk-Olasik. 2013. "New Tacrine Analogs as Acetylcholinesterase Inhibitors — Theoretical Study with Chemometric Analysis" Molecules 18, no. 3: 2878-2894. https://doi.org/10.3390/molecules18032878