Stereocontrolled Synthesis and Functionalization of Cyclobutanes and Cyclobutanones


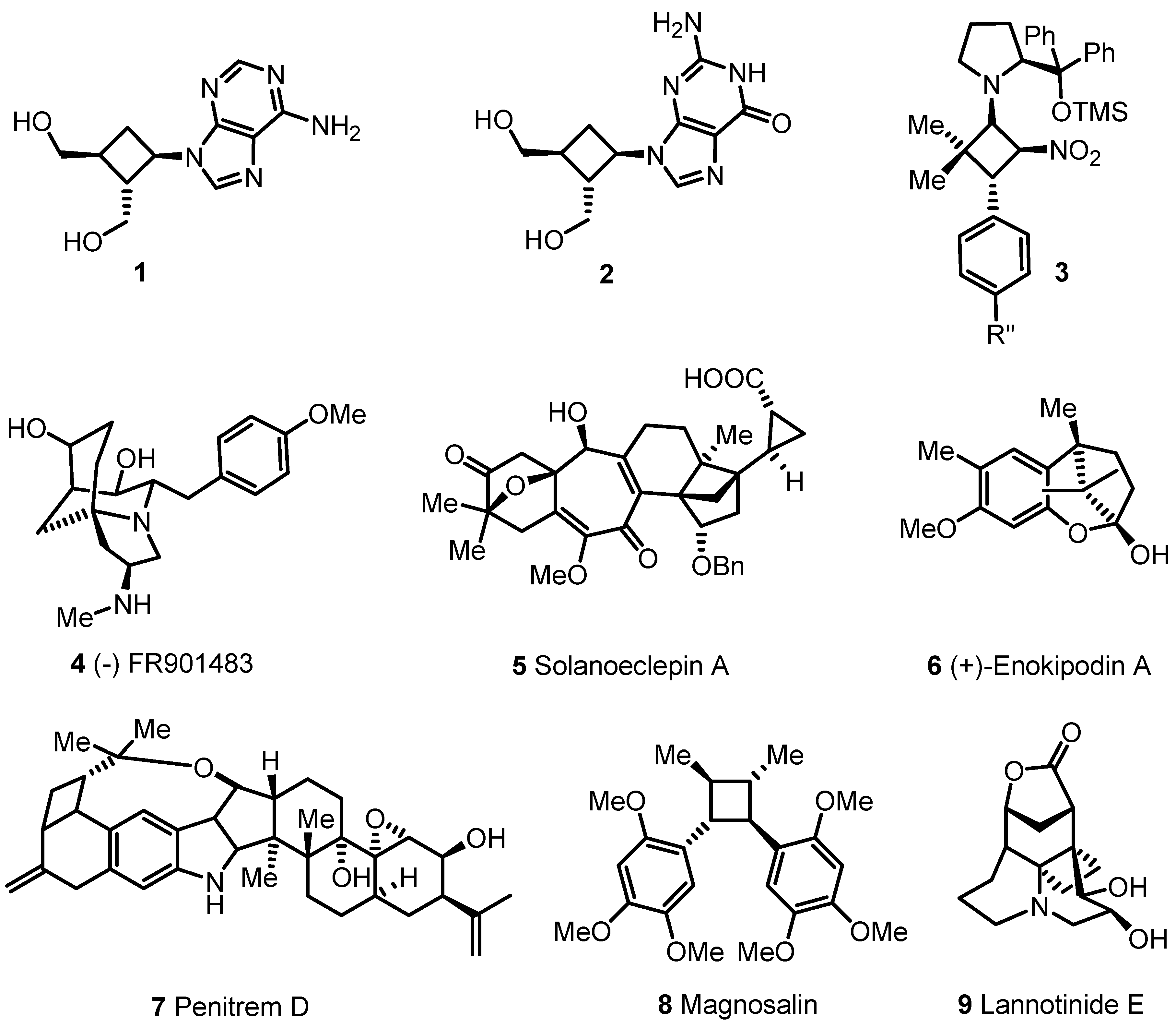{kind=link}
{kind=link}
{kind=link}
{kind=link}
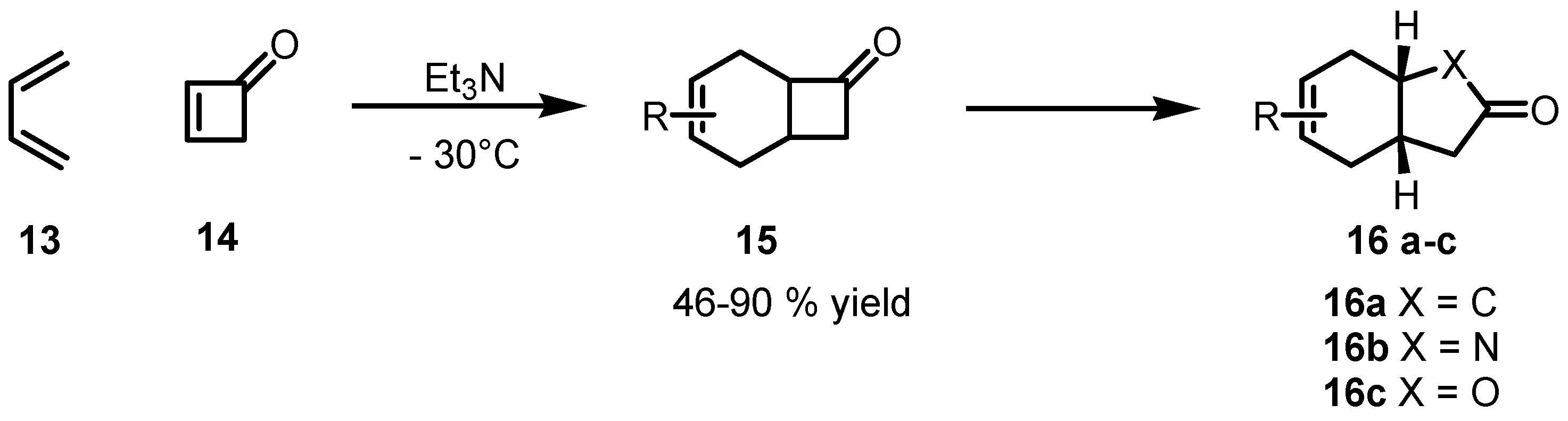{kind=link}
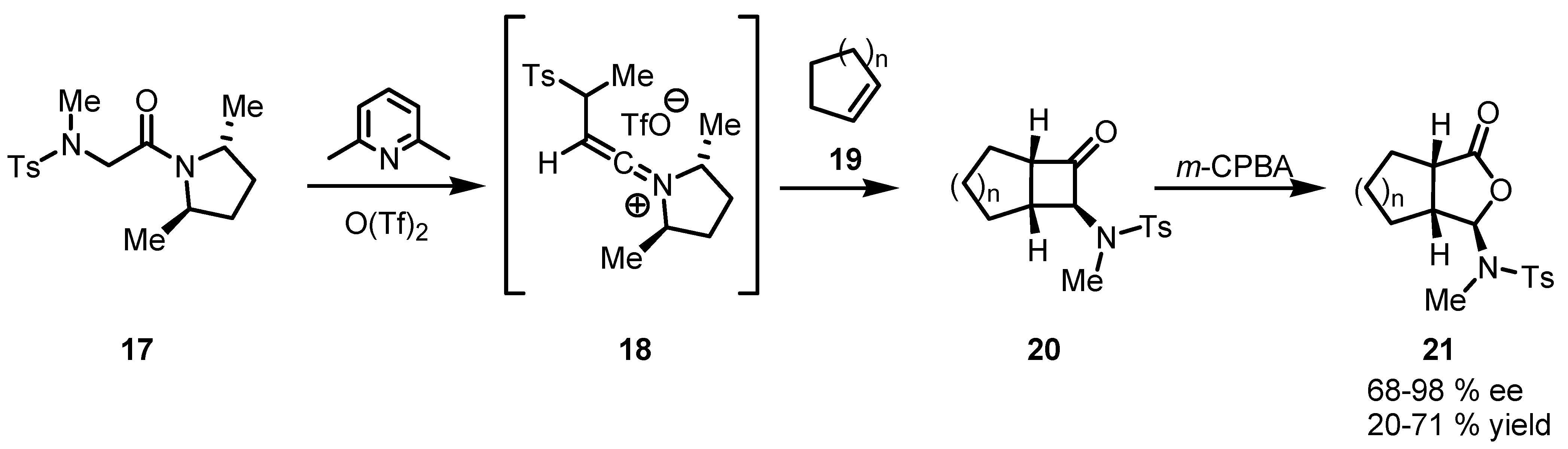{kind=link}
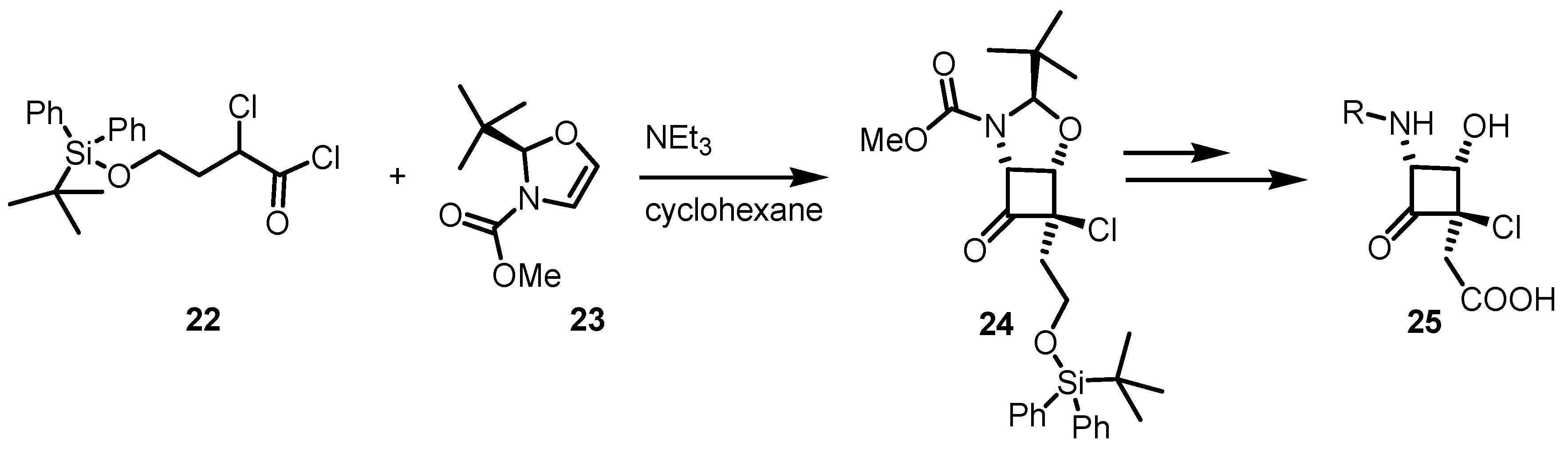{kind=link}
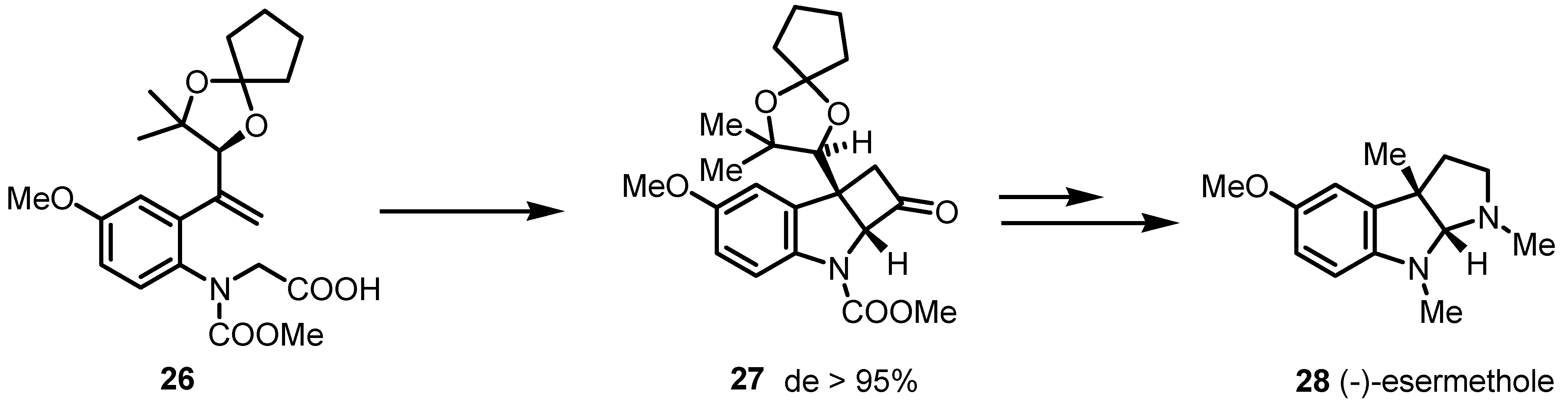{kind=link}
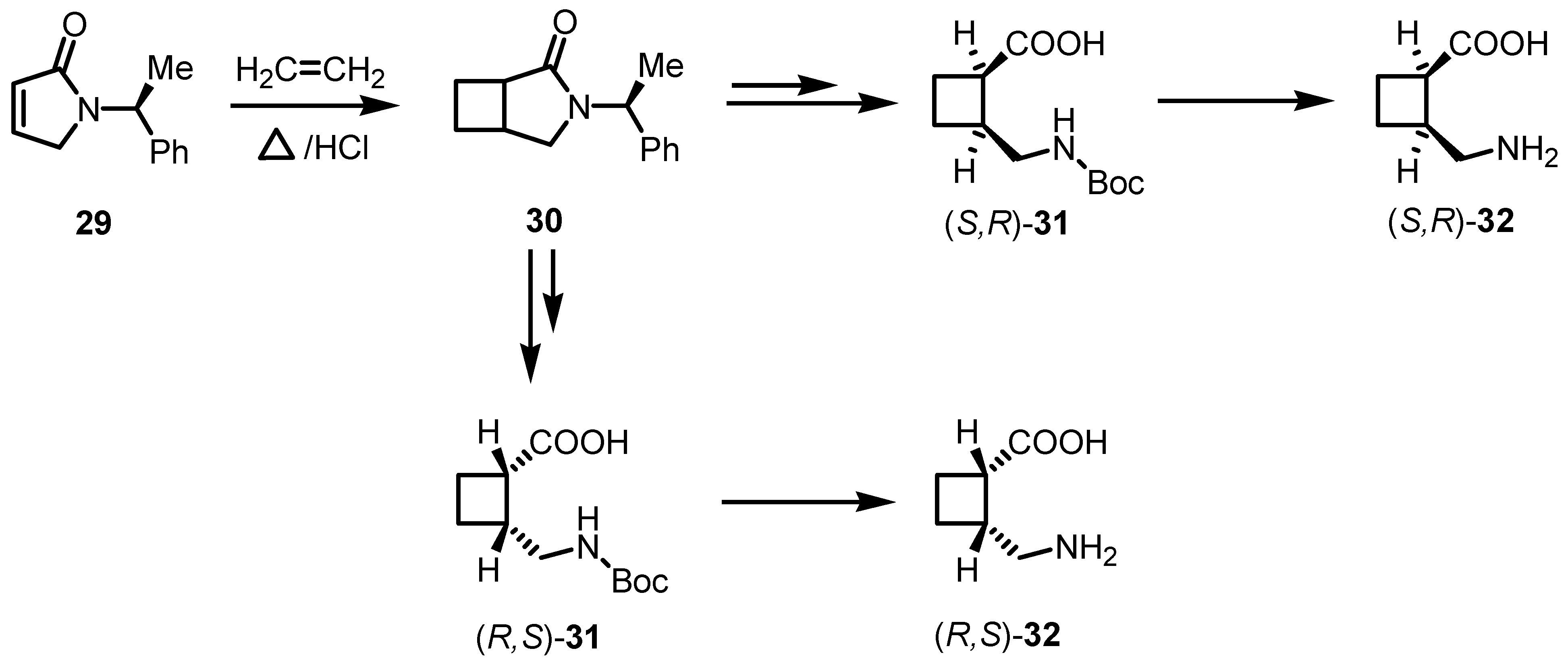{kind=link}
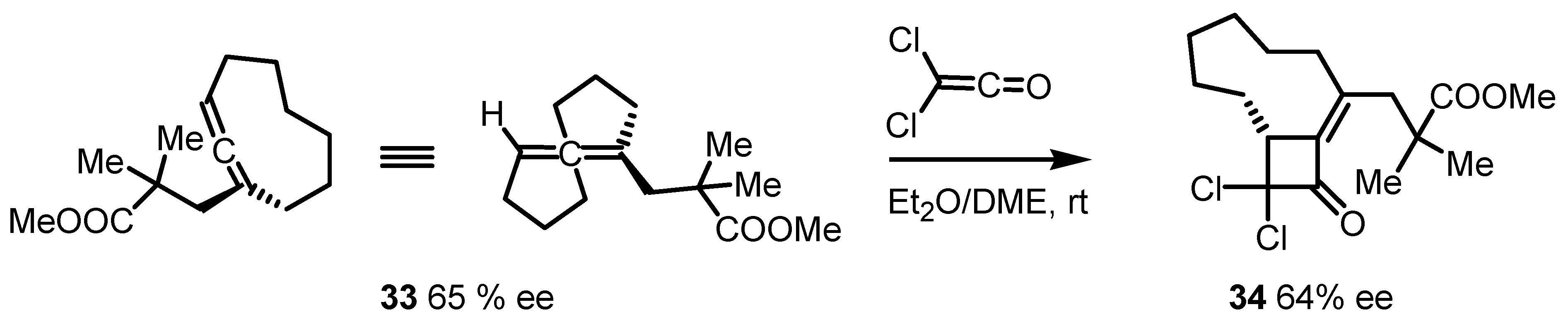{kind=link}
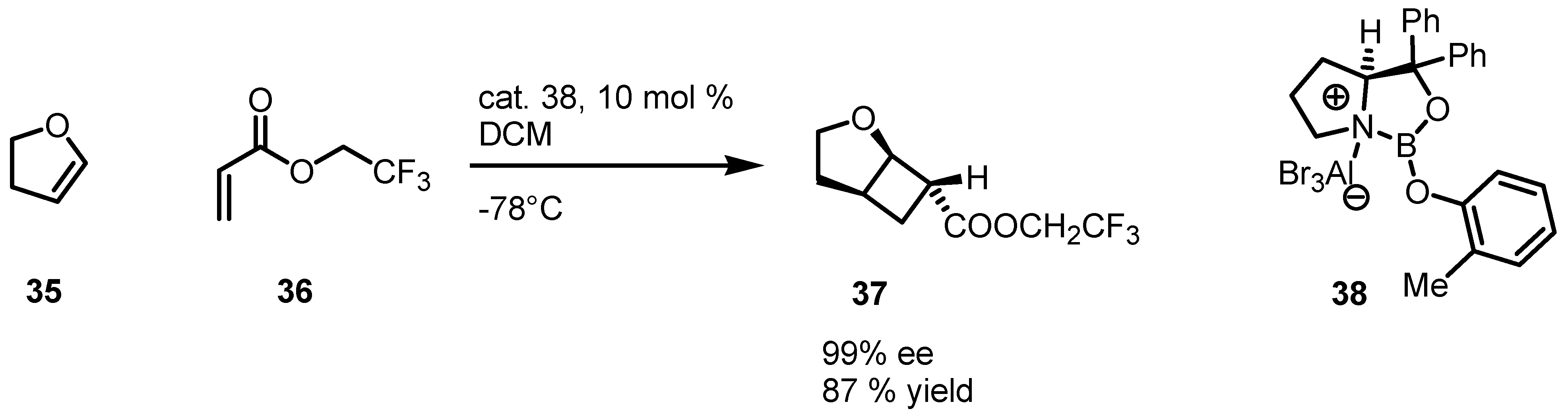{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
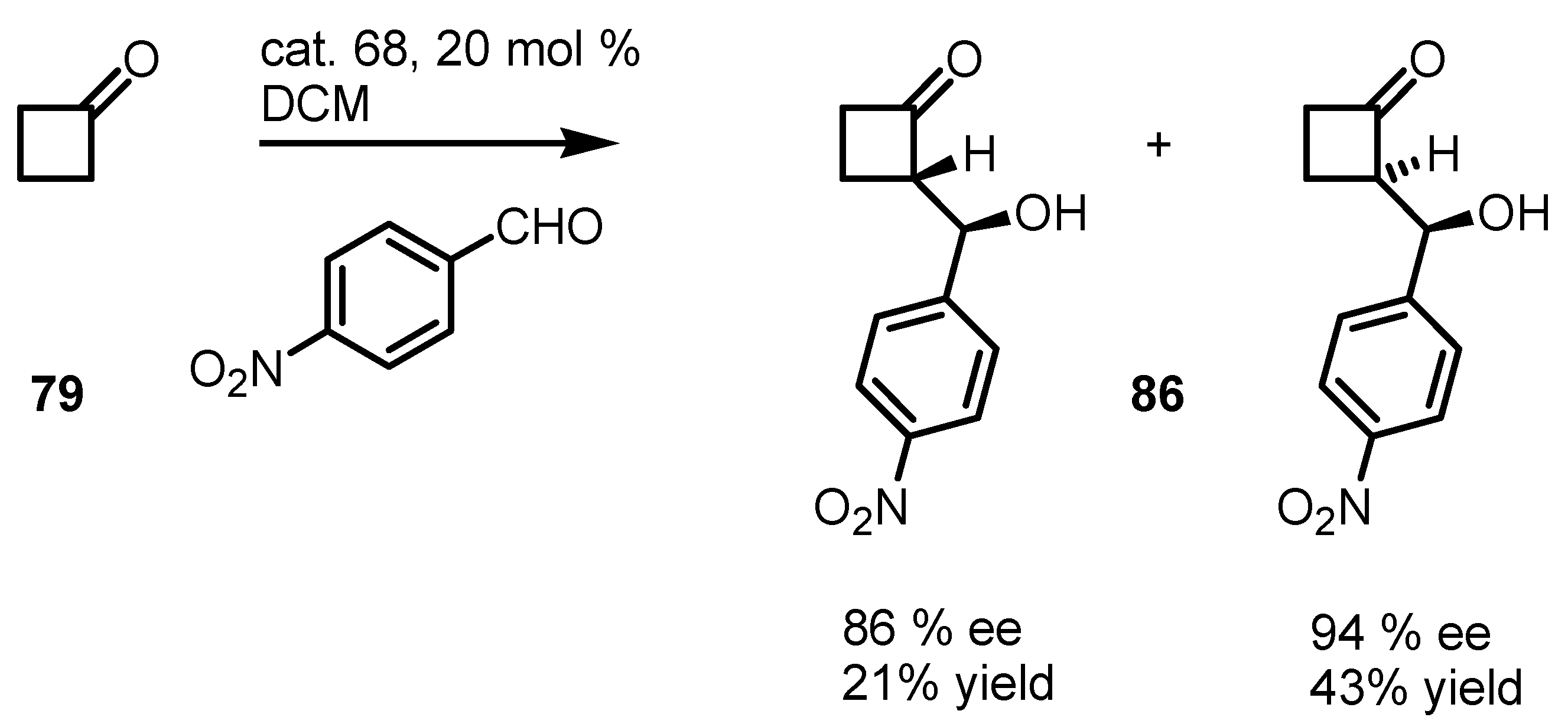{kind=link}
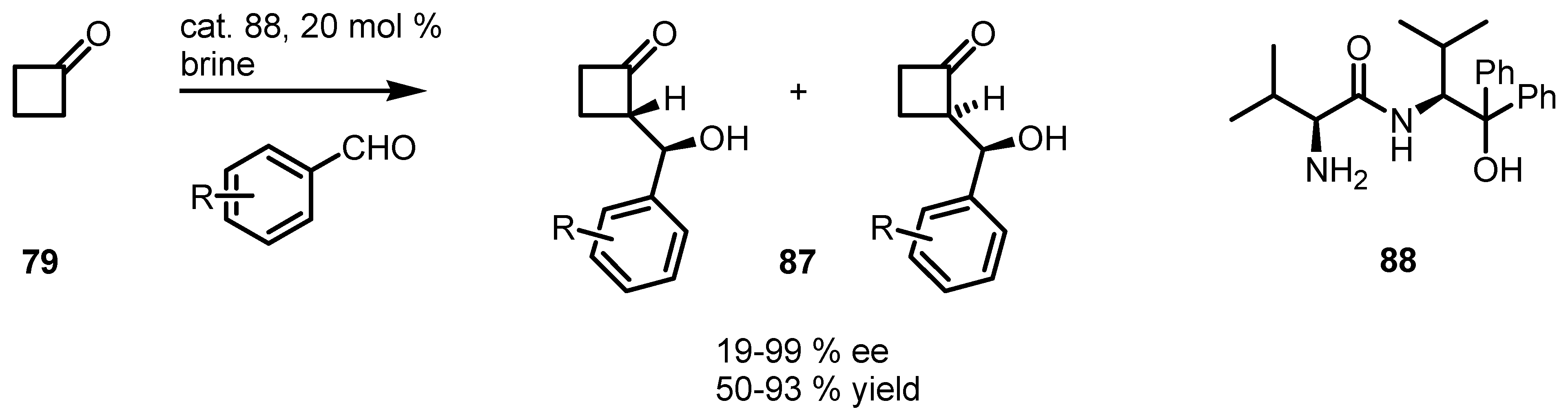{kind=link}
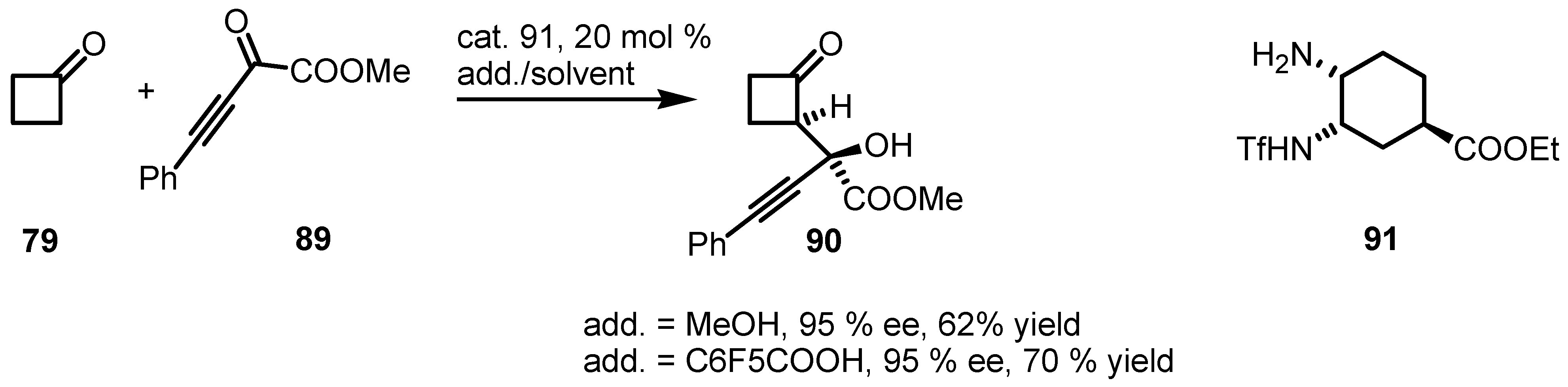{kind=link}
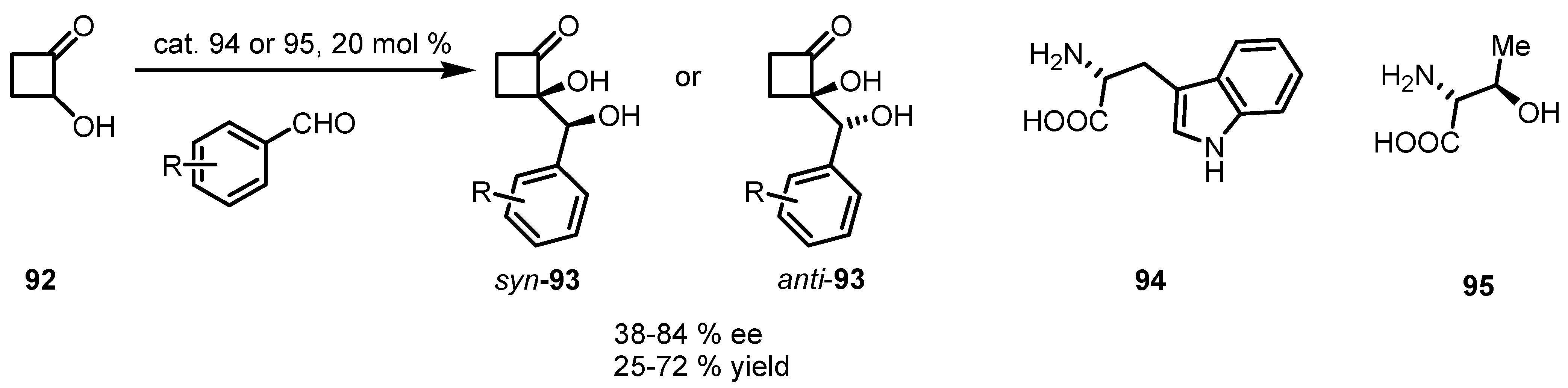{kind=link}
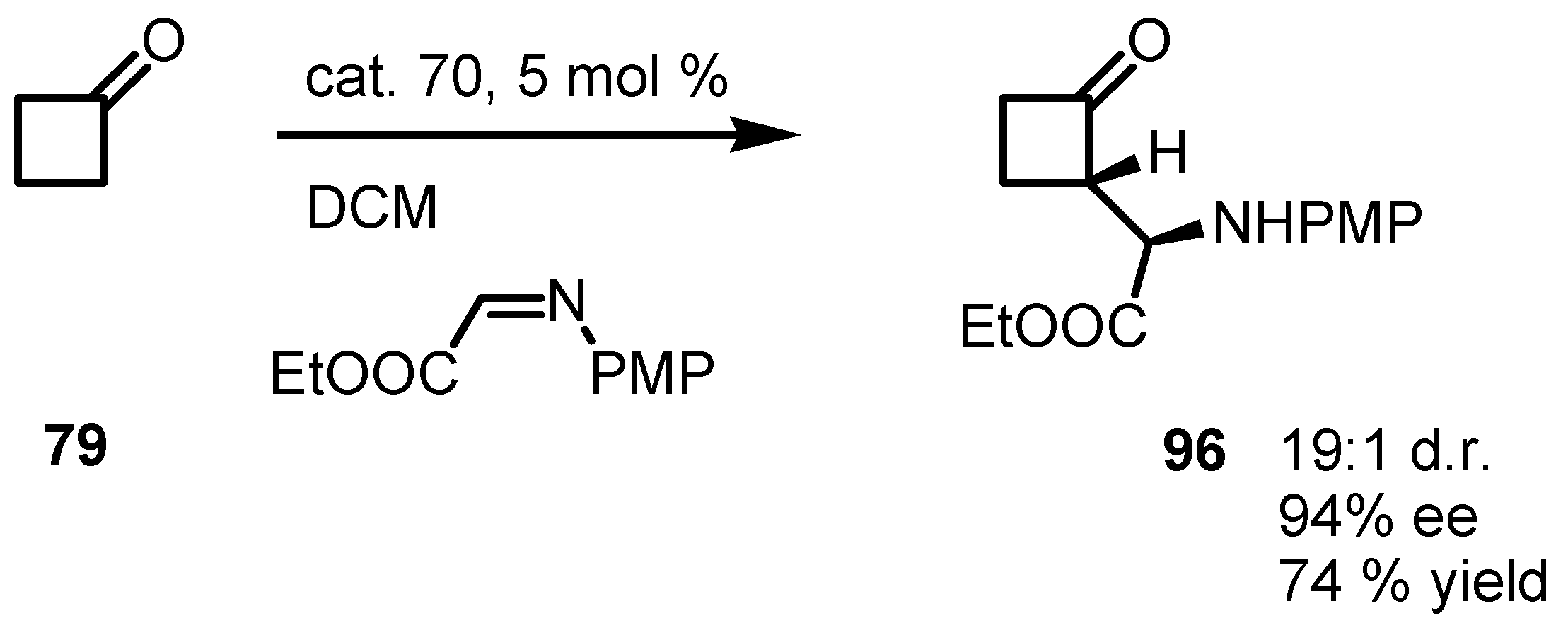{kind=link}
{kind=link}
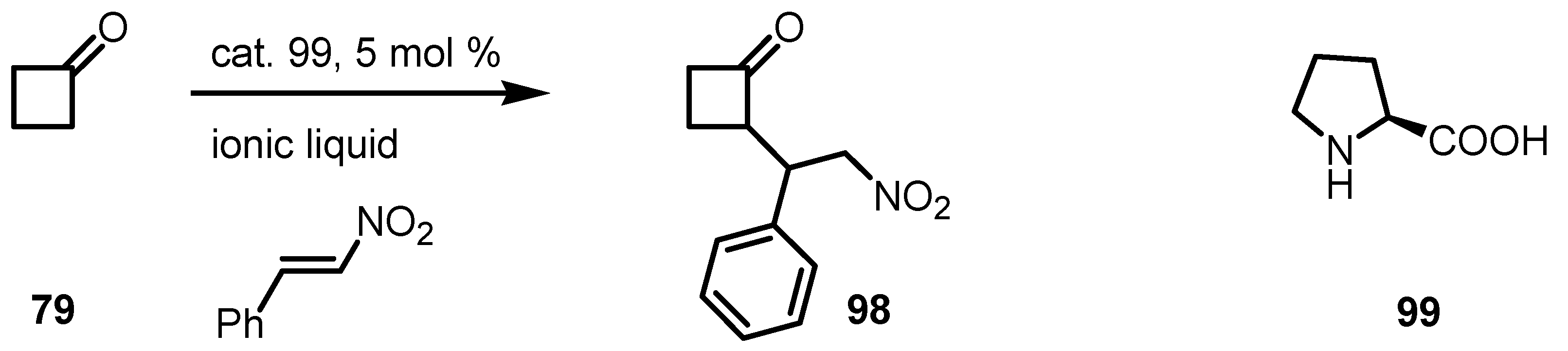{kind=link}
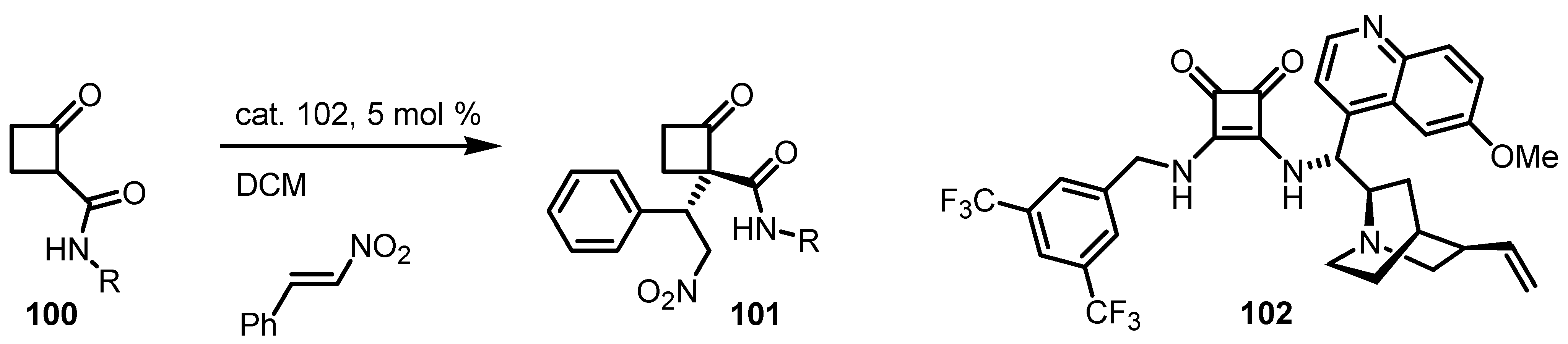{kind=link}
{kind=link}
{kind=link}
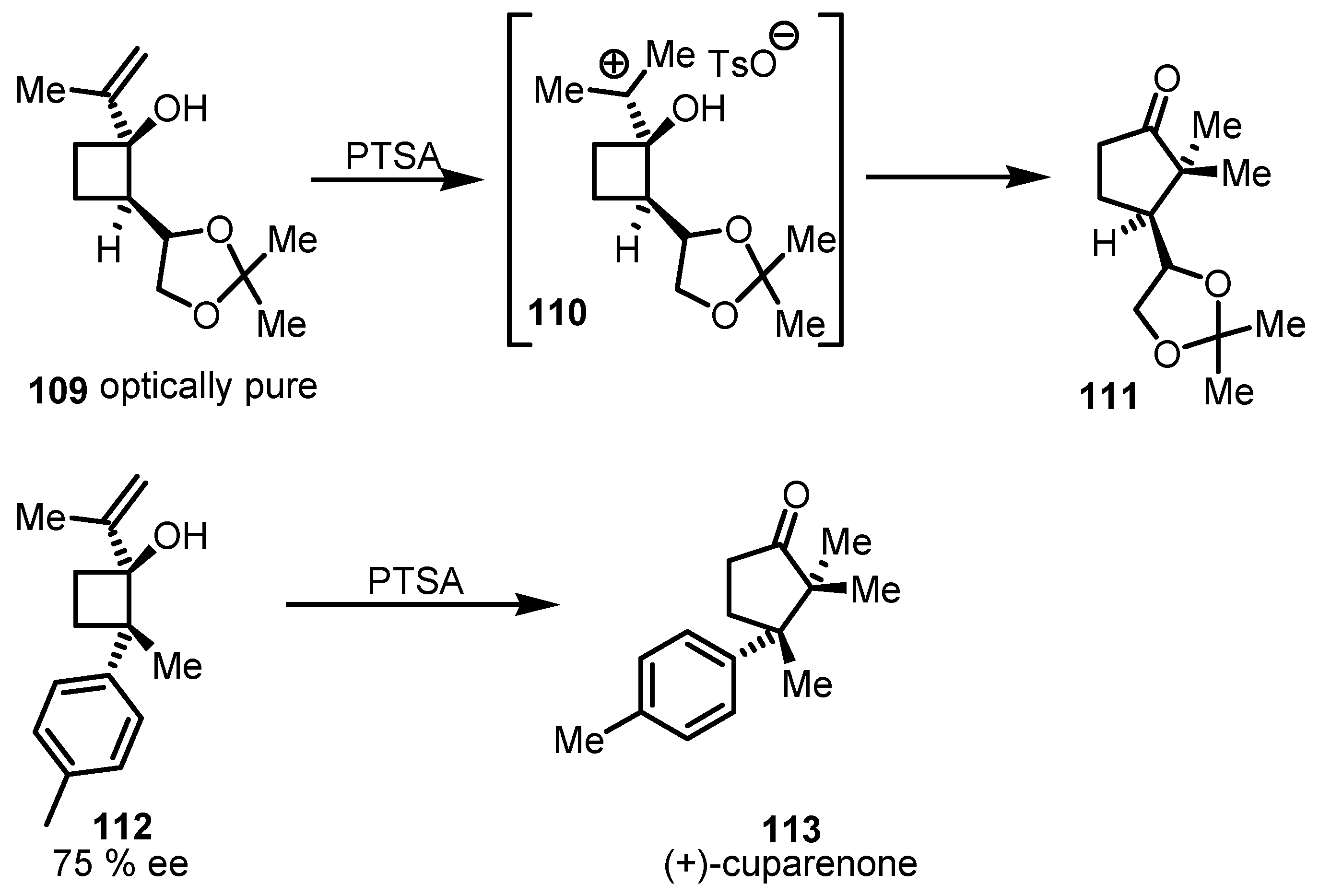{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
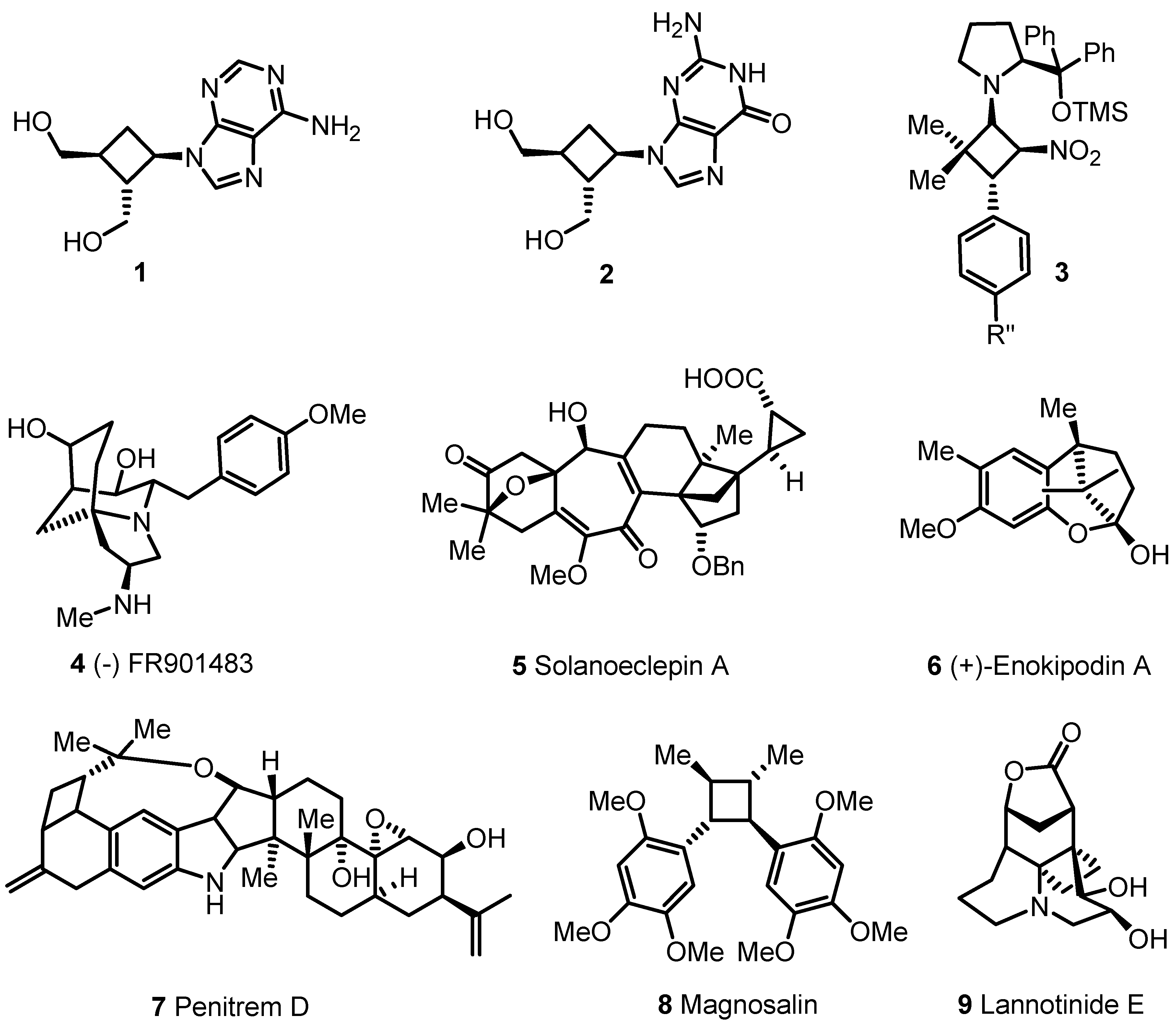
- 2.
- Stereoselective [2+2] Cycloaddition Synthesis of Cyclobutane Derivatives
- 2.1.
- Stereoselective Synthesis of Cyclobutane Amino Acids
- 2.1.
- Stereoselective Diels-Alder Reactions using Cyclobutenones
- 3.
- Enantioselective Stoichiometric Synthesis of Cyclobutane Derivatives
- 3.1.
- Enantioselective Stoichiometric [2+2] Cycloadditions
- 3.2.
- Chiral Allene based [2+2] Asymmetric Cycloadditions
- 3.3.
- Organocatalyzed Enantioselective [2+2] Cycloadditions
- 3.4.
- Iminium-Ion Intermediated [2+2] Cycloaddition of Enals
- 3.5.
- Hydrogen-Bonding Mediated [2+2] Asymmetric Cycloaddition
- 4.
- Desymmetrization of cyclobutane and cyclobutanone derivatives
- 4.1.
- Organocatalyzed Bronsted Acids based Desymmetrizations
- 4.2.
- Organocatalyzed aldol based Desymmetrizations Reactions
- 4.3.
- Enaminocatalyzed Reactions of Cyclobutanones with Nitrosobenzene
- 5.
- Biocatalytic Resolution of Cyclobutane Derivatives
- 5.1.
- Biocatalytic PPL based Cyclobutanols Resolution by Esterification and Hydrolysis
- 6.
- Cyclobutanone α-Functionalization
- 6.1.
- α-Functionalization of Cyclobutanones via SOMO Catalysis
- 6.2.
- Asymmetric SN1 Alkylation of Cyclobutanones
- 6.3.
- Organocatalyzed Aldol Reactions
- 6.4.
- Organocatalyzed Mannich Addition of Cyclobutanones to Glycolates
- 6.5.
- Organocatalyzed Michael Addition of Cyclobutanones to Nitrostyrenes
- 6.6.
- Cyclobutanone α-Heteroatom Functionalization
- 7.
- Cyclobutane Ring Enlargement
- 7.1.
- Chiral Non Racemic Cyclobutanes Ring Expansion. Synthesis of Chiral Cyclopentanones
- 7.2.
- Chiral Non Racemic Oxaspirohexanes Ring Enlargement
- 7.3.
- Organocatalyzed Enantioselective Cyclobutanols Ring Enlargement
- 7.4.
- Organocatalyzed Enantioselective Fluorination-Induced Cyclobutanes and Cyclopropanes Ring Expansions
- 8.
- Enantioselective Bayer-Villiger Oxidation
- 8.1.
- Enantio- and Diastereoselective Bayer-Villiger Oxidation of Chiral Cyclobutanones
- 8.2.
- Organocatalyzed Enantioselective Cyclobutanone Bayer-Villiger Oxidation
- 8.3.
- Biocatalytic Enantioselective Cyclobutanone Bayer-Villiger
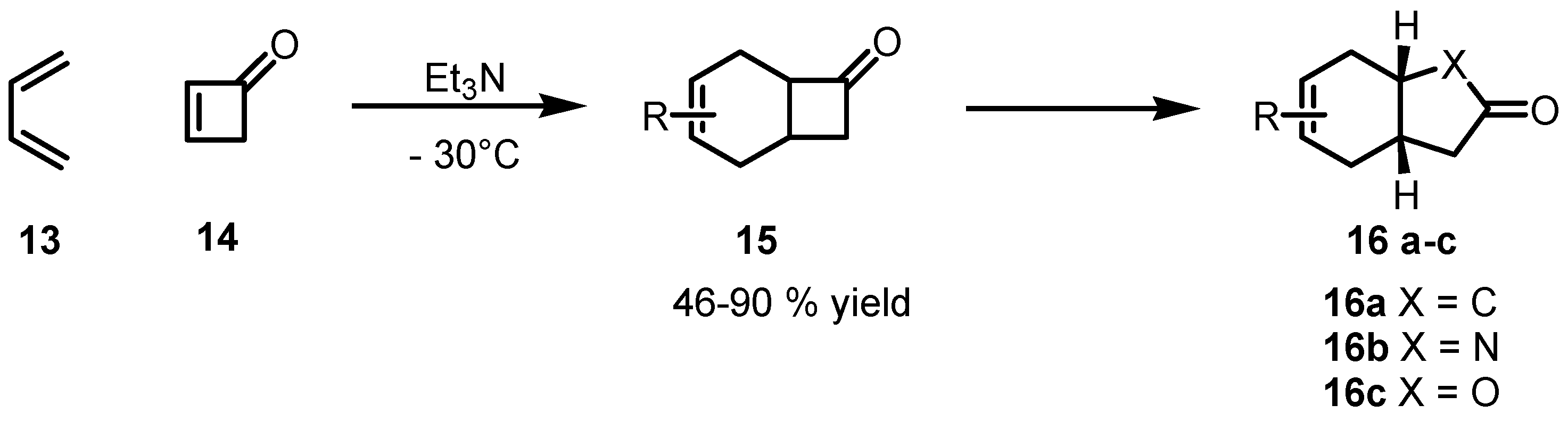
2. Stereoselective [2+2] Cycloaddition Synthesis of Cyclobutane Derivatives
2.1. Stereoselective Synthesis of Cyclobutane Amino Acids
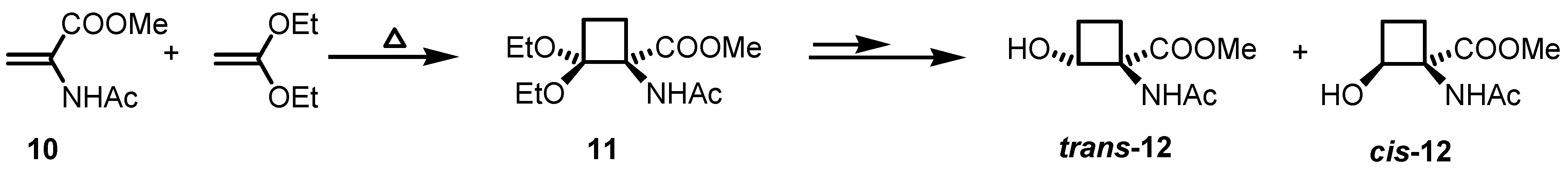
2.2. Stereoselective Diels-Alder Reactions using Cyclobutenones
3. Enantioselective Stoichiometric Synthesis of Cyclobutane Derivatives
3.1. Enantioselective Stoichiometric [2+2] Cycloadditions

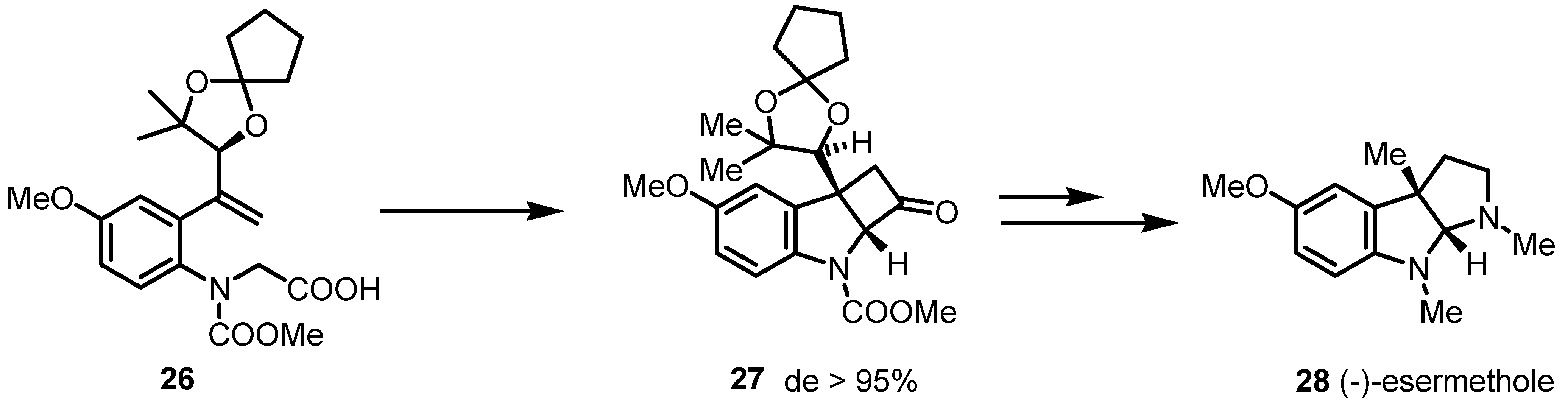
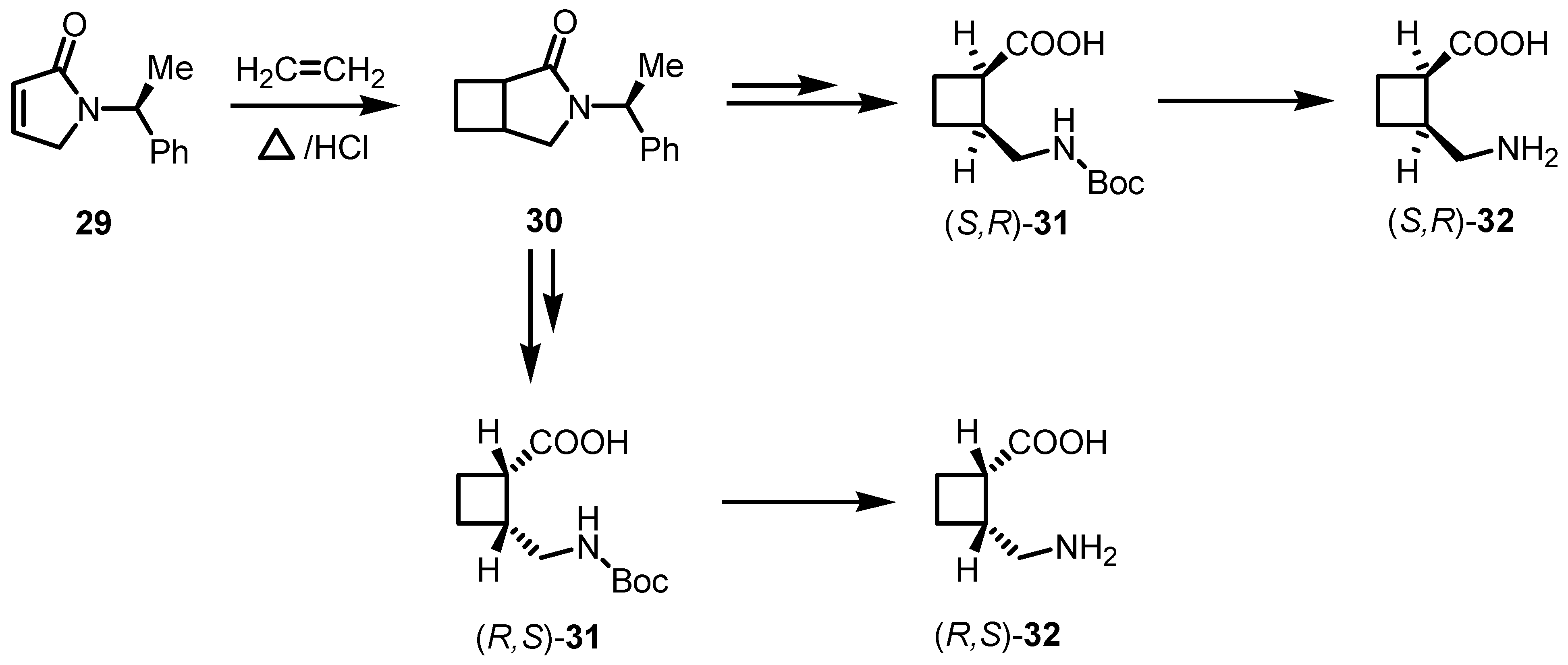
3.2. Chiral Allene-Based [2+2] Asymmetric Cycloadditions

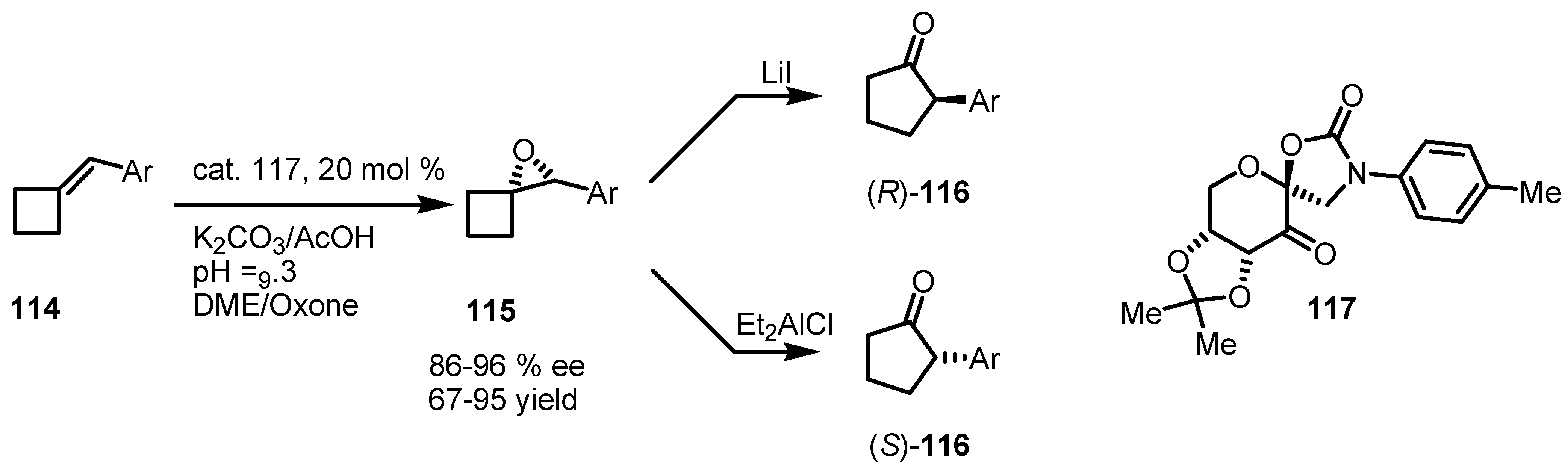
3.3. Organocatalyzed Enantioselective [2+2] Cycloadditions

3.4. Iminium-Ion Intermediated [2+2] Cycloaddition of Enals
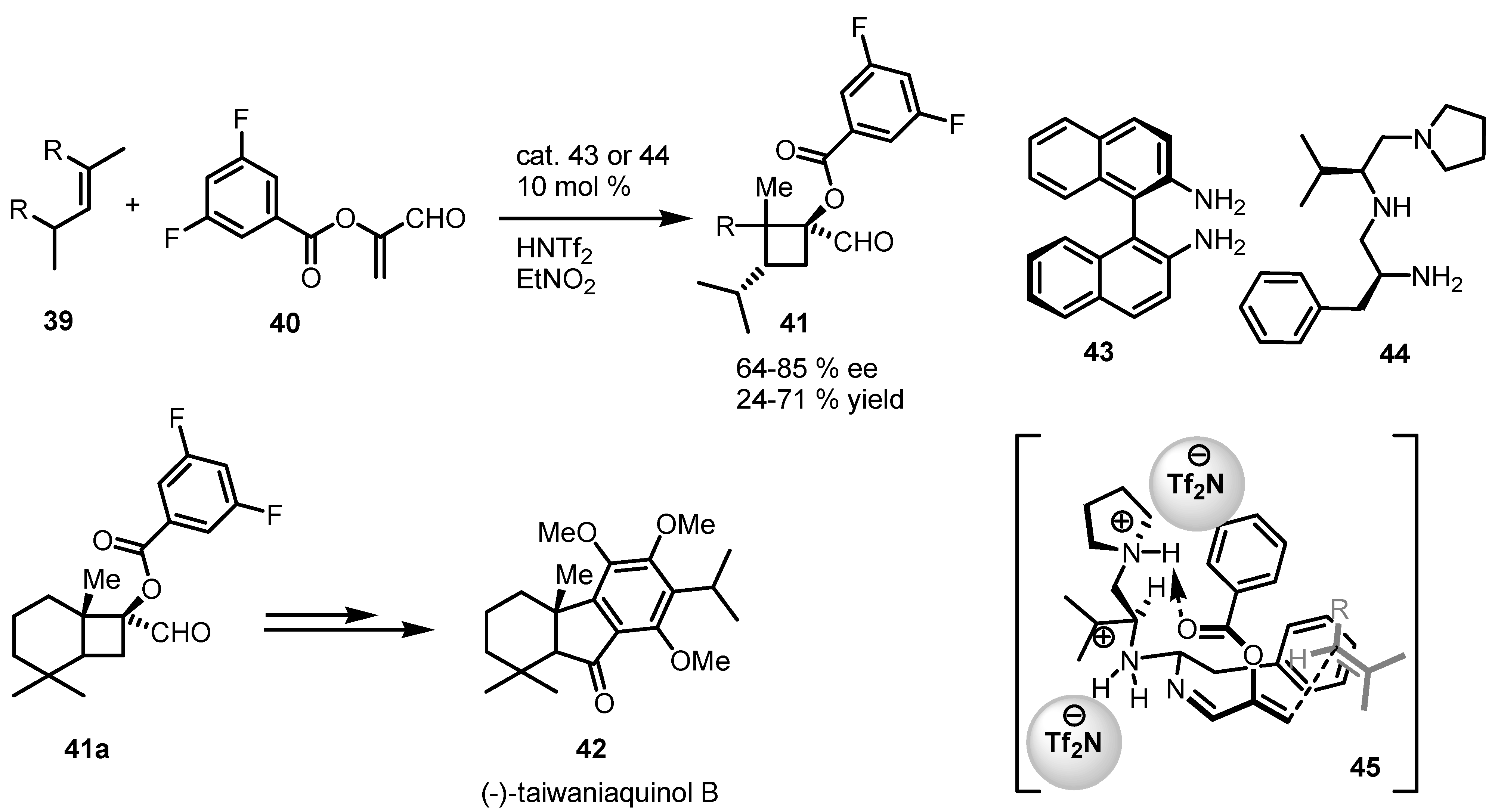
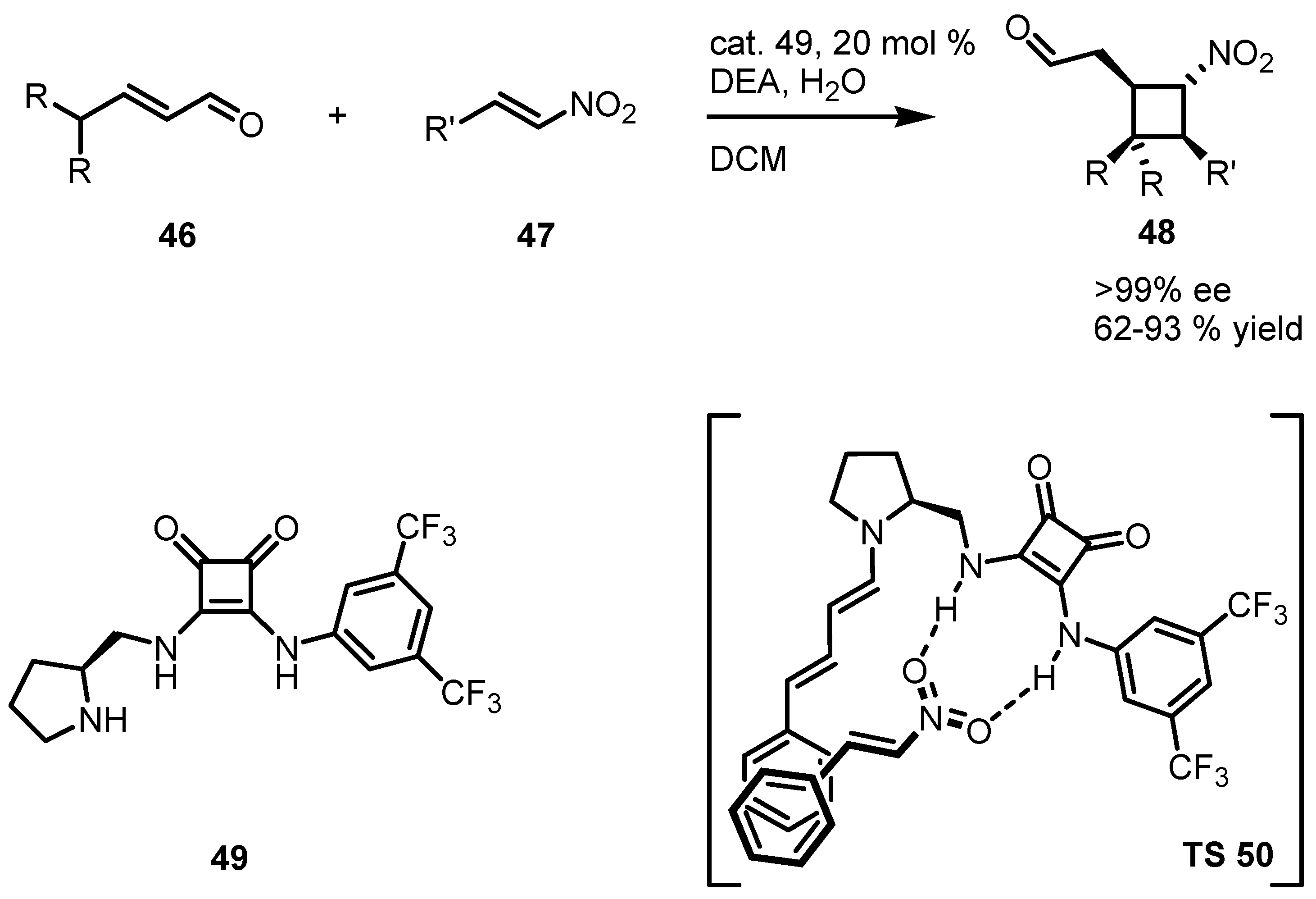
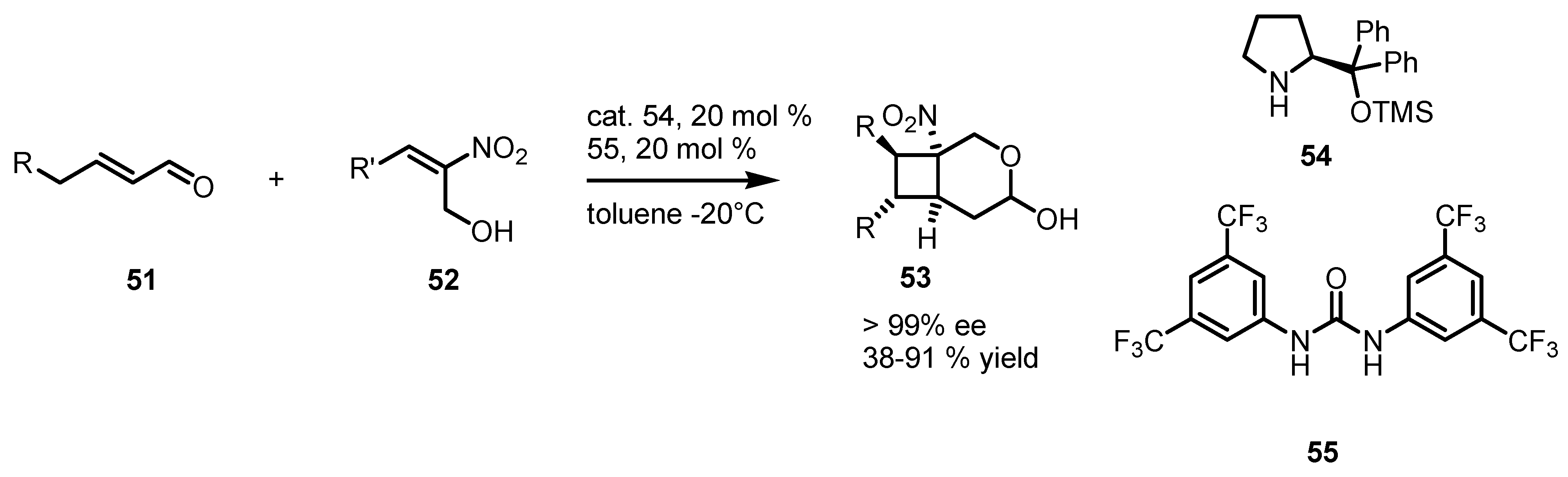
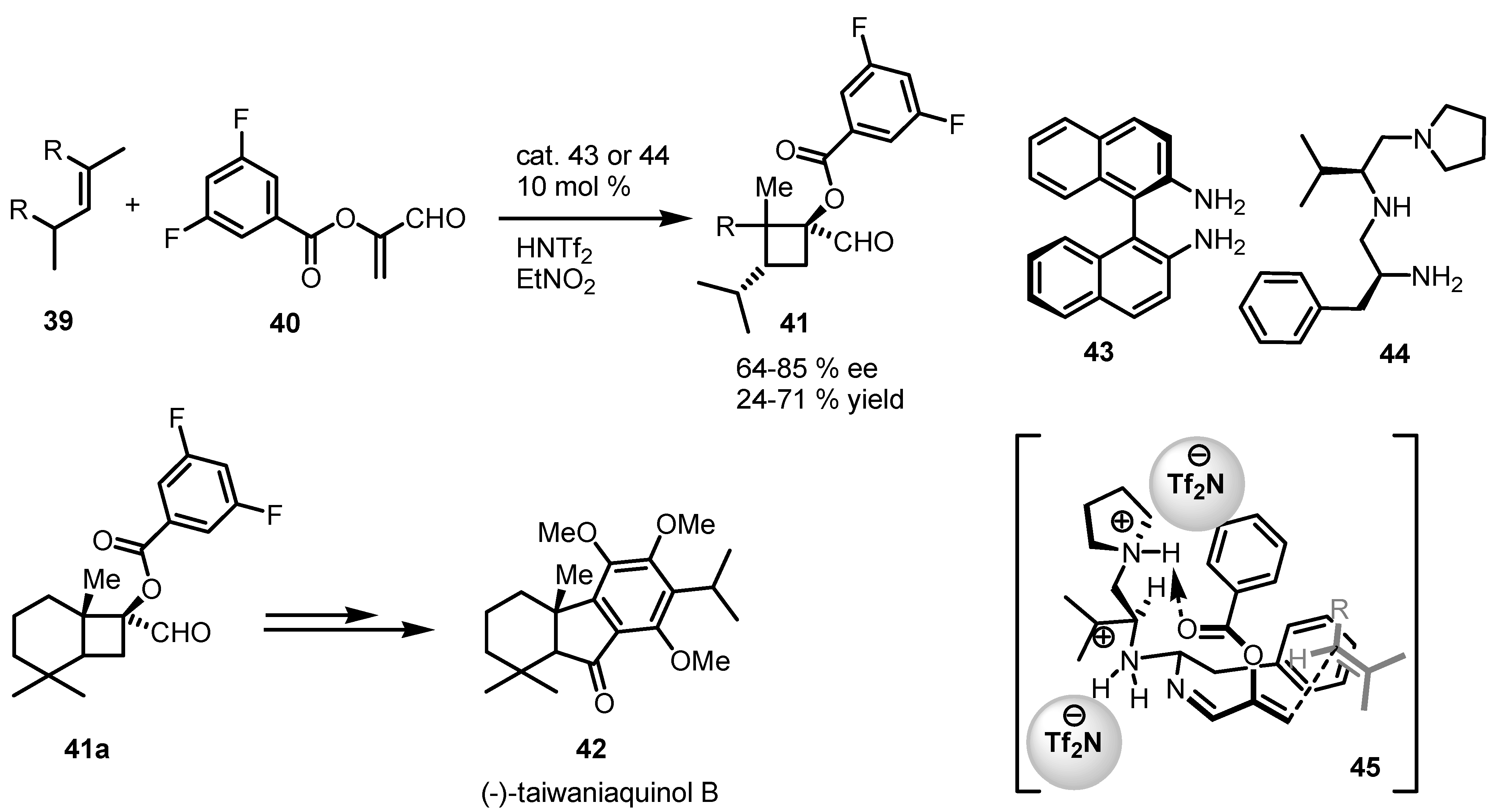
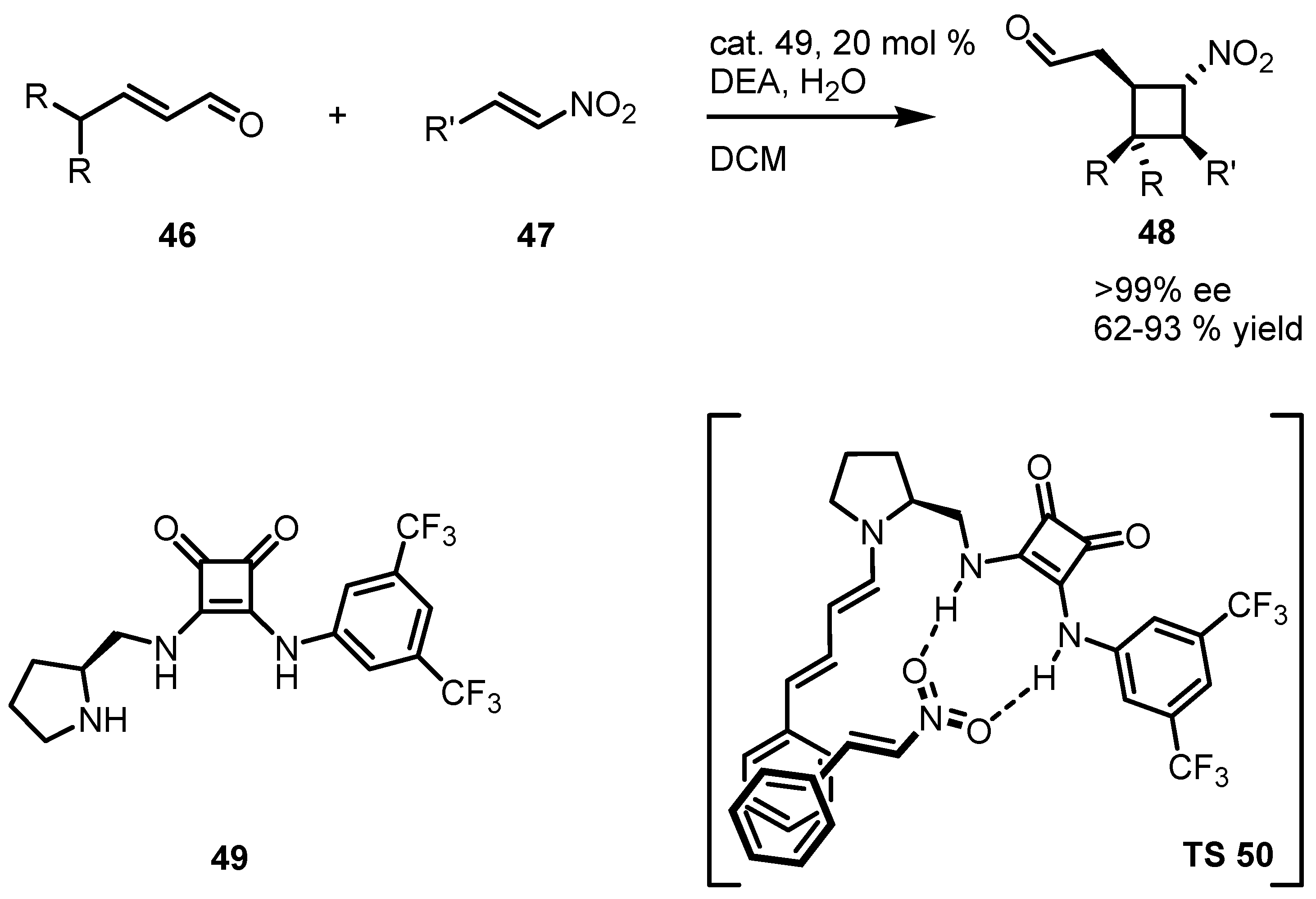
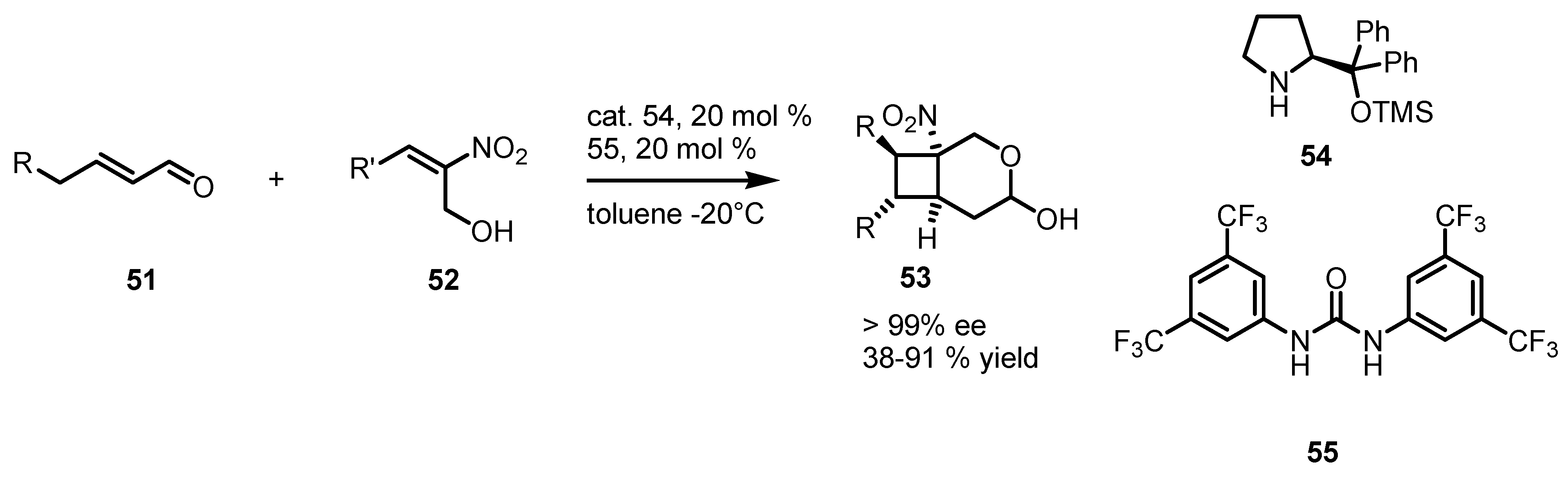
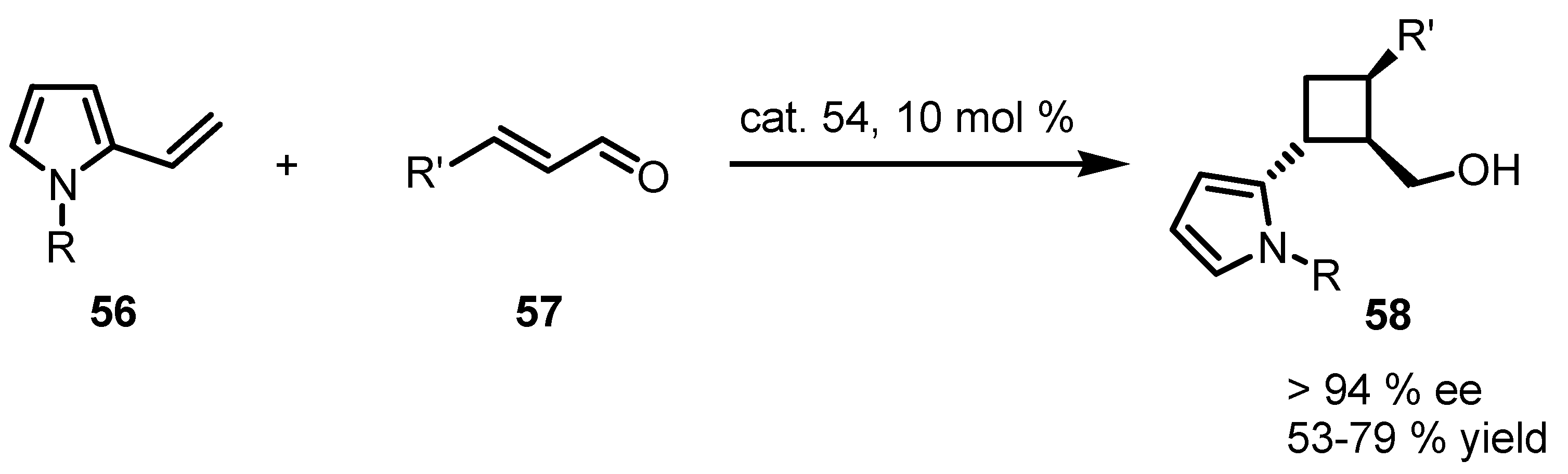
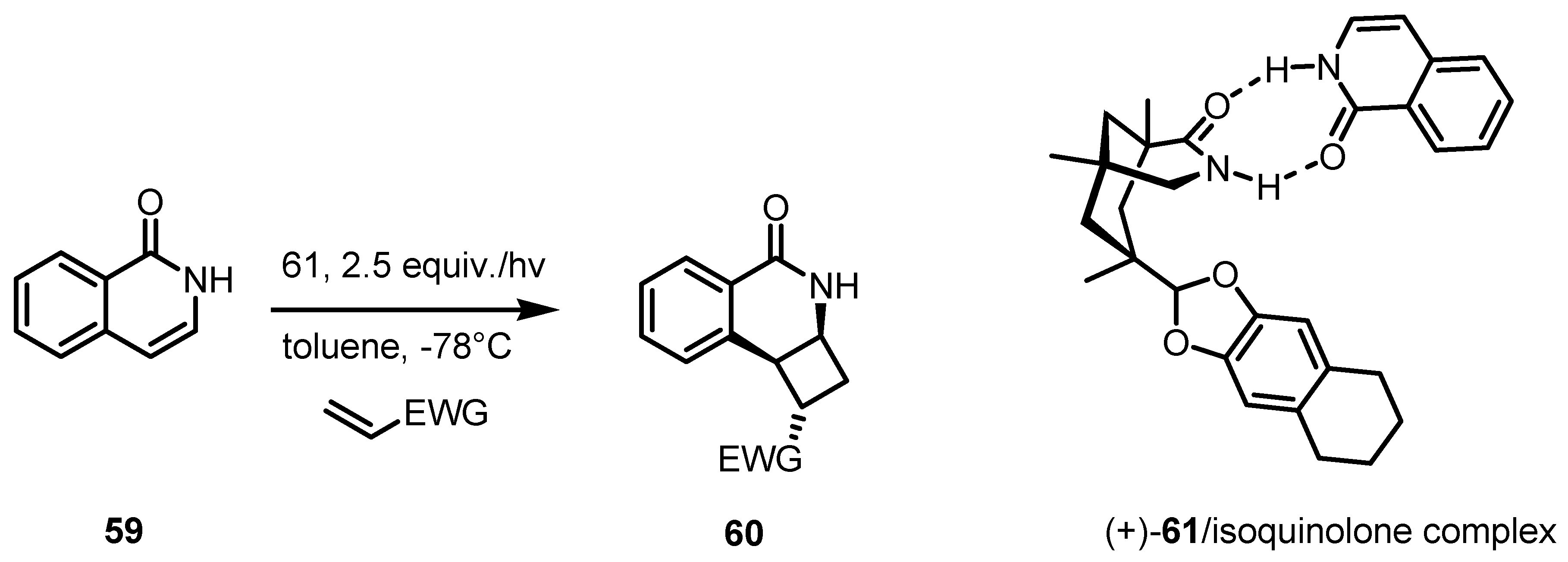
3.5. Hydrogen-Bonding Mediated [2+2] Asymmetric Cycloaddition
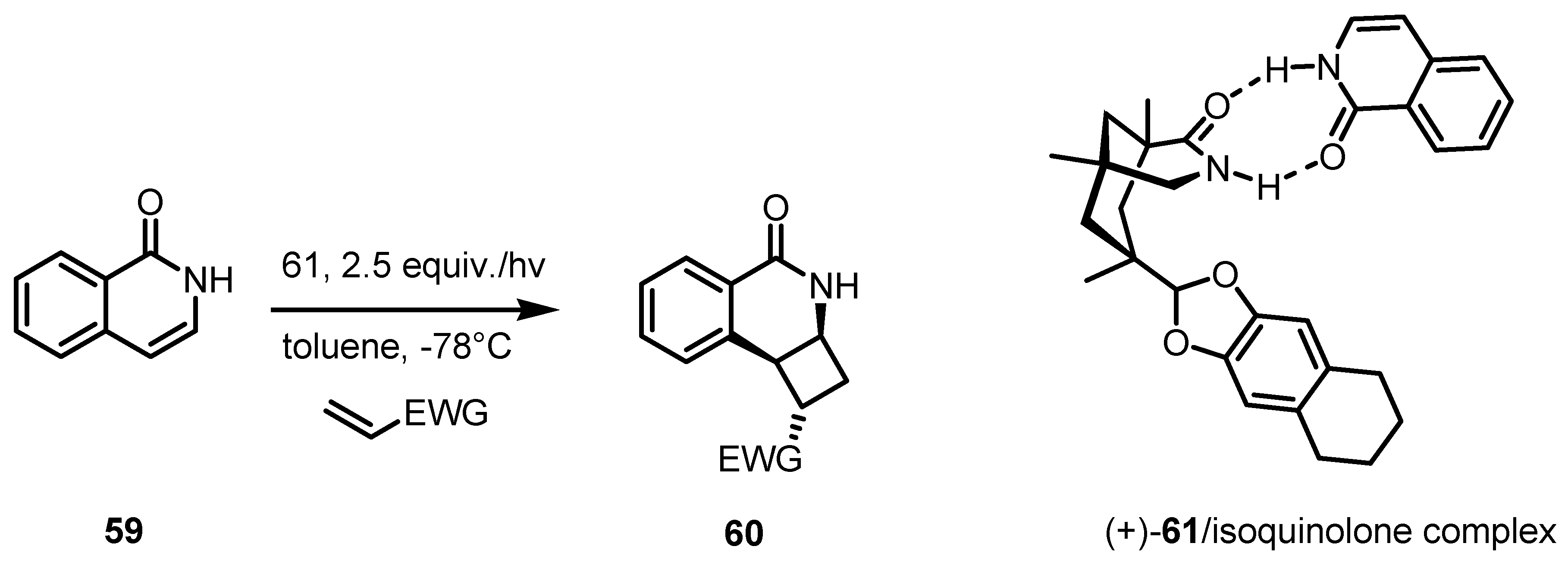
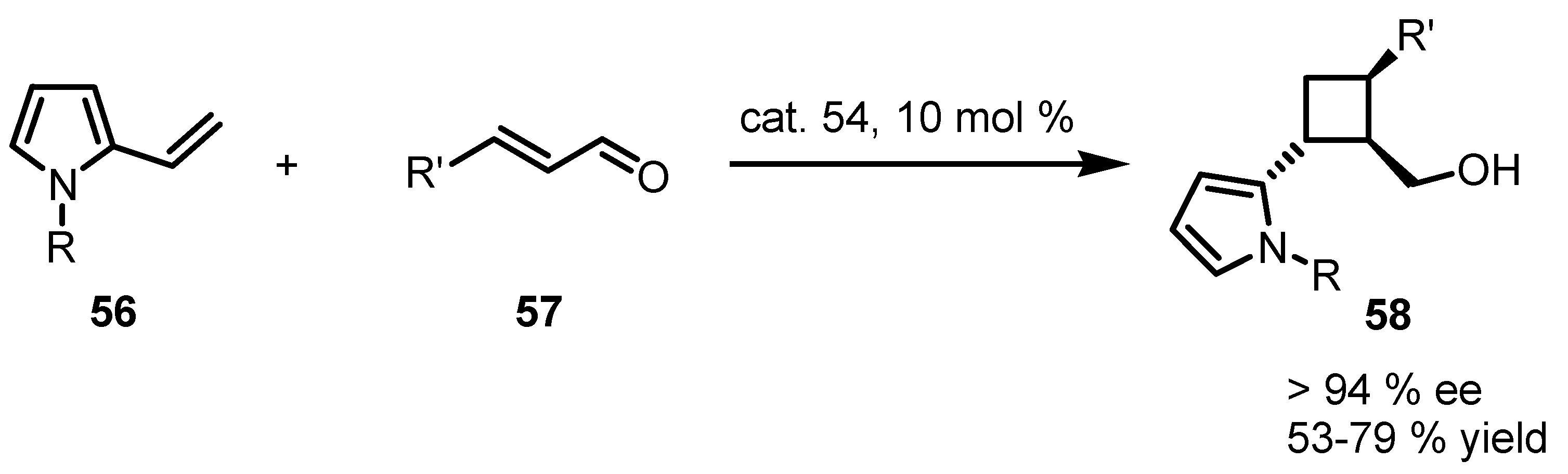
4. Desymmetrization of Cyclobutane and Cyclobutanone Derivatives
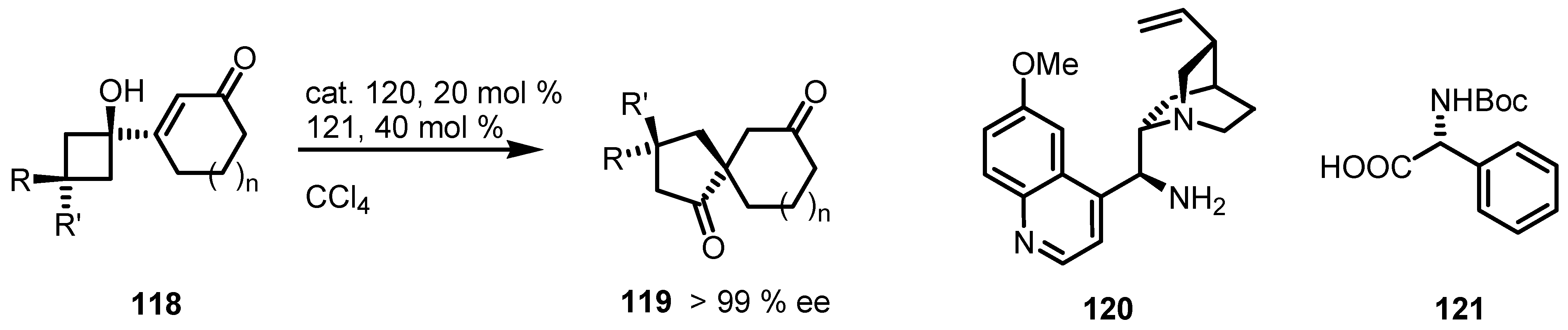
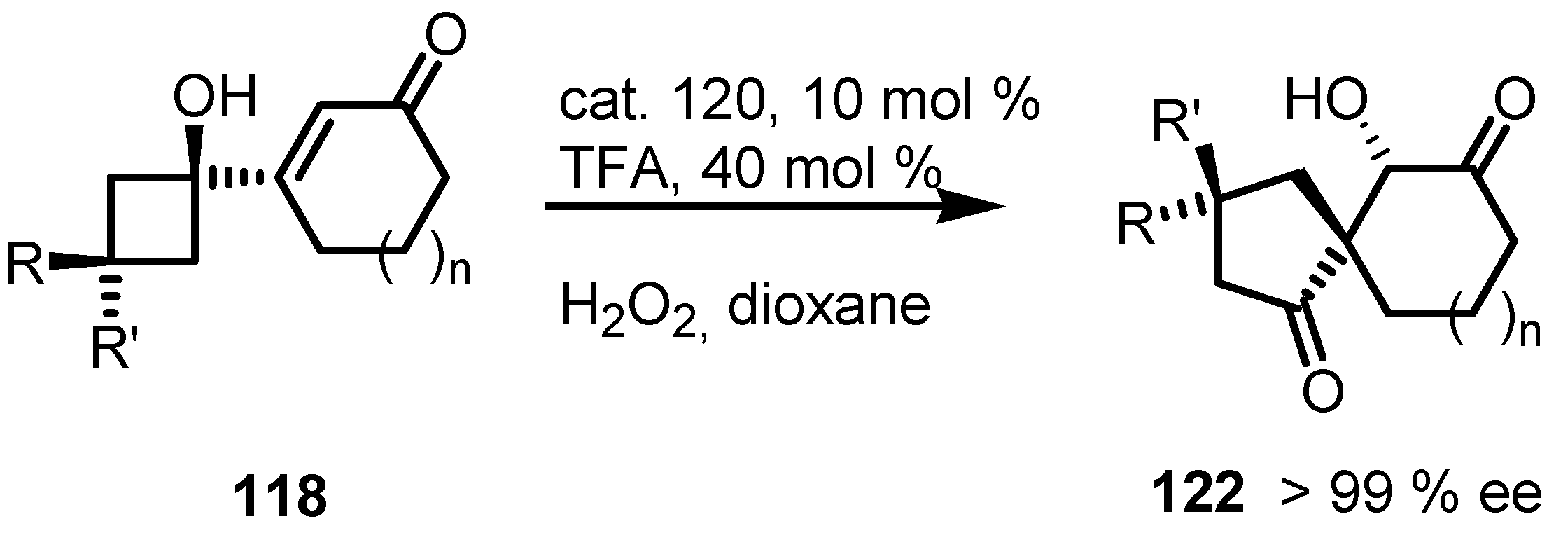
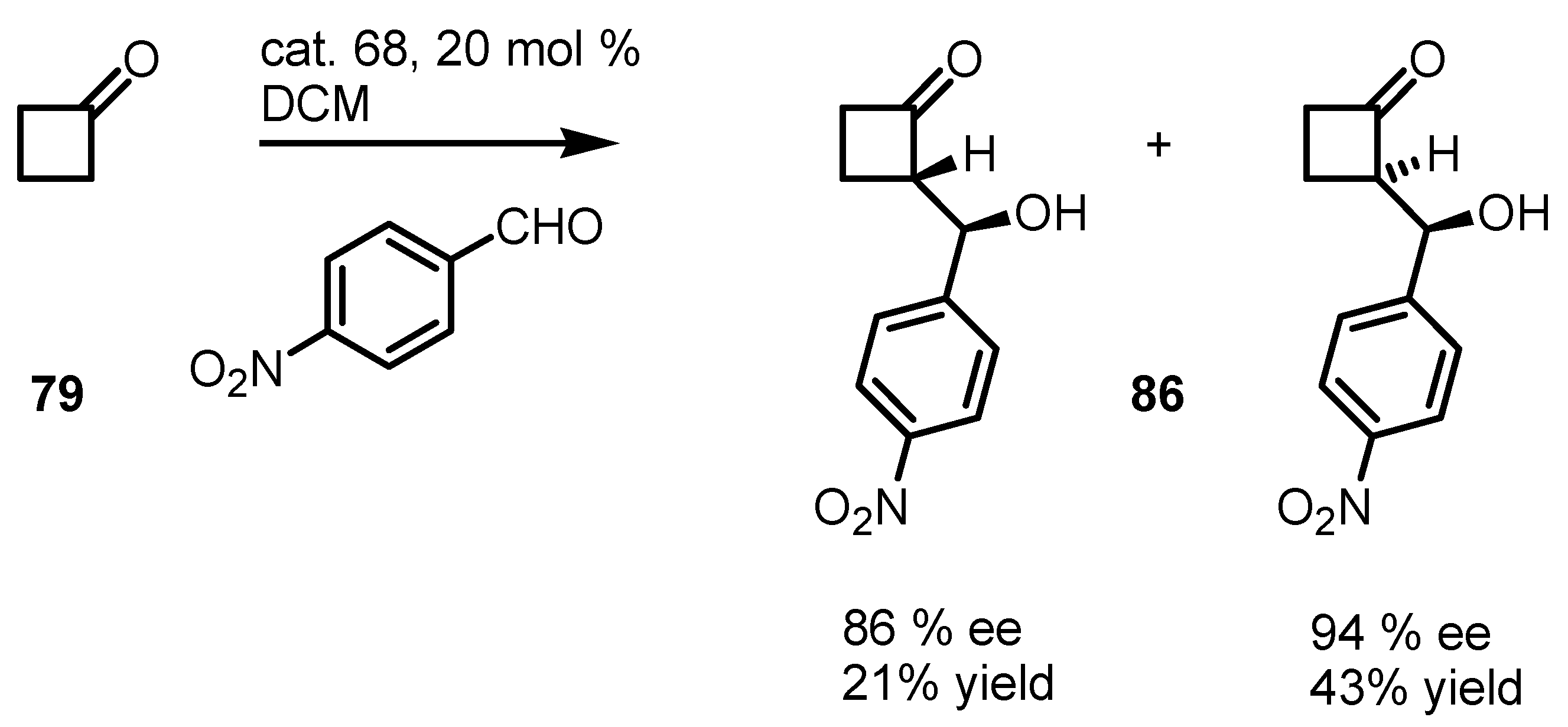
4.1. Organocatalyzed Bronsted Acids based Desymmetrizations
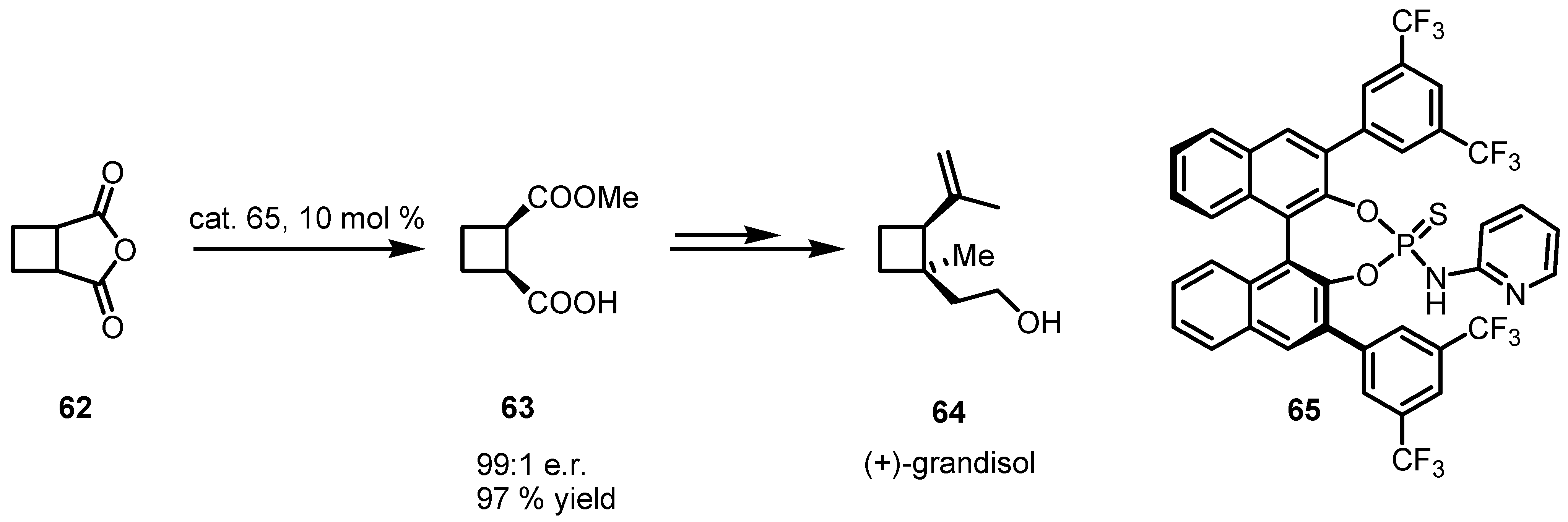
4.2. Organocatalyzed Aldol based Desymmetrization Reactions

4.3. Enaminocatalyzed Reactions of Cyclobutanones with Nitrosobenzene
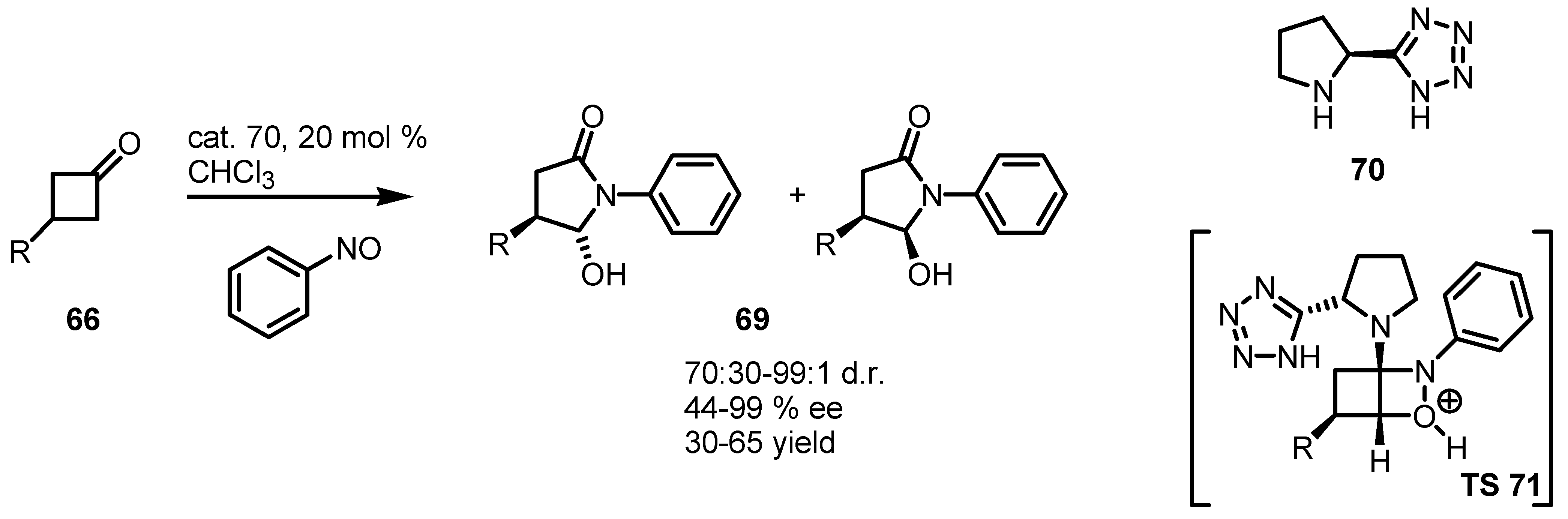
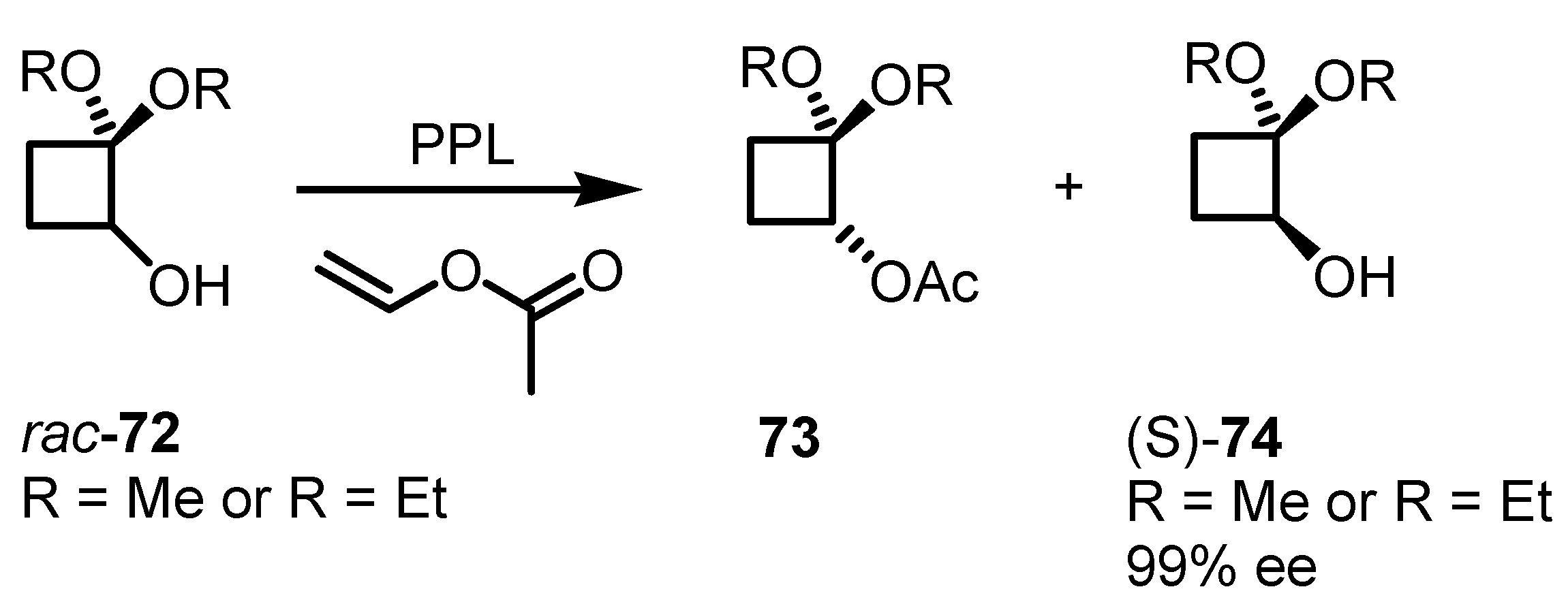
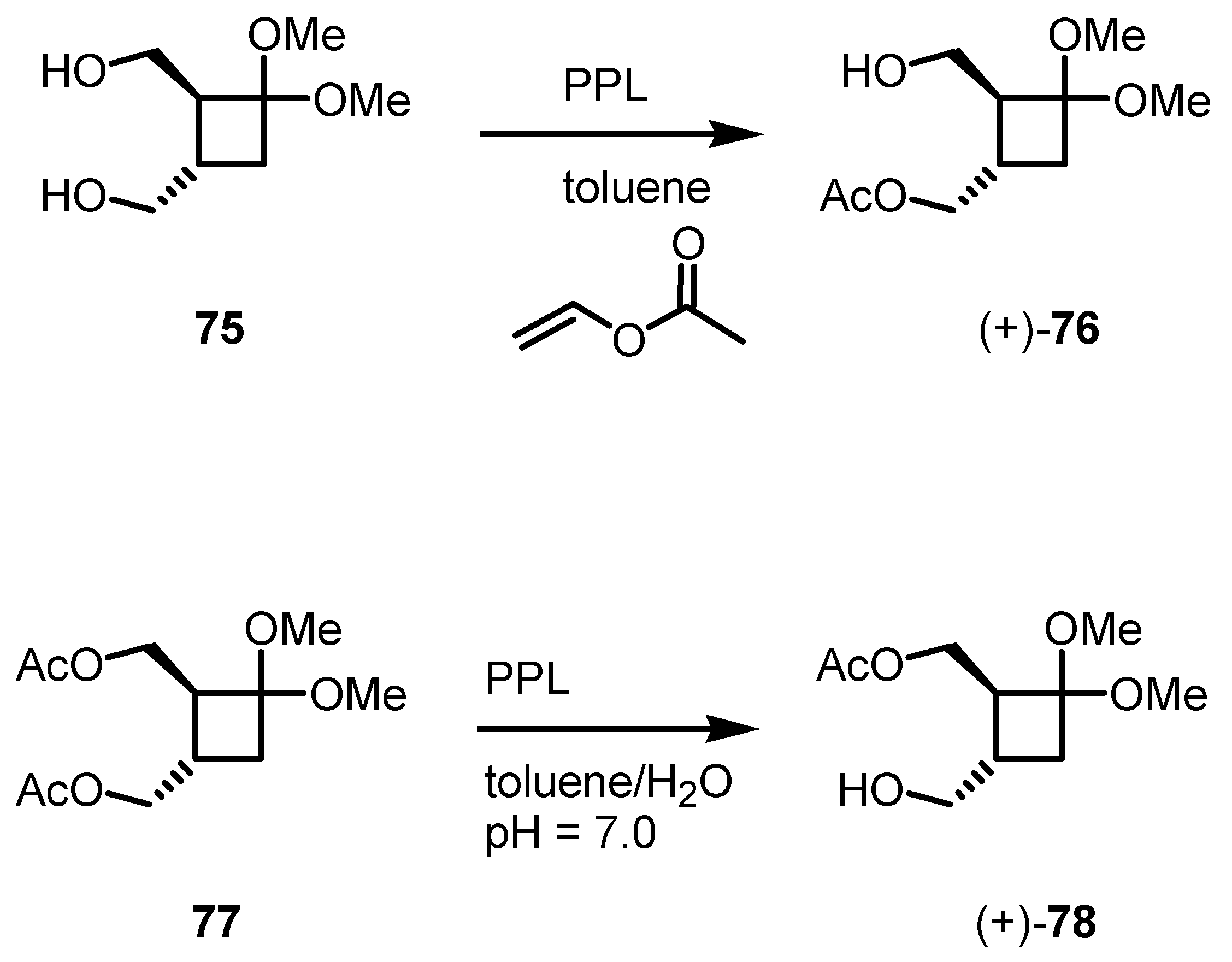
5. Biocatalytic Resolution of Cyclobutanes
5.1. Biocatalytic PPL based Cyclobutanol Resolution by Esterification and Hydrolysis


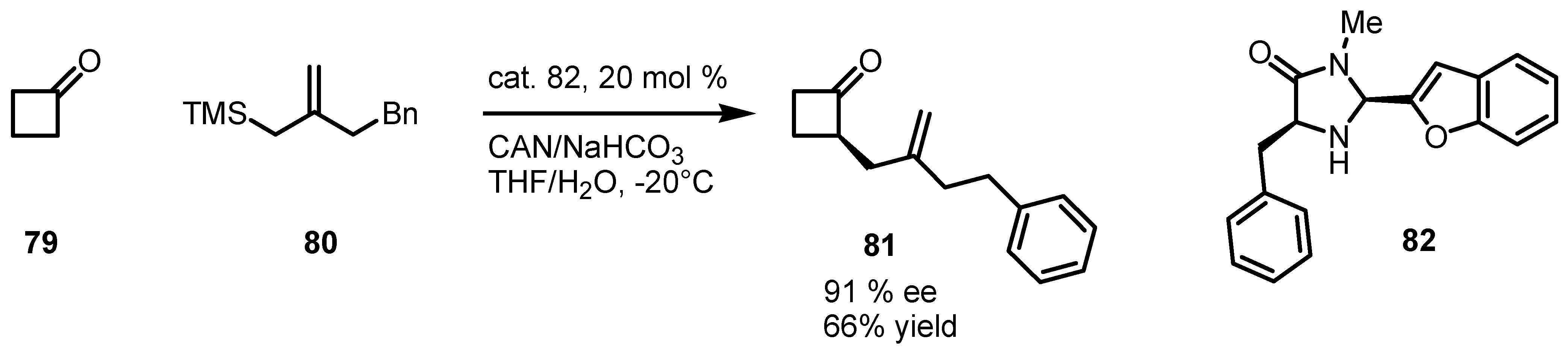
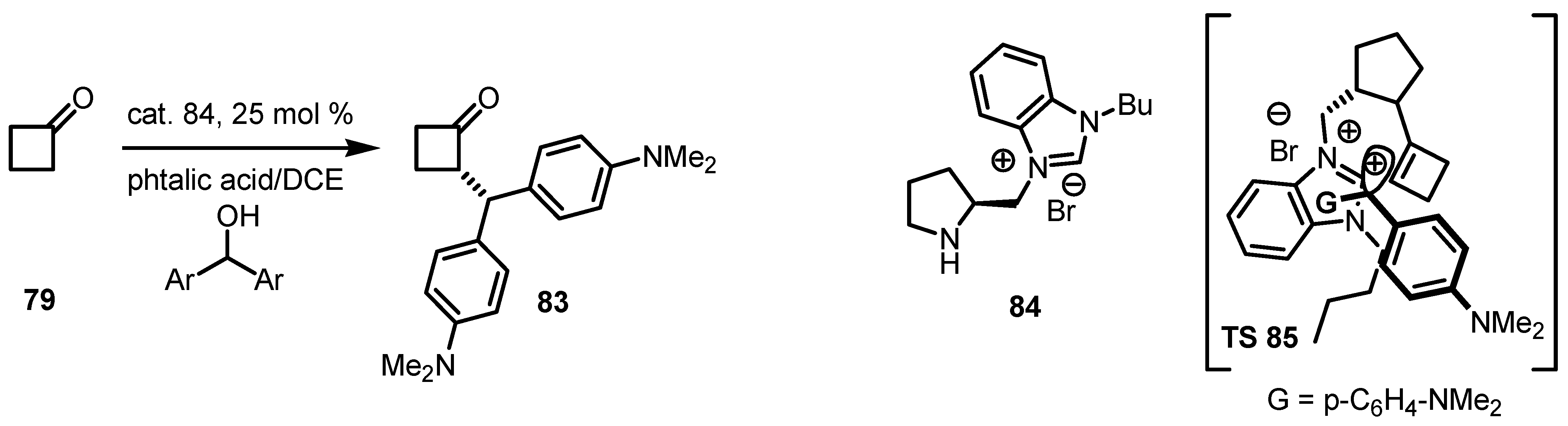
6. Cyclobutanone α-Functionalization
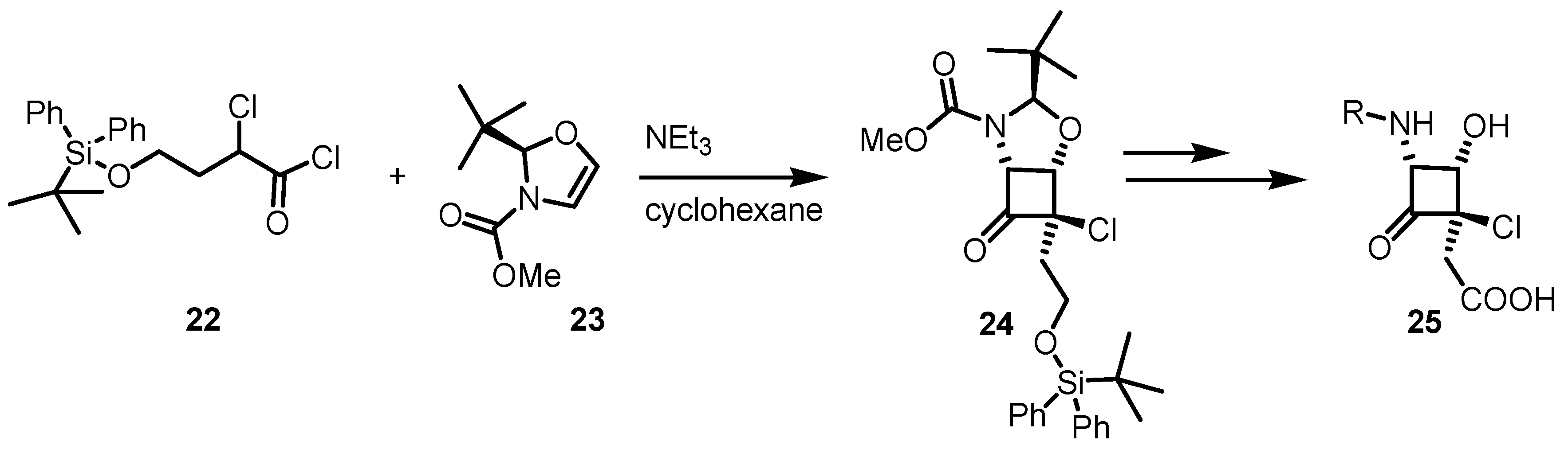
6.1. α-Functionalization of Cyclobutanones via SOMO Catalysis
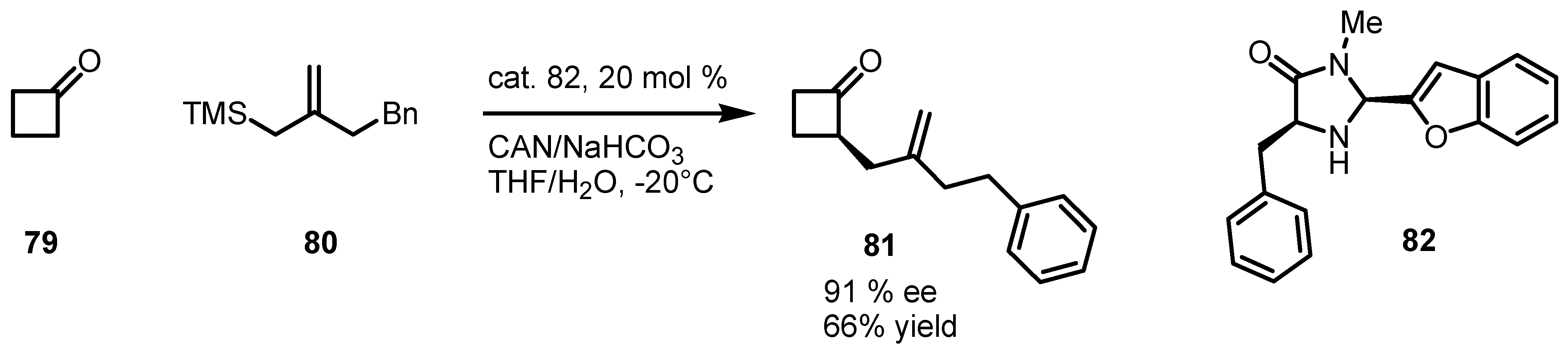
6.2. Asymmetric SN1 Alkylation of Cyclobutanones
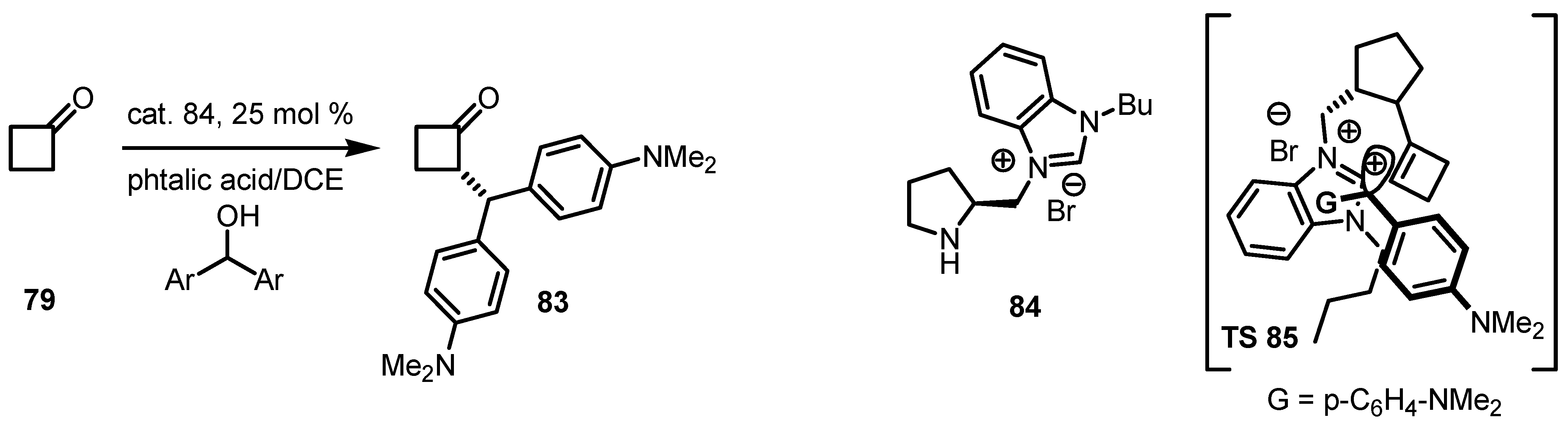
6.3. Organocatalyzed Aldol Reactions

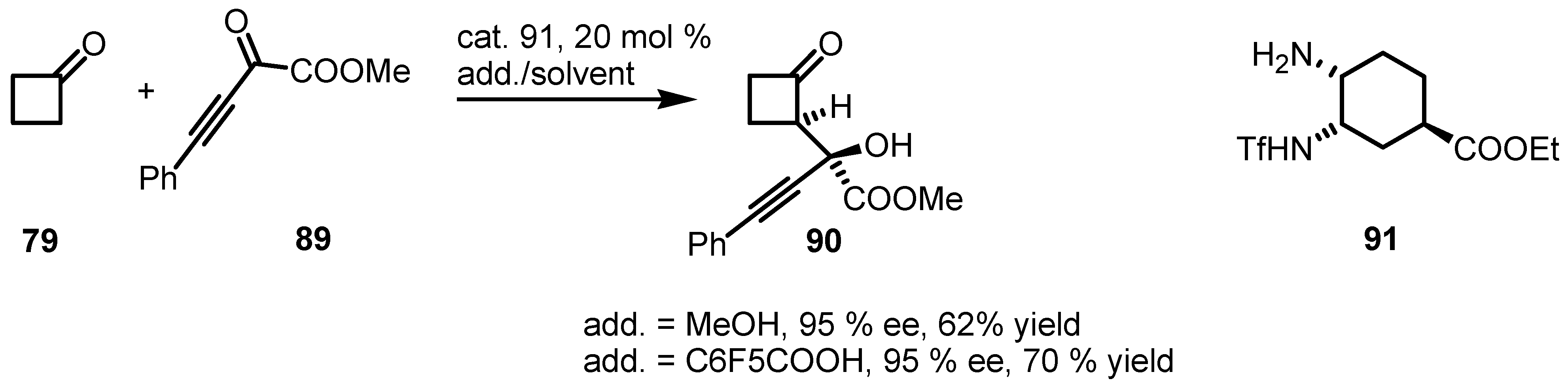
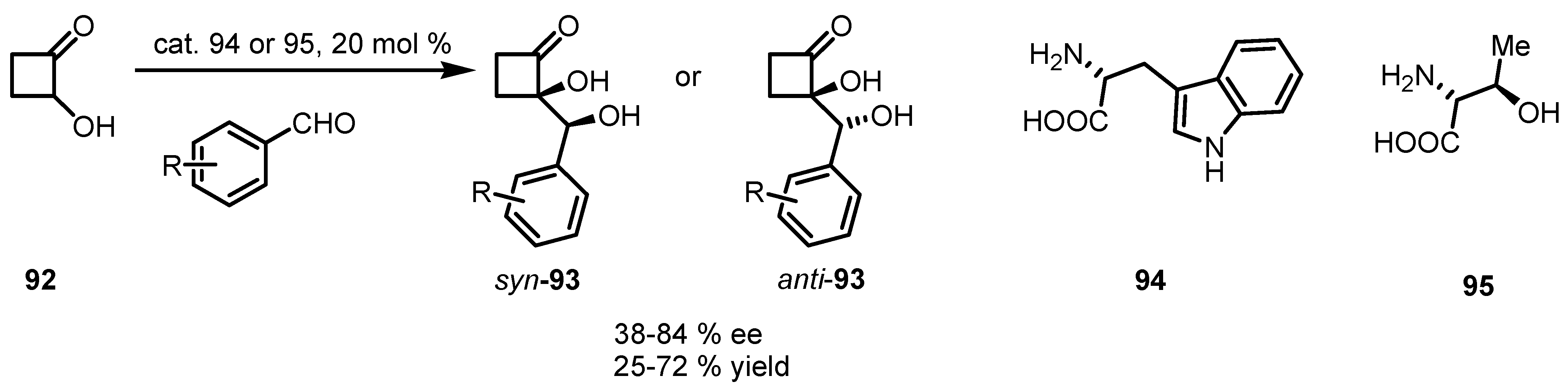
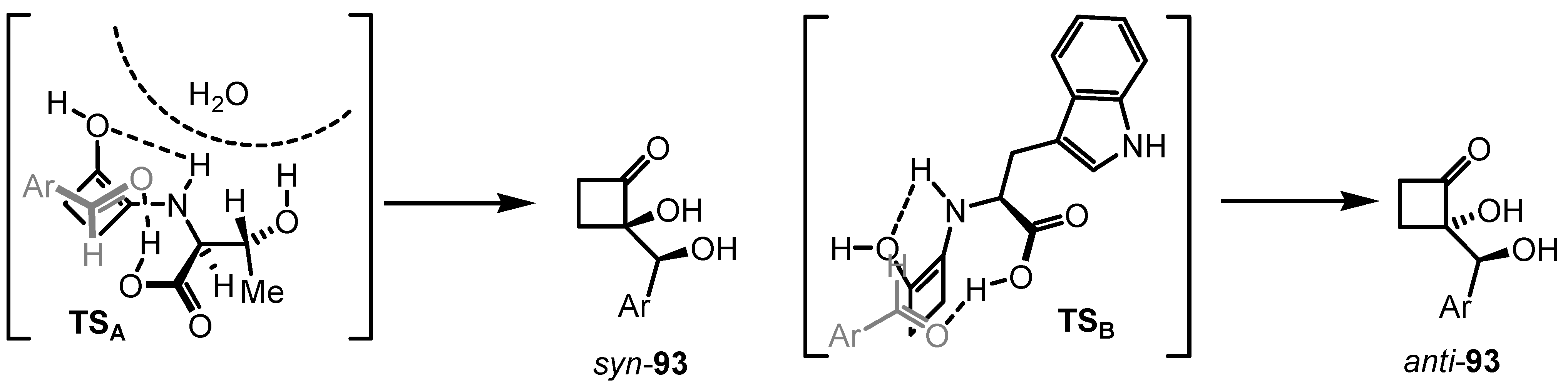
6.4. Organocatalyzed Mannich Addition of Cyclobutanones to Glycolates
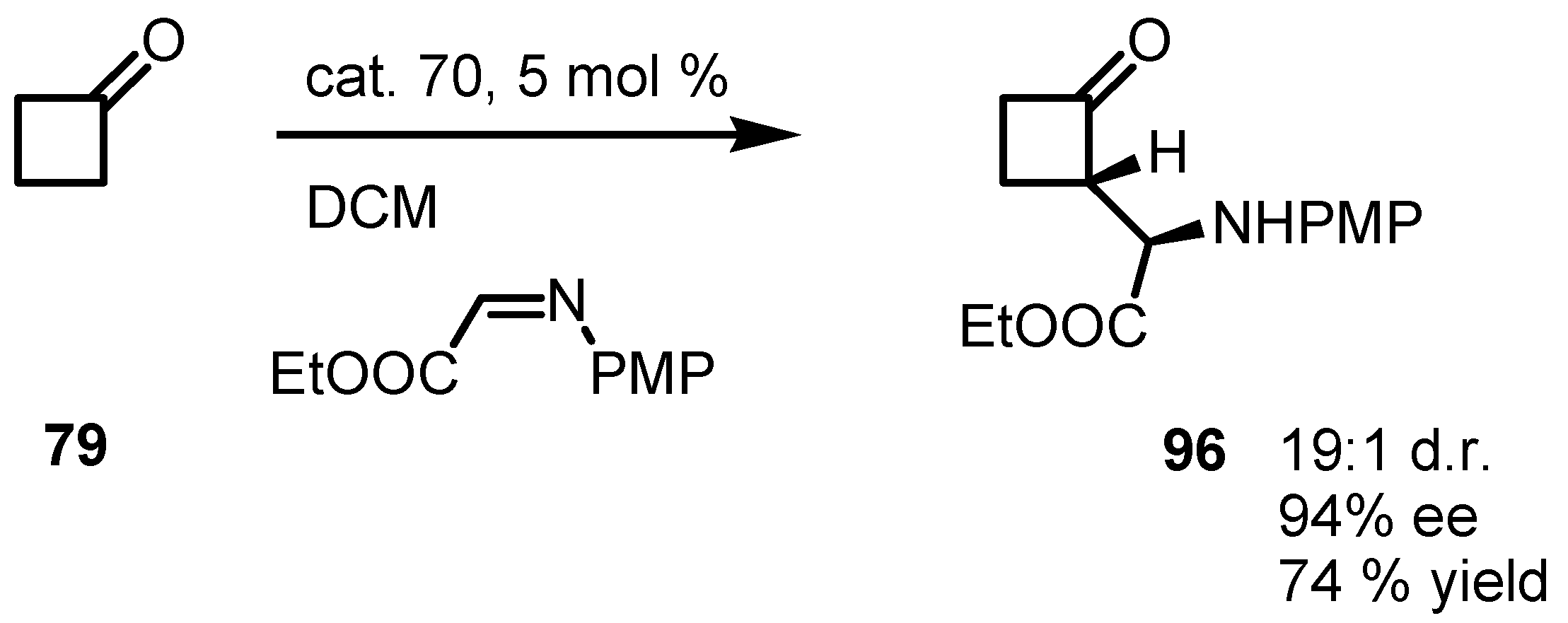
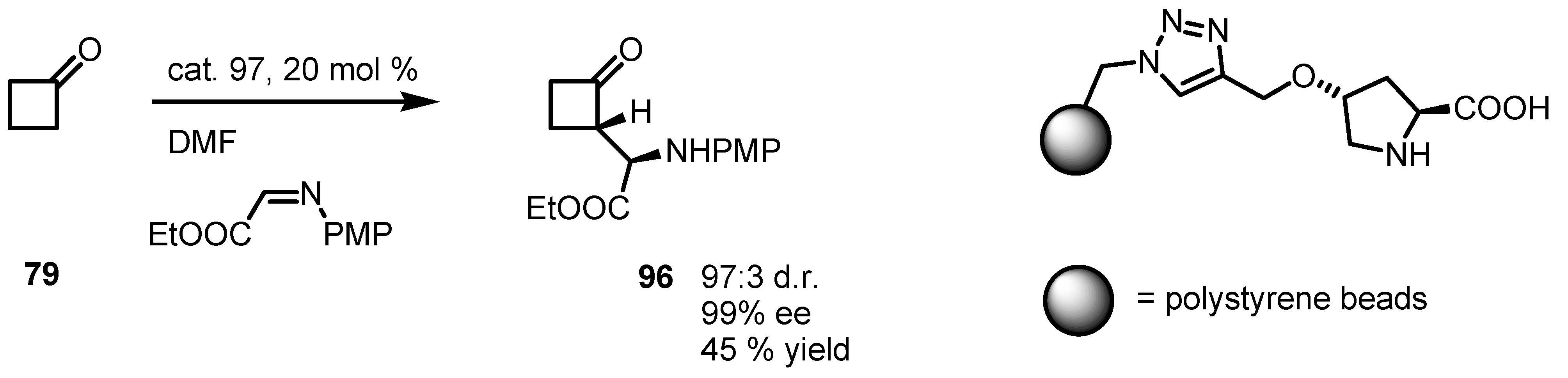
6.5. Organocatalyzed Michael addition of Cyclobutanones to Nitrostyrenes

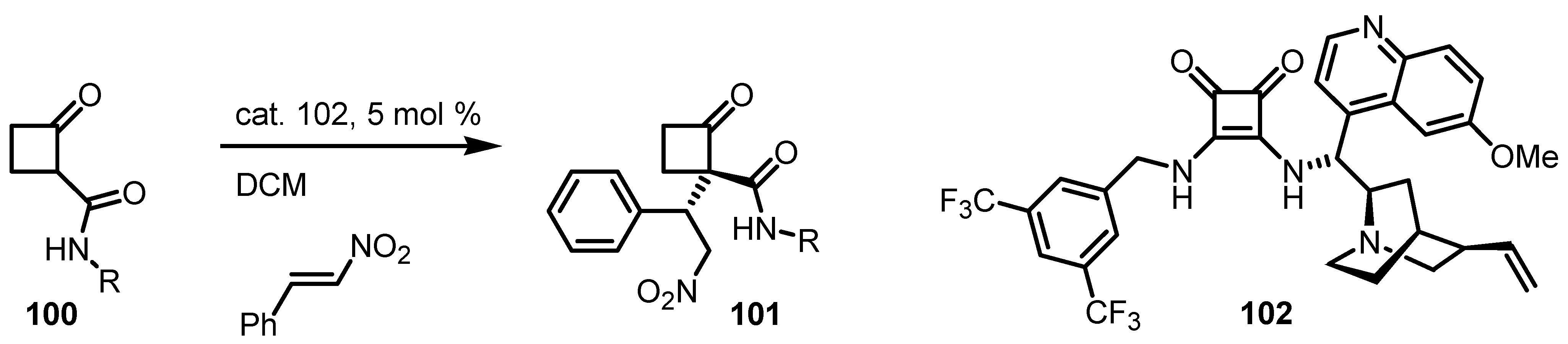
6.6. Cyclobutanone α-Heteroatom Functionalization
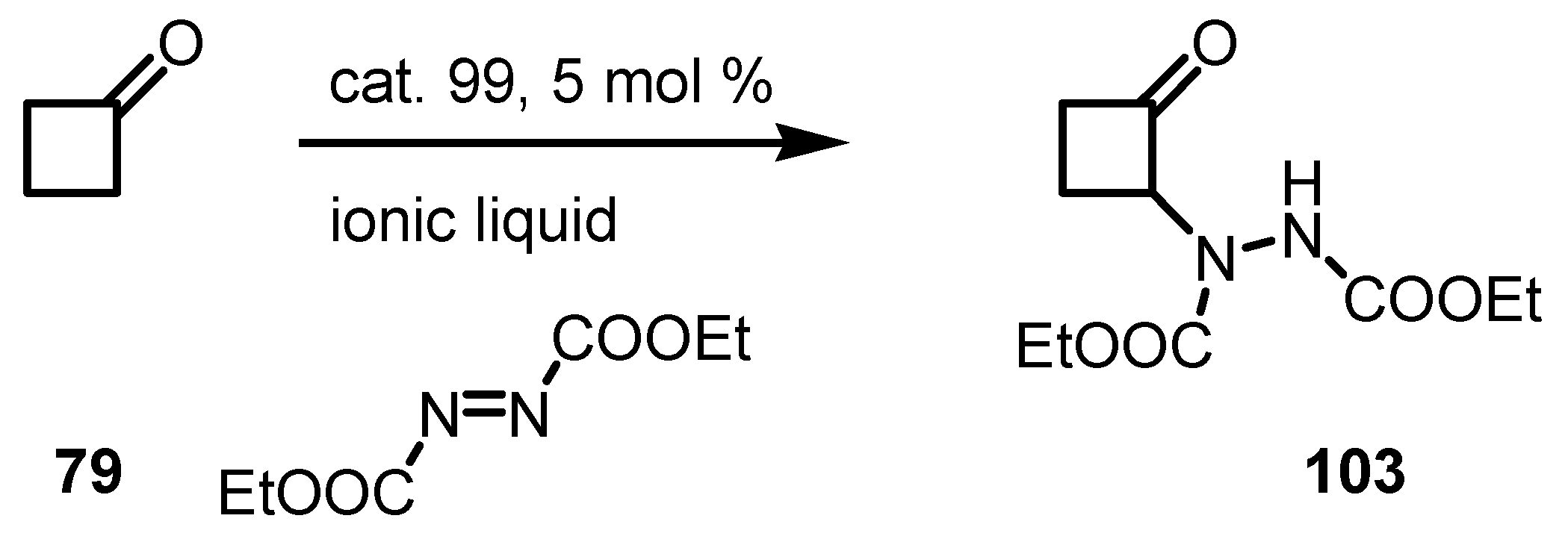
7. Cyclobutane Ring Enlargement
7.1. Chiral Non Racemic Cyclobutanes Ring Expansion
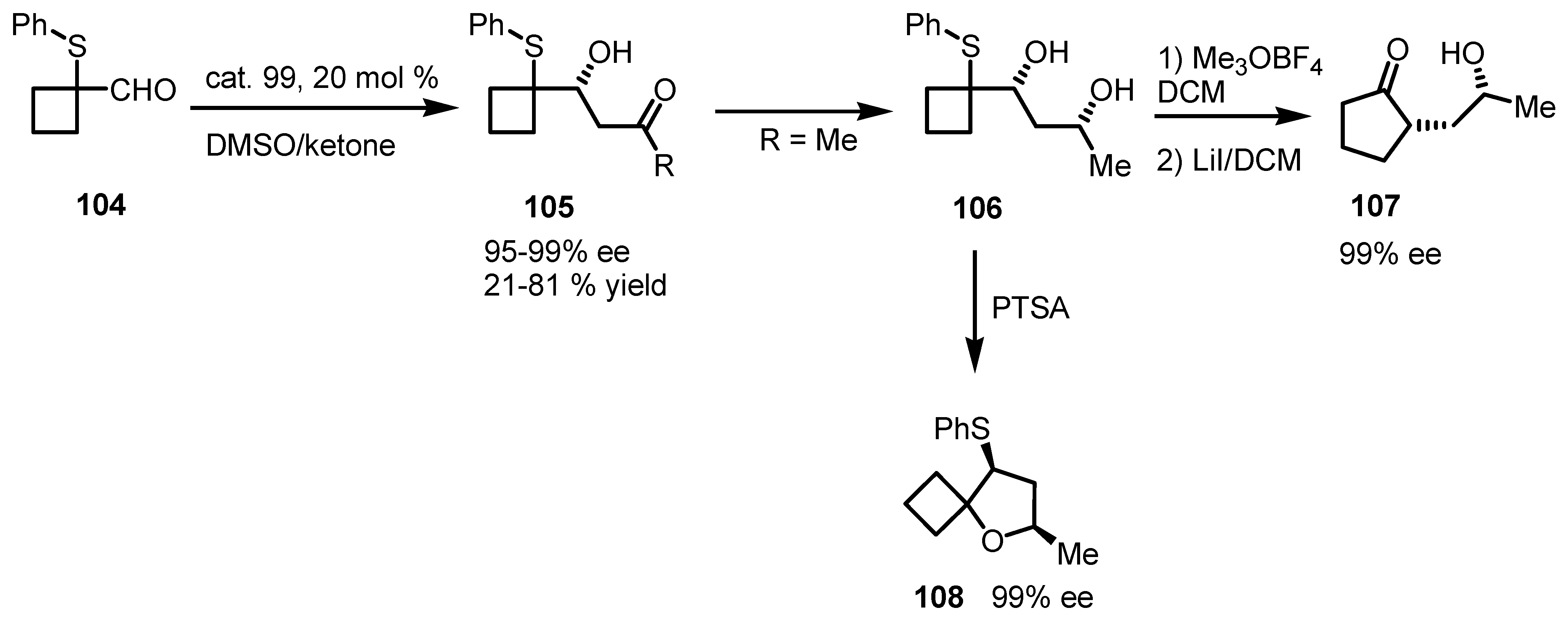
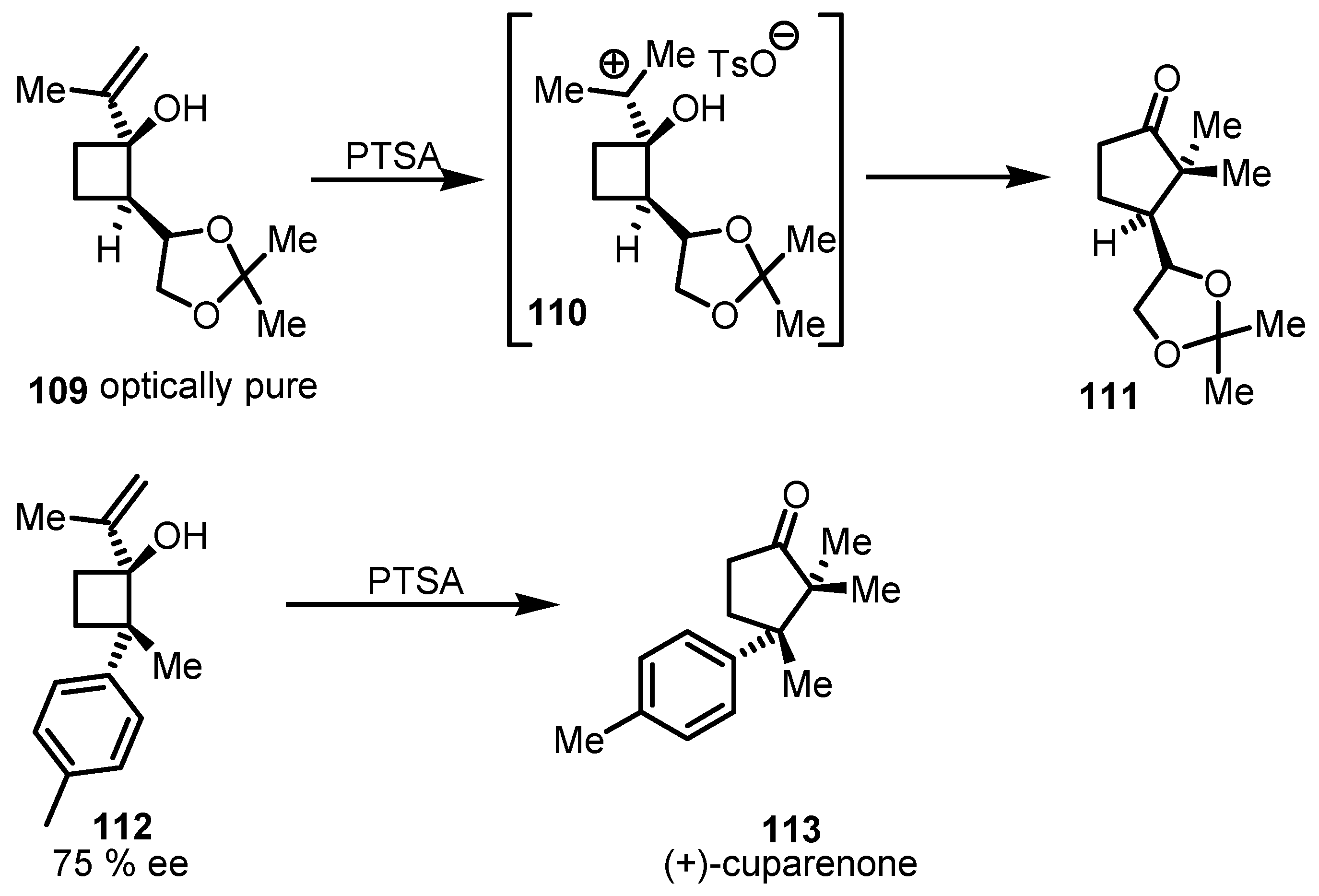
7.2. Chiral Non Racemic Oxaspirohexanes Ring Enlargement
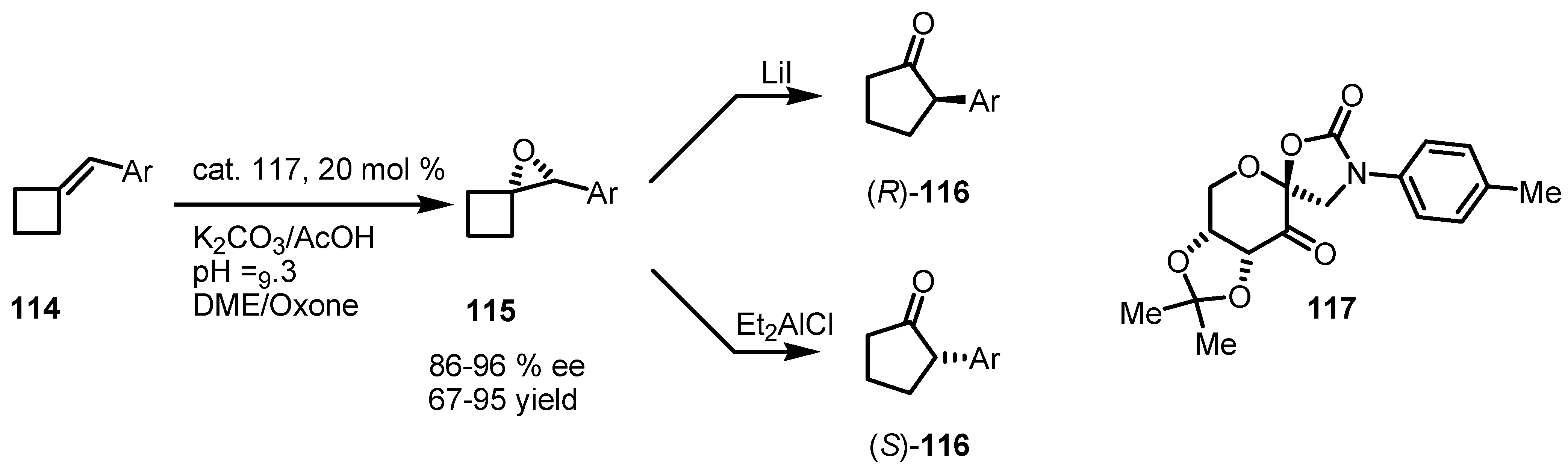
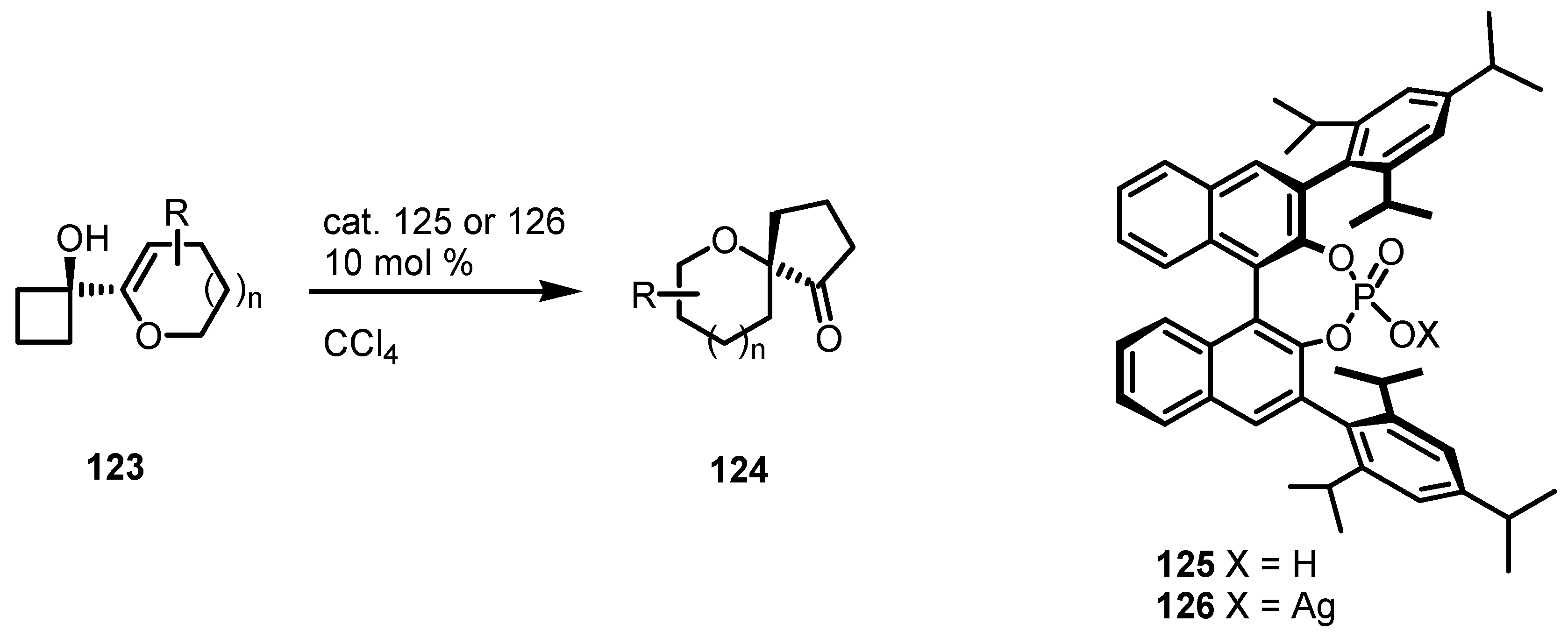
7.3. Organocatalyzed Enantioselective Cyclobutane Ring Expansions
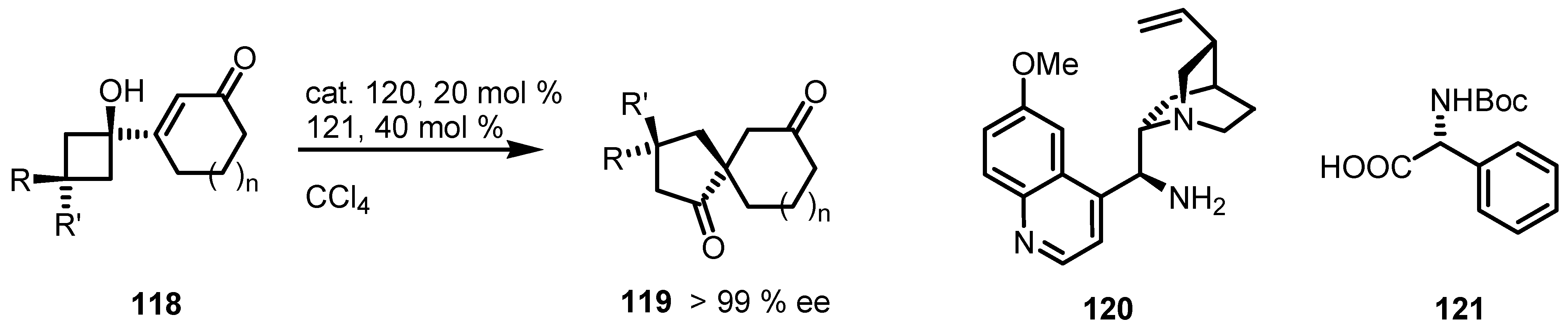
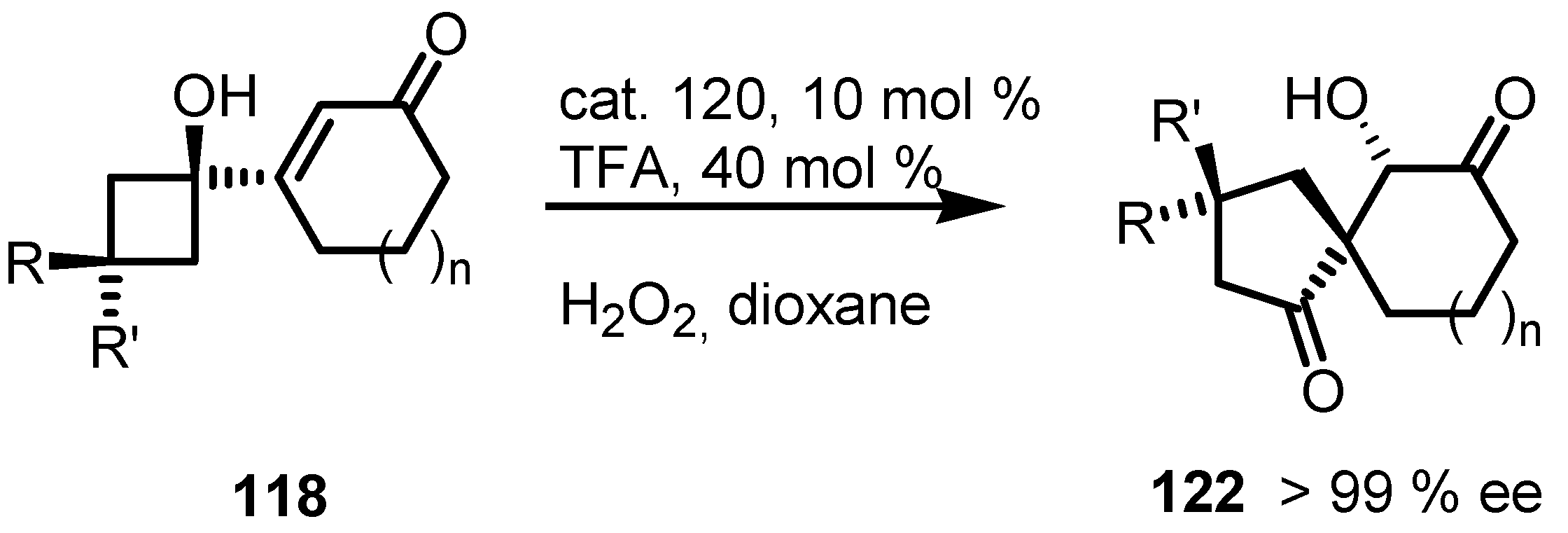

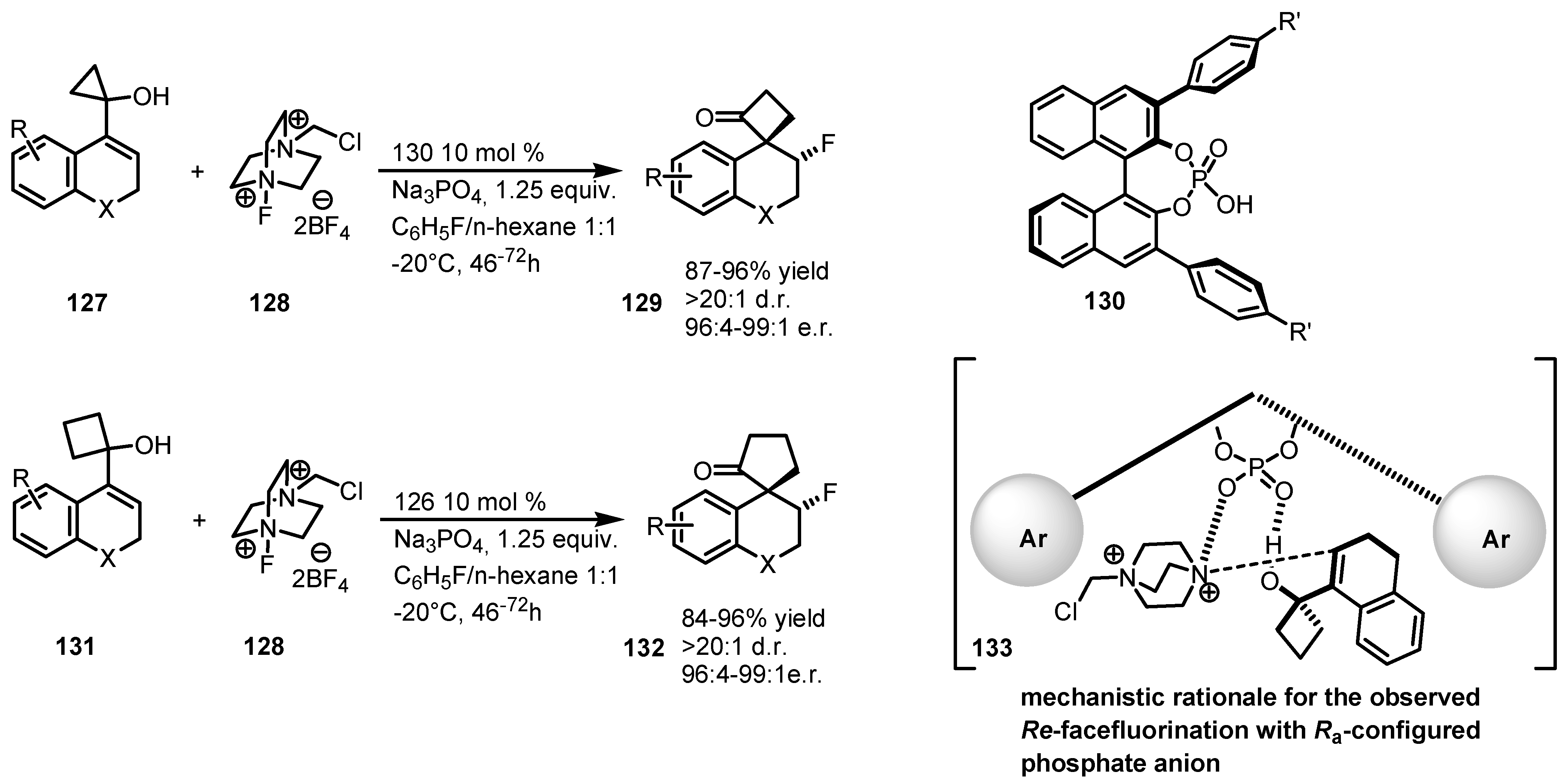
7.4. Organocatalyzed Enantioselective Fluorination-Induced Cyclobutanes and Cyclopropanes Ring Expansion

8. Enantioselective Bayer-Villiger Oxidation of Cyclobutanones
8.1. Enantio- and Diastereoselective Bayer-Villiger Oxidation of Chiral Cyclobutanones
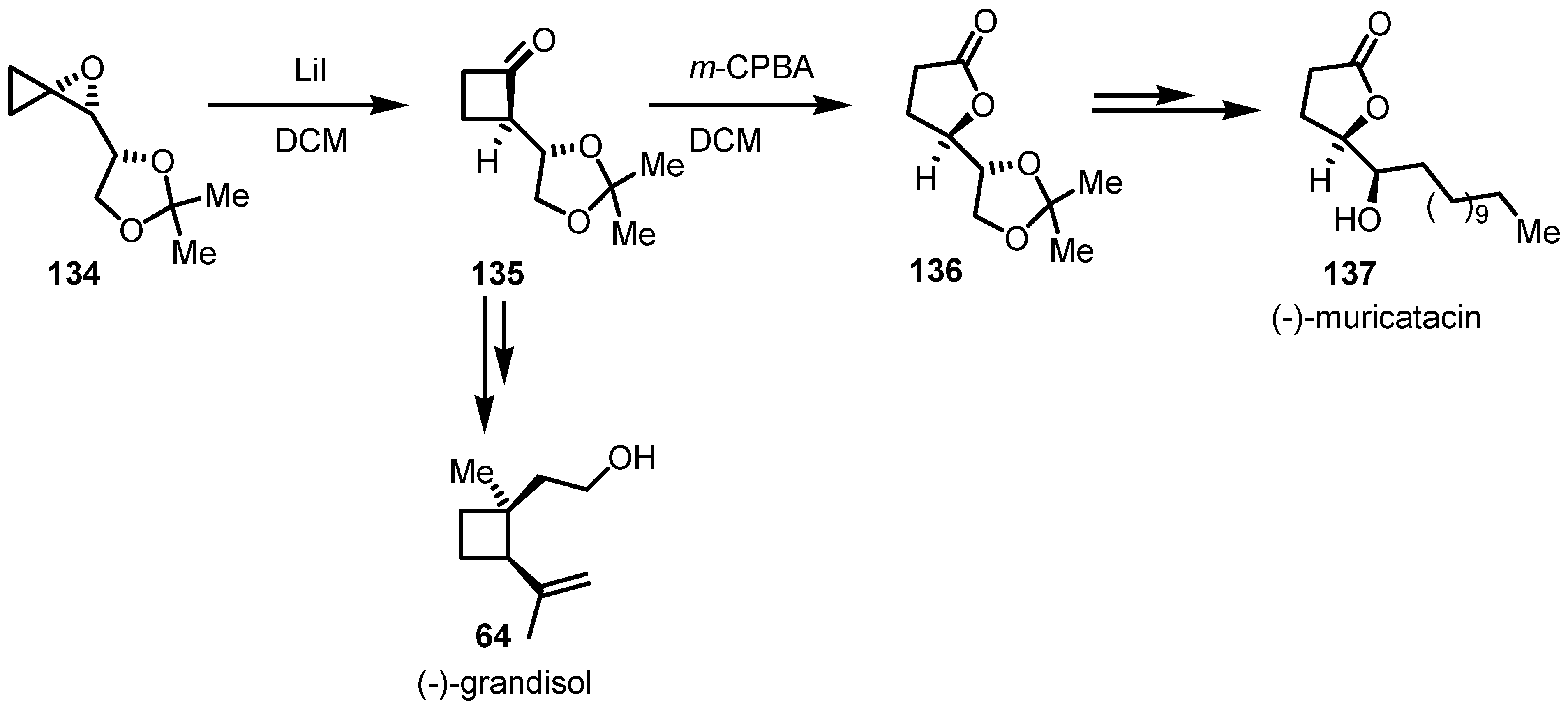
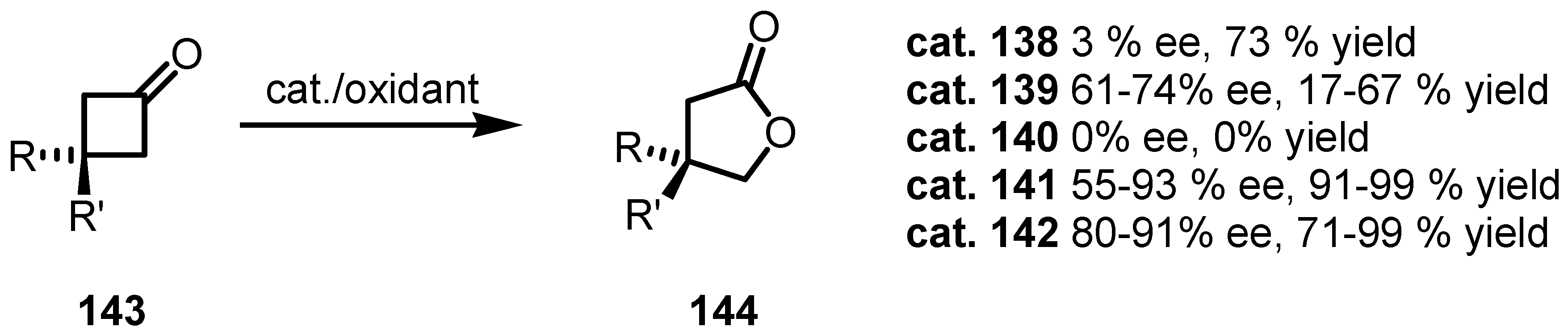
8.2. Organocatalyzed Enantioselective Cyclobutanone Bayer-Villiger Oxidation
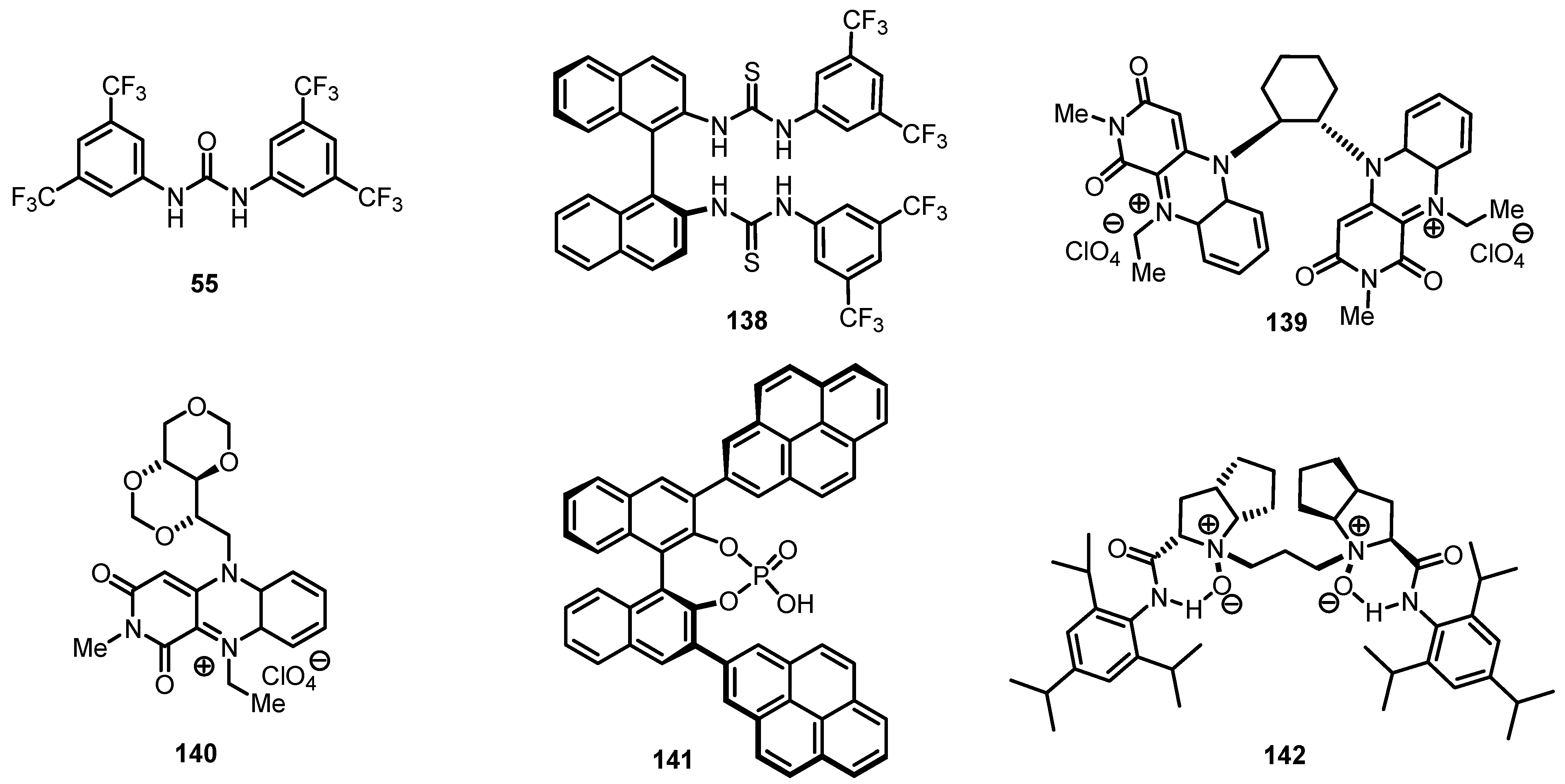
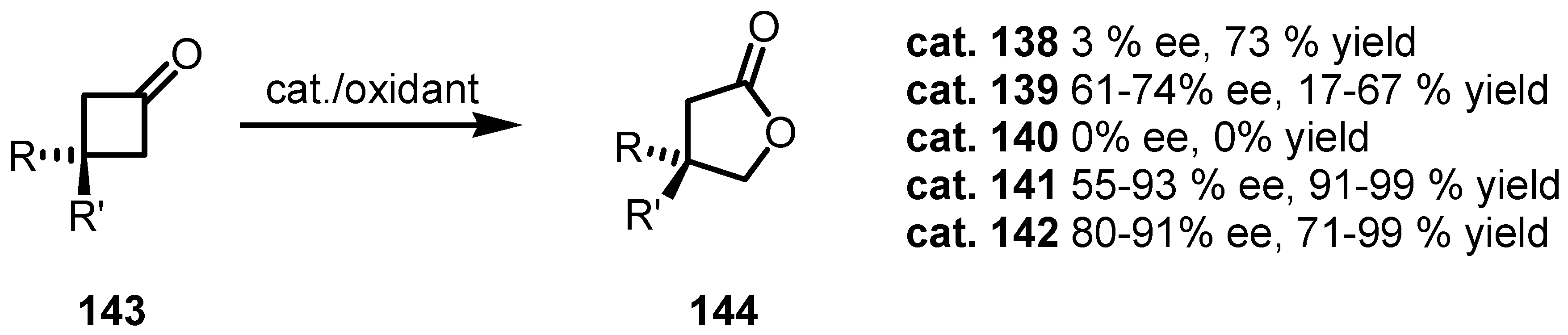
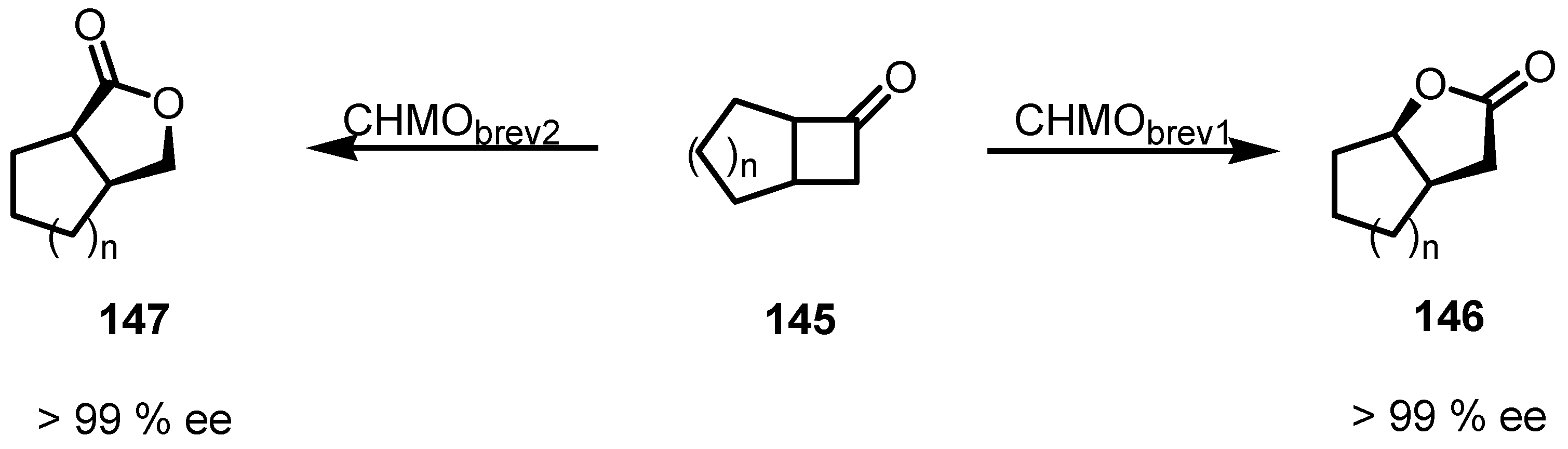
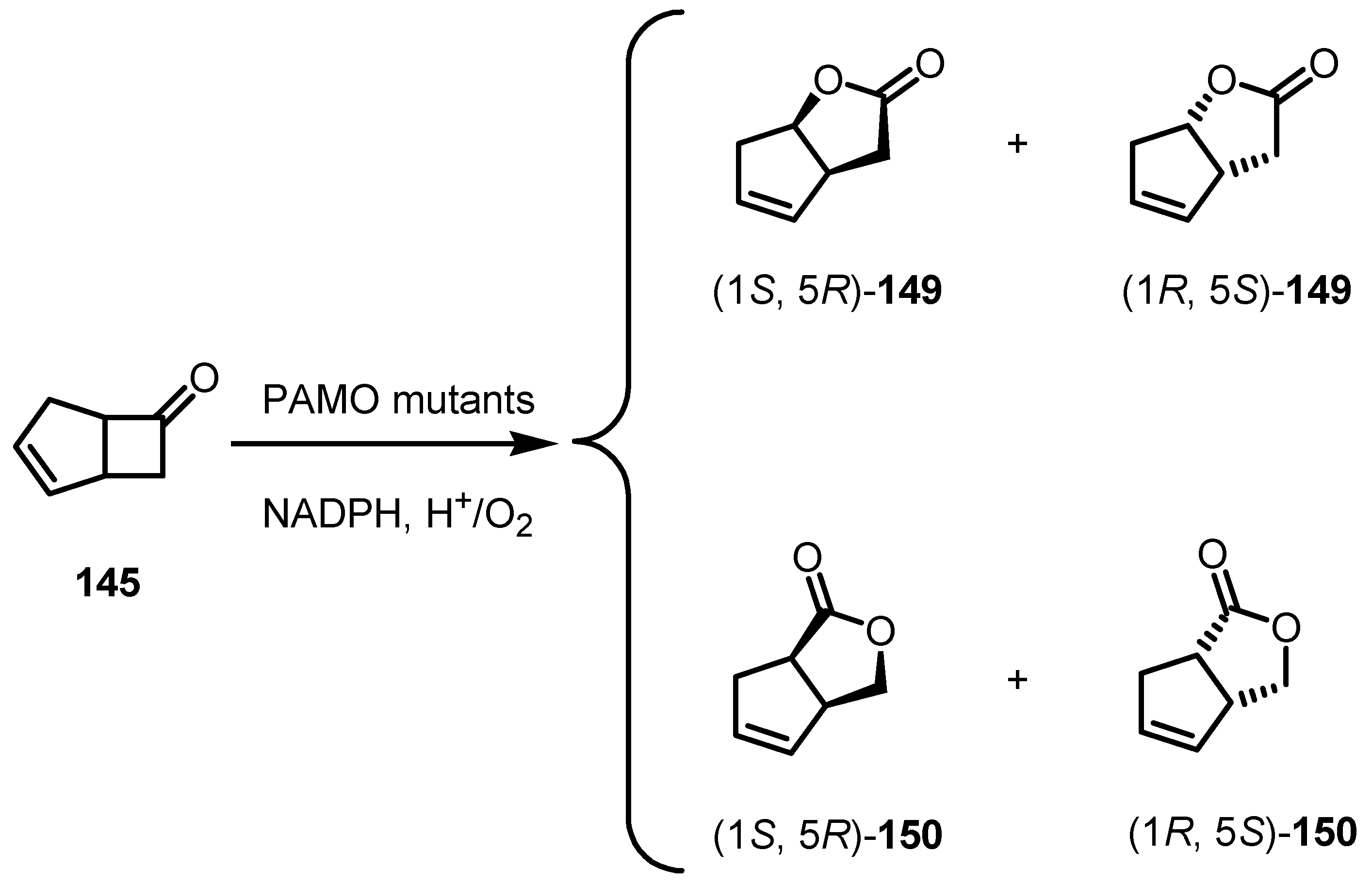
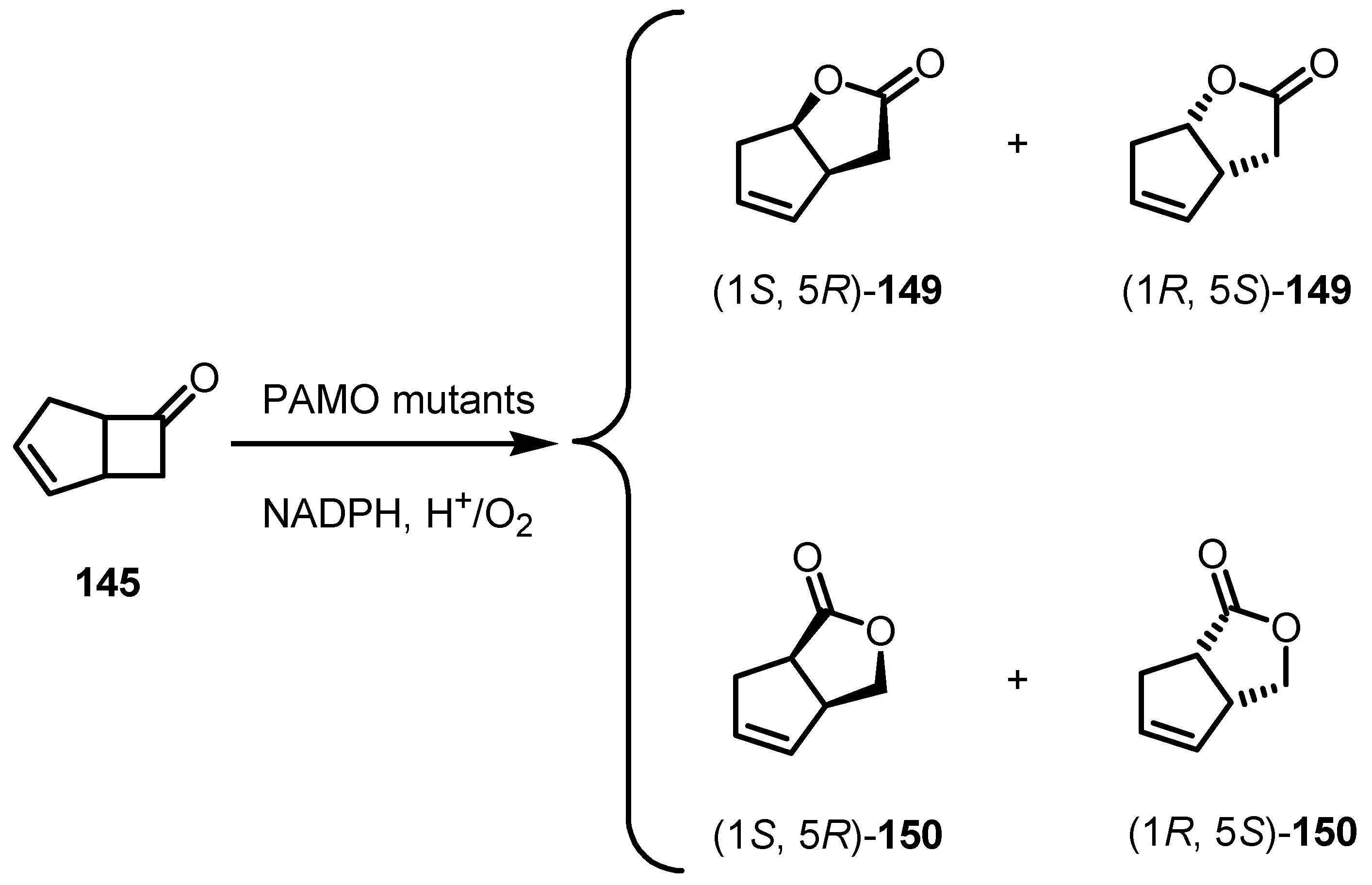
8.3. Biocatalytic Enantioselective Cyclobutanone Bayer-Villiger Oxidation
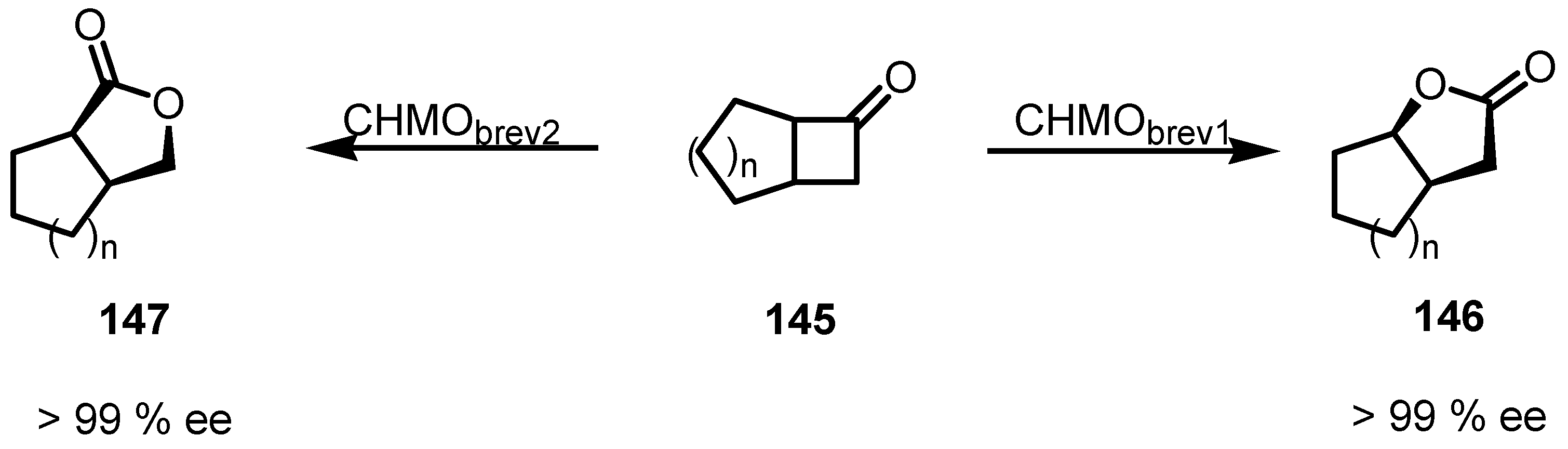
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rappaport, Z.; Liebman, J.F. The Chemistry of Cyclobutanes Part I; Wiley: Chichester, UK, 2005. [Google Scholar]
- Namyslo, J.C.; Kaufmann, D.E. The application of cyclobutane derivatives in organic synthesis. Chem. Rev. 2003, 103, 1485–1537. [Google Scholar] [CrossRef]
- Seiser, T.; Saget, T.; Tran, D.N.; Cramer, N. Cyclobutanes in catalysis. Angew. Chem. Int. Ed. 2011, 50, 7740–7752. [Google Scholar] [CrossRef]
- Dabrowski, J.A.; Moebius, D.C.; Wommack, A.J.; Kornahrens, A.F.; Kingsbury, J.S. Catalytic and regioselective ring expansion of arylcyclobutanones with trimethylsilyldiazomethane. Ligand-dependent entry to β-ketosilane or enolsilane adducts. Org. Lett. 2010, 12, 3598–3601. [Google Scholar] [CrossRef]
- Nordvik, T.; Brinker, U.H. A novel route to geminal dibromocyclobutanes: Syntheses of 2-substituted cyclobutanone acetals and Their reaction with boron tribromide. J. Org. Chem. 2003, 68, 9394–9399. [Google Scholar] [CrossRef]
- Crepin, D.; Dawick, J.; Aissa, L. Combined rhodium-catalyzed carbon–hydrogen activation and β-carbon elimination to access eight-membered rings. Angew. Chem. Int. Ed. 2010, 49, 620–623. [Google Scholar] [CrossRef]
- Bach, T.; Hehn, J.P. Photochemical reactions as key steps in natural product synthesis. Angew. Chem. Int. Ed. 2011, 50, 1000–1045. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. Exploiting [2+2] cycloaddition chemistry: Achievements with allenes. Chem. Soc. Rev. 2010, 39, 783–816. [Google Scholar] [CrossRef]
- De Nanteuil, F.; Waser, J. Synthesis of aminocyclobutanes by iron-catalyzed [2+2] cycloaddition. Angew. Chem. Int. Ed. 2013, 52, 9009–9013. [Google Scholar] [CrossRef]
- Randall, M.L.; Lo, P.C.-K.; Bonitatebus, P.J.; Snapper, M.L. [2+2] Photocycloaddition/thermal retrocycloaddition. A new entry into functionalized 5–8-5 ring systems. J. Am. Chem. Soc. 1999, 121, 4534–4535. [Google Scholar]
- Shipe, W.D.; Sorensen, E.J. Convergent, enantioselective syntheses of guanacastepenes A and E featuring cyclobutane framentation. J. Am. Chem. Soc. 2006, 128, 7025–7035. [Google Scholar] [CrossRef]
- Cho, S.Y.; Cha, J.K. Enantioselective synthesis of 2-substituted cyclobutanones. Org. Lett. 2000, 2, 1337–1340. [Google Scholar] [CrossRef]
- Nemoto, H.; Miyata, J.; Hakamata, H.; Fukumoto, K. The first example of asymmetric dihydroxylation of cyciopropylidene derivatives. An enantioenriched formal total synthesis of (−)-filformin. Tetrahedron Lett. 1995, 36, 1055–1058. [Google Scholar] [CrossRef]
- Bekish, A.V.; Kulinkovich, O.G. Differentiation between the ethoxycarbonyl groups in diethyl malate via their titanium-catalyzed reductive cyclopropanation with ethylmagnesium bromide and subsequent site-selective three-carbon ring cleavage. Tetrahedron Lett. 2005, 46, 6975–6978. [Google Scholar] [CrossRef]
- Ollivier, J.; Salaun, J. 1-Hydroxycyclopropanecarboxaldehyde tetrahydropyranyl ether. Preparation and rearrangement of functionalized 1-vinylcyclopropanols. Tetrahedron Lett. 1984, 25, 1269–1272. [Google Scholar] [CrossRef]
- Alberti, G.; Bernard, A.M.; Frongia, A.; Piras, P.P.; Secci, F.; Spiga, M. Intramolecular capture of a cyclobutylthionium ion for the synthesis of new strained heterocycles and carbocycles: A rapid assembly of the BCD ring sequence of penitrems. Synlett 2006, 2006, 2241–2245. [Google Scholar]
- Honda, M.; Nishizawa, T.; Nishii, Y.; Fujinami, S.; Segi, M. Reaction behavior of cyclopropylmethyl cations derived 1-phenylselenocyclopropylmethanols with acids. Tetrahedron 2009, 65, 9403–9411. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Shobe, D.; Nelson, G.L. Substituent effects on cyclobutyl and cyclopropylcarbinyl cations. J. Am. Chem. Soc. 1993, 115, 10645–10652. [Google Scholar] [CrossRef]
- Bernard, A.M.; Floris, C.; Frongia, A.; Piras, P.P. The first facile synthesis of some 1,2a, 3,8b-tetrahydro-2H-cyclobuta[c] chromenes through intramolecular alkylation of an aromatic ring by a cyclobutanone. Synlett 2002, 2002, 796–798. [Google Scholar]
- Trost, B.M.; Bogdanowicz, M.J. New synthetic reactions X. Versatile cyclobutanone (spiroannelation) and .gamma.-butyrolactone (lactone annelation) synthesis. J. Am. Chem. Soc. 1973, 95, 5321–5334. [Google Scholar] [CrossRef]
- Nemoto, H.; Miyata, J.; Yoshida, M.; Raku, N.; Fukumoto, K. A novel palladium-mediated cascade reaction triggered by strain release of the cyclobutane system. A new general route to benzo- and naphthohydrindans. J. Org. Chem. 1997, 62, 7850–7857. [Google Scholar] [CrossRef]
- Salaun, J. Optically active cyclopropanes. Chem. Rev. 1989, 89, 1247–1270. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Ruijter, E. Strategies for total and diversity-oriented synthesis of natural product-(like) macrocycles. Top. Curr. Chem. 2005, 137, 137–184. [Google Scholar]
- Fleck, M.; Bach, T. Total synthesis of the tetracyclic sesquiterpene punctaporonin C. Angew. Chem. Int. Ed. 2008, 47, 6189–6191. [Google Scholar] [CrossRef]
- Mascitti, V.; Corey, E.J. Total synthesis of (±)-pentacycloanammoxic Acid. J. Am. Chem. Soc. 2004, 126, 15664–15665. [Google Scholar] [CrossRef]
- Mangion, I.K.; MacMillan, D.W.C. Total synthesis of brasoside and littoralisone. J. Am. Chem. Soc. 2005, 127, 3696–3697. [Google Scholar] [CrossRef]
- Ma, A.-J.; Tu, Y.-Q.; Peng, J.-B.; Dou, Q.-Y.; Hou, S.-H.; Zhang, F.-M. Total synthesis of (−)-FR901483. Org. Lett. 2012, 14, 3604–3607. [Google Scholar] [CrossRef]
- Tanino, K.; Takahashi, M.; Tomata, Y.; Tokura, H.; Uehara, T.; Narabu, T.; Miyashita, M. Total synthesis of solanoeclepin A. Nat. Chem. 2011, 3, 484–488. [Google Scholar] [CrossRef]
- Hue, B.T.B.; Dijkink, J.; Kuiper, S.; Larson, K.K.; Guziec, F.S.; Goubitz, J.K.; Fraanje, J.; Schenk, H.; van Maarseveen, J.H.; Hiemstra, H. Synthesis of the cyclobutanone core of solanoeclepin A via intramolecular allene butenolide photocycloaddition. Org. Biomol. Chem. 2003, 1, 4364–4366. [Google Scholar] [CrossRef]
- Fournier, E.A.; Lee-Ruff, E. Synthesis of cyclobutane nucleosides. Nucleos. Nucleot. Nucleic Acids 2011, 30, 391–404. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Mladenova, G. Enantiomerically pure cyclobutane derivatives and their use in organic synthesis. Chem. Rev. 2003, 103, 1449–1484. [Google Scholar] [CrossRef]
- Darses, B.; Greene, A.E.; Coote, S.C.; Poisson, J.-F. Expedient approach to chiral cyclobutanones: Asymmetric synthesis of Cyclobut-G. Org. Lett. 2008, 10, 821–824. [Google Scholar] [CrossRef]
- Liu, H.; Tomooka, C.S.; Xu, B.R.; Yerxa, R.W.; Sullivan, Y.X.; Moore, H.W. Dimethyl squarate and its conversion to 3-ethenyl-4-methoxycyclobutene-1,2-dione and 2-butyl-6-ethenyl-5-methoxy-1,4-benzoquinone. Org. Synth. 2004, 10, 178–183. [Google Scholar]
- Xu, S.; Yerxa, B.R.; Sullivan, R.W.; Moore, H.W. Synthesis of substituted cyclobutenediones from 3-ethenyl-4-methoxy-cyclobutene-1,2-dione. Tetrahedron Lett. 1991, 32, 1129–1132. [Google Scholar] [CrossRef]
- Xiong, Y.; Xia, H.; Moore, H.W. Ring expansion of 4-Alkynylcyclobutenones. Synthesis of enantiomerically pure pyranoquinones from 4-(-4-Oxo-1,6-enynyl)-4-hydroxycyclobutenones and 4-(4–0xo-1,6-dialkynyl)-4-hydroxycyclobutenones. J. Org. Chem. 1995, 60, 6460–6467. [Google Scholar] [CrossRef]
- Law, K.-W. Organic photoconductive materials: Recent trends and developments. Chem. Rev. 1993, 93, 449–406. [Google Scholar] [CrossRef]
- Xie, J.; Comeau, A.B.; Seto, C.T. Squaric acids: A new motif for designing inhibitors of protein tyrosine phosphatase. Org. Lett. 2004, 6, 83–86. [Google Scholar]
- Liu, H.; Tomooka, C.S.; Moore, H.W. An efficient general synthesis of squarate esters. Synth. Commun. 1997, 27, 2177–2180. [Google Scholar] [CrossRef]
- Potora-Komisarska, K.; Benohoud, M.; Ishikawa, H.; Seebach, D.; Hayash, Y. Organocatalyzed Michael addition of aldehydes to nitro alkenes. Generally accepted mechanism revisited and revised. Helv. Chim. Acta 2011, 94, 719–745. [Google Scholar] [CrossRef]
- Bures, J.; Armstrong, A.; Blackmond, D.G. Mechanistic rationalization of organocatalyzed conjugate addition of linear aldehydes to nitro-olefins. J. Am. Chem. Soc. 2011, 133, 8822–8825. [Google Scholar] [CrossRef]
- Dembitsky, V.D. Bioactive cyclobutane-containing alkaloids. J. Nat. Med. 2008, 62, 1–33. [Google Scholar] [CrossRef]
- Secci, F.; Frongia, A.; Ollivier, J.; Piras, P.P. Convenient formal synthesis of (±)-cuparene, (±)-enokipodins A and B, and (±)-cuparene-1,4-quinone. Synthesis 2007, 7, 999–1002. [Google Scholar]
- Sinninghe Damsté, J.S.; Strous, M.; Rijpstra, W.I.C.; Hopmans, E.C.; Geenevasen, J.A.J.; van Duin, A.C.T.; van Niftrik, L.A.; Jetten, M.S.M. Linearly concatenated cyclobutane lipids form a dense bacterial membrane. Nature 2002, 419, 708–712. [Google Scholar] [CrossRef]
- Vidal, J.; Huet, F. Synthesis of .alpha.-methylenecyclobutanones. First preparation of norsarkomycin methyl ester. J. Org. Chem. 1988, 53, 611–616. [Google Scholar] [CrossRef]
- Honda, T.; Kimura, N. An enantioselective synthesis of 3,4-disubstituted butyrolactones. J. Chem. Soc. Chem. Commun. 1994, 1, 77–78. [Google Scholar] [CrossRef]
- Li, Y.; Mao, S.; Hager, M.W.; Becnel, K.D.; Schinazi, R.F.; Liotta, D.C. Synthesis and evaluation of 2'-substituted cyclobutyl nucleosides and nucleotides as potential anti-HIV agents. Bioorg. Med. Chem. Lett. 2007, 17, 3398–3401. [Google Scholar] [CrossRef]
- Reeves, M.; Eidamshaus, C.; Kim, J.; Stoltz, B.M. Enantioselective construction of α-quaternary cyclobutanones by catalytic asymmetric allylic alkylation. Angew. Chem. Int. Ed. 2013, 52, 6718–6721. [Google Scholar] [CrossRef]
- Cope, S.M.; Tailor, D.; Nagorski, R.W.J. Determination of the pKa of cyclobutanone: Brønsted correlation of the general base-catalyzed enolization in aqueous solution and the effect of ring strain. J. Org. Chem. 2010, 76, 380–390. [Google Scholar]
- Carter, C.A.G.; Greidanus, G.; Chen, J.-X.; Stryker, J.M. A new synthesis of cyclobutanones: Highly selective carbonylation of titanacyclobutane complexes prepared by free radical alkylation. J. Am. Chem. Soc. 2001, 123, 8872–8873. [Google Scholar] [CrossRef]
- Resende, P.; Almeida, W.P.; Coelho, F. An efficient synthesis of (R)-(−)-baclofen. Tetrahedron Asymmetry 1999, 10, 2113–2118. [Google Scholar] [CrossRef]
- Avenosa, A.; Busto, J.H.; Canal, N.; Peregrina, J.M. Selective Michael-aldol reaction by use of sterically hindered aluminum aryloxides as Lewis acids: An easy approach to cyclobutane amino acids. Org. Lett. 2005, 7, 3597–3600. [Google Scholar] [CrossRef]
- Li, X.; Danishefsky, S.J. Intramolecular Diels–Alder reactions of cycloalkenones: Translation of high endo selectivity to trans-junctions. J. Am. Chem. Soc. 2010, 132, 11004–11005. [Google Scholar] [CrossRef]
- Ghosez, L.; Mahuteau-Betzer, F.; Gericot, C.; Vallribera, A.; Cordier, J.-F. Enantioselective vicinal bis-acylation of olefins. Chem. Eur. J. 2002, 8, 3411–3422. [Google Scholar] [CrossRef]
- Yang, G.; Ghosez, L. Synthesis of enantiopure α-chlorocyclobutanones and cyclobutanols as scaffolds for the diverted synthesis of serine protease inhibitors. J. Org. Chem. 2009, 74, 1738–1748. [Google Scholar]
- Araki, T.; Ozawa, T.; Yokoe, H.; Kanematsu, M.; Yoshida, M.; Shishido, K. Diastereoselective intramolecular carbamoylketene/alkene [2+2] cycloaddition: Enantioselective access to pyrrolidinoindoline alkaloids. Org. Lett. 2013, 15, 200–203. [Google Scholar] [CrossRef]
- André, V.; Vidal, A.; Ollivier, J.; Robin, S.; Aitken, D.J. Rapid access to cis-cyclobutane γ-amino acids in enantiomerically pure form. Tetrahedron Lett. 2011, 52, 1253–1255. [Google Scholar] [CrossRef]
- Declerck, V.; Aitken, D.J. A refined synthesis of enantiomerically pure 2-aminocyclobutanecarboxylic acids. Amino Acids 2011, 41, 587–595. [Google Scholar] [CrossRef]
- Fernandes, C.; Faure, S.; Pereira, E.; Thery, V.; Declerck, V.; Guillot, R.; Aitken, D.J. 12-Helix folding of cyclobutane β-amino acid oligomers. Org. Lett. 2010, 12, 3606–3609. [Google Scholar] [CrossRef]
- Ogasawara, M.; Okada, A.; Nakajima, K.; Takahashi, T. Palladium-catalyzed synthesis of endocyclic allenes and their application in stereoselective [2+2]-cycloaddition with Ketenes. Org. Lett. 2009, 11, 177–180. [Google Scholar] [CrossRef]
- Canales, E.; Corey, E.J. Highly enantioselective [2+2]-cycloaddition reactions catalyzed by a chiral aluminum bromide complex. J. Am. Chem. Soc. 2007, 129, 12686–12687. [Google Scholar]
- Ishihara, K.; Nakano, K. Enantioselective [2+2] cycloaddition of unactivated alkenes with α-acyloxyacroleins catalyzed by chiral organoammonium salts. J. Am. Chem. Soc. 2007, 129, 8930–8931. [Google Scholar] [CrossRef]
- Fillion, E.; Fishlock, D. Total synthesis of (±)-taiwaniaquinol B via a domino intramolecular Friedel−Crafts acylation/carbonyl α-tert-alkylation reaction. J. Am. Chem. Soc. 2005, 127, 13144–13145. [Google Scholar] [CrossRef]
- Albrecht, L.; Dickmeiss, G.; Cruz, F.; Rodriguez-Eschlich, C.; Davis, R.L.; Jorgensen, K.A. Asymmetric organocatalytic formal [2+2]-cycloadditions via bifunctional H-bond directing dienamine catalysis. J. Am. Chem. Soc. 2012, 134, 2543–2546. [Google Scholar] [CrossRef]
- Talavera, G.; Reyes, E.; Vicario, J.L.; Carrillo, L. Cooperative dienamine/hydrogen-bonding catalysis: Enantioselective formal [2+2]-cycloaddition of enals with nitroalkenes. Angew. Chem. Int. Ed. 2012, 51, 4104–4107. [Google Scholar] [CrossRef]
- Duan, G.-J.; Ling, J.-B.; Wang, W.-P.; Luo, Y.-C.; Xu, P.-F. Organocatalytic formal [2+2] cycloaddition initiated by vinylogous Friedel–Crafts alkylation: Enantioselective synthesis of substituted cyclobutane derivatives. Chem. Commun. 2013, 49, 4625–4627. [Google Scholar] [CrossRef]
- Coote, S.C.; Bach, T. Enantioselective intermolecular [2+2]-photocycloadditions of isoquinolone mediated by a chiral hydrogen-bonding template. J. Am. Chem. Soc. 2013, 135, 14948–14951. [Google Scholar] [CrossRef]
- Wakchaure, V.N.; List, B. A new structural motif for bifunctional Brønsted acid/base organocatalysis. Angew. Chem. Int. Ed. 2010, 49, 4136–4139. [Google Scholar] [CrossRef]
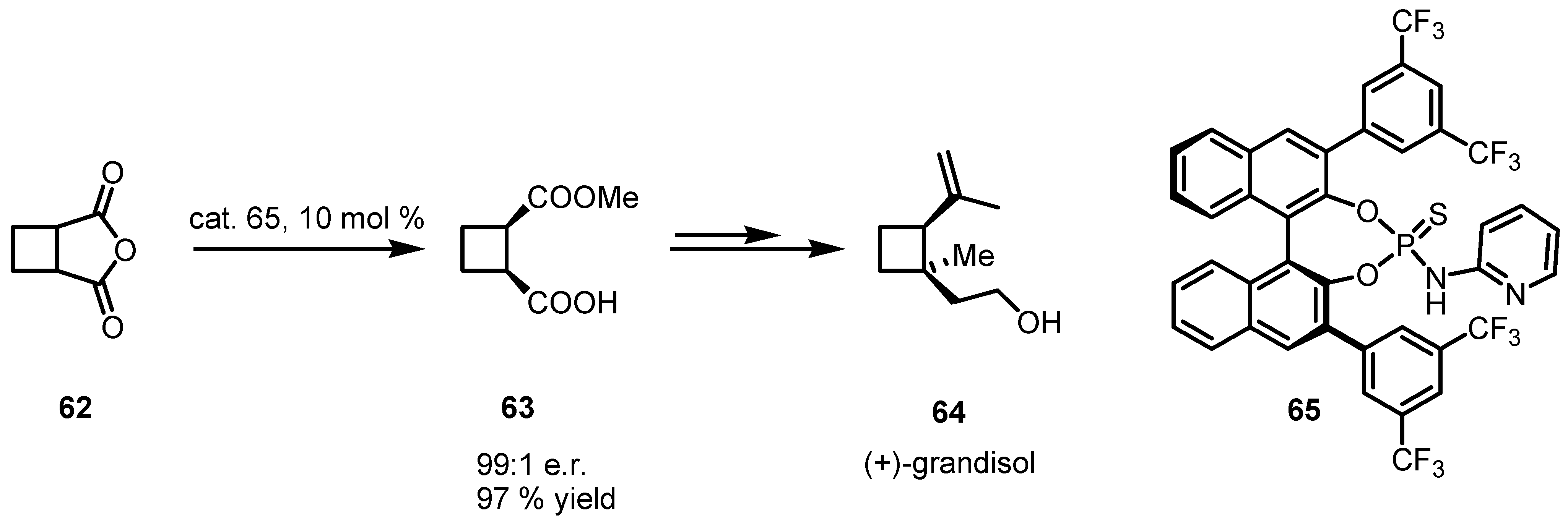
- Frongia, A.; Girard, C.; Ollivier, J.; Piras, P.P.; Secci, F. Convenient formal synthesis of (+)-grandisol through Lewis acid promoted enantioselective pinacolic rearrangement. Synlett 2008, 2008, 2823–2825. [Google Scholar]
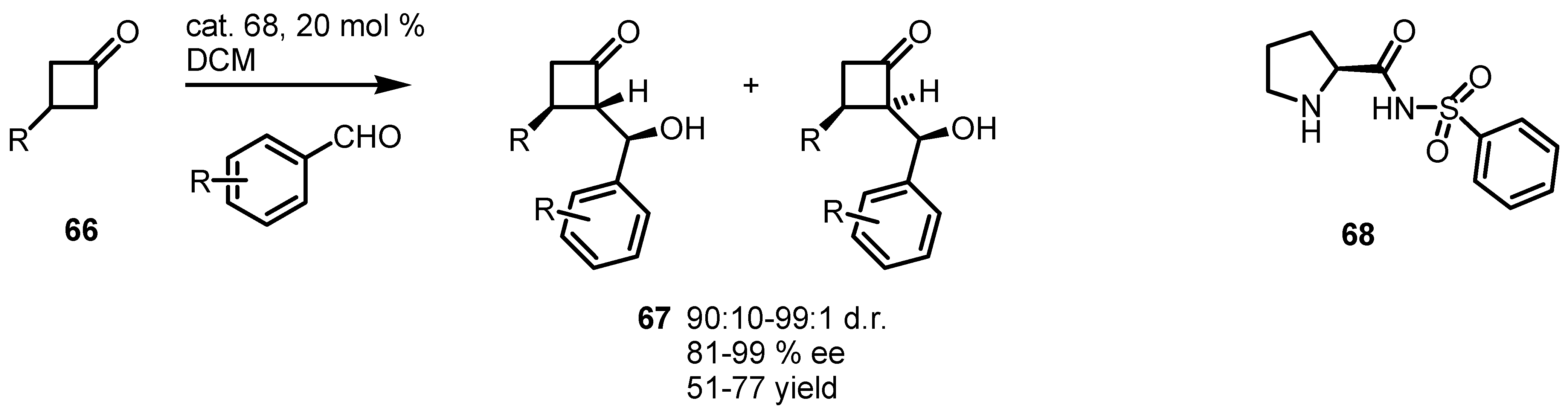
- Aitken, D.J.; Bernard, A.M.; Capitta, F.; Frongia, A.; Guillot, R.; Ollivier, J.; Piras, P.P.; Secci, F.; Spiga, M. Very high stereoselectivity in organocatalyzed desymmetrizing aldol reactions of 3-substituted cyclobutanones. Org. Biomol. Chem. 2012, 10, 5045–4048. [Google Scholar] [CrossRef]
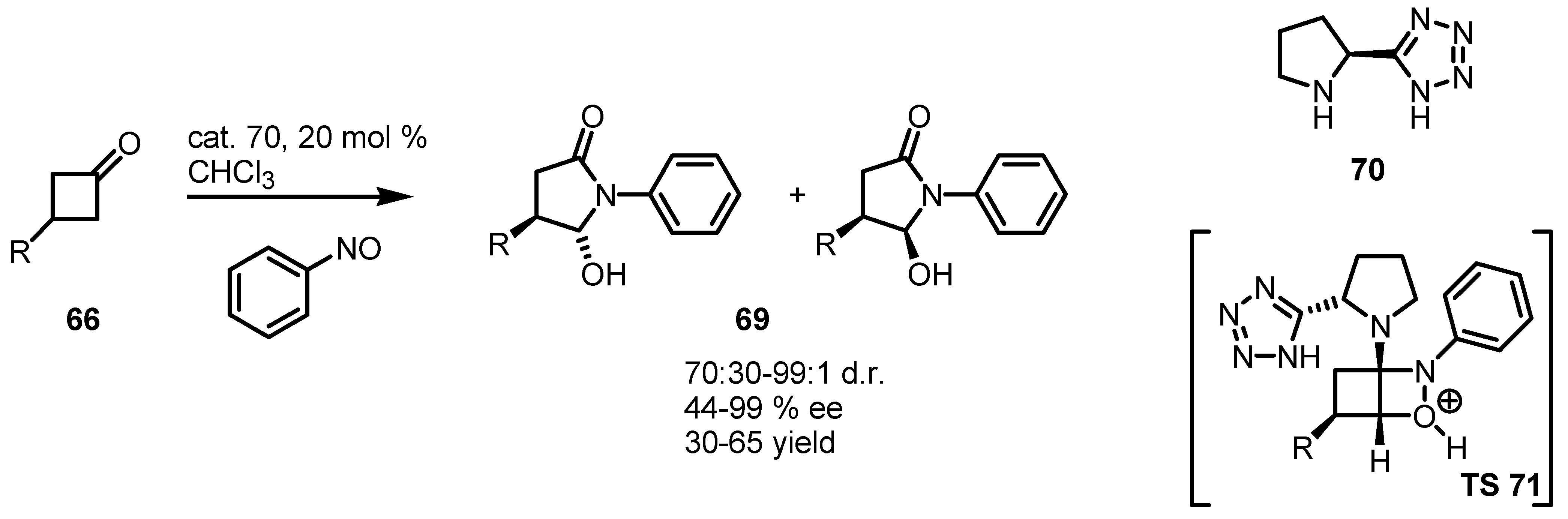
- Capitta, F.; Frongia, A.; Ollivier, J.; Piras, P.P.; Secci, F. Unexpected formation of optically active 4-substituted 5-hydroxy-γ-lactams by organocatalyzed reaction of 3-substituted cyclobutanones with nitrosobenzene. Synlett 2011, 2011, 89–93. [Google Scholar]
- Aubé, J.; Wang, Y.; Ghosh, S.; Langhans, K. Oxaziridine-mediated ring expansions of substituted cyclobutanones: Synthesis of (−)-γ-amino-hydroxybutyric acid (GABOB). Synth. Commun. 1991, 21, 693–701. [Google Scholar] [CrossRef]
- Sahasrabudhe, K.; Gracias, V.; Furness, K.; Smith, B.T.; Katz, C.E.; Reddy, D.S.; Aubé, J. Asymmetric schmidt reaction of hydroxyalkyl azides with ketones. J. Am. Chem. Soc. 2003, 125, 7914–7922. [Google Scholar]
- Hazelard, D.; Fadel, A.; Morgant, G. The first synthesis of both enantiomers of 2-hydroxycyclobutanone acetals by enzymatic transesterification: Preparation of (R)-(+)-2-benzyloxycyclobutanone and its antipode. Tetrahedron Asymmetry 2004, 15, 1711–1718. [Google Scholar] [CrossRef]
- Salezadeh-Asl, R.; Lee-Ruff, E. Enantiomeric resolution of cyclobutanones and related derivatives by enzyme-catalyzed acylation and hydrolysis. Tetrahedron Asymmetry 2005, 16, 3986–3991. [Google Scholar] [CrossRef]
- Avenoza, A.; Busto, J.H.; Canal, N.; Corzana, F.; Peregrina, J.M.; Pérez-Fernández, M.; Rodríguez, F. Cyclobutane amino acid analogues of furanomycin obtained by a formal [2+2] cycloaddition strategy promoted by methylaluminoxane. J. Org. Chem. 2010, 75, 545–552. [Google Scholar] [CrossRef]
- Burgess, K.; Li, S.; Rebenspies, J. Chiral 1,3-cyclobutane amino acids: Syntheses and extended conformations. Tetrahedron Lett. 1997, 38, 1681–1684. [Google Scholar] [CrossRef]
- Fernández-Tejada, A.; Corzana, F.; Busto, J.H.; Avenoza, A.; Peregrina, J.M. Stabilizing unusual conformations in small peptides and glucopeptides using a hydroxylated cyclobutane amino acid. Org. Biomol. Chem. 2009, 7, 2885–2893. [Google Scholar] [CrossRef]
- Mastracchio, A.; Warkentin, A.A.; Walji, A.M.; MacMillan, D.W.C. Direct and enantioselective α-allylation of ketones via singly occupied molecular orbital (SOMO) catalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 20648–20651. [Google Scholar] [CrossRef]
- Um, J.M.; Gutierrez, O.; Schoenebeck, F.; Houk, K.N.; MacMillan, D.W.C. Nature of intermediates in organo-SOMO catalysis of α-arylation of aldehydes. J. Am. Chem. Soc. 2010, 132, 6001–6005. [Google Scholar]
- Rendler, S.; MacMillan, D.W.C. Enantioselective polyene cyclization via organo-SOMO catalysis. J. Am. Chem. Soc. 2010, 132, 5027–5029. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, L.; Li, X.; Li, J.; Luo, S.; Cheng, J.-P. Asymmetric SN1 α-alkylation of cyclic ketones catalyzed by functionalized chiral ionic liquid (FCIL) organocatalysts. Chem. Eur. J. 2010, 16, 2045–2049. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Benfatti, F.; Zoli, L. Organocatalytic asymmetric alkylation of aldehydes by SN1-type reaction of alcohols. Angew. Chem. Int. Ed. 2009, 48, 1313–1316. [Google Scholar] [CrossRef]
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2013; Volume 1. [Google Scholar]
- Yang, H.; Carter, R.G. Proline sulfonamide-based organocatalysis: Better late than never. Synlett 2010, 2010, 2827–2838. [Google Scholar]
- Ma, X.; Da, C.-S.; Yi, L.; Jia, Y.-N.; Guo, Q.-P.; Che, L.-P.; Wu, F.-C.; Wang, J.-R.; Li, W.-P. Highly efficient primary amine organocatalysts for the direct asymmetric aldol reaction in brine. Tetrahedron Asymmetry 2009, 20, 1419–1424. [Google Scholar] [CrossRef]
- Moteki, S.A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. An achiral-acid-induced switch in the enantioselectivity of a chiral cis-diamine-based organocatalyst for asymmetric aldol and Mannich reactions. Angew. Chem. Int. Ed. 2012, 51, 1187–1190. [Google Scholar] [CrossRef]
- Aitken, D.J.; Capitta, F.; Frongia, A.; Gori, D.; Guillot, R.; Ollivier, J.; Piras, P.P.; Secci, F.; Spiga, M. The first organocatalysed direct aldol reaction of 2-hydroxycyclobutanone. Synlett 2011, 2011, 712–716. [Google Scholar]
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Aitken, D.J.; Capitta, F.; Frongia, A.; Ollivier, J.; Piras, P.P.; Secci, F. Solvent-free stereoselective organocatalyzed aldol reaction of 2-hydroxycyclobutanone. Synlett 2012, 2012, 727–730. [Google Scholar]
- Cobb, A.J.A.; Ley, S.V.; Shaw, D.M. 5-pyrrolidin-2-yltetrazole: A new, catalytic, more soluble alternative to proline in an organocatalytic asymmetric Mannich-type reaction. Synlett 2004, 2004, 558–560. [Google Scholar]
- Veverkova, E.; Strasserova, J.; Sebesta, R.; Toma, S. Asymmetric Mannich reaction catalyzed by N-arylsulfonyl-L-proline amides. Tetrahedron Asymmetry 2010, 21, 58–61. [Google Scholar] [CrossRef]
- Alza, E.; Rodriguez-Escrich, C.; Sayaleno, S.; Bastero, A.; Pericàs, M.A. A solid-supported organocatalyst for highly stereoselective, Batch, and continuous-flow Mannich reactions. Chem. Eur. J. 2009, 15, 10167–10172. [Google Scholar] [CrossRef]
- Kotrusz, P.; Toma, S.; Schmalz, H.-G.; Adler, A. Michael addition of aldehydes and ketones to β-nitrostyrenes in an ionic liquid. Eur. J. Org. Chem. 2004, 2004, 1577–1583. [Google Scholar]
- Mailhol, D.; del Mar Sanchez Duque, M.; Raimondi, W.; Bonne, D.; Costantieux, T.; Coquerel, T.; Rodriguez, J. Enantioselective organocatalytic Michael addition of cyclobutanones to nitroalkenes. Adv. Synth. Catal. 2012, 354, 3523–3532. [Google Scholar] [CrossRef]
- Kotrusz, P.; Alemayehu, S.; Toma, S.; Schmalz, H.-G.; Alder, A. Enantioselective organocatalysis in ionic liquids: Addition of aliphatic aldehydes and ketones to diethyl azodicarboxylate. Eur. J. Org. Chem. 2005, 2005, 4904–4911. [Google Scholar] [CrossRef]
- Bernard, A.M.; Frongia, A.; Guillot, R.; Piras, P.P.; Secci, F.; Spiga, M. L-Proline-catalyzed direct intermolecular asymmetric aldol reactions of 1-phenylthiocycloalkyl carboxaldehydes with ketones. Easy access to spiro- and fused-cyclobutyltetrahydrofurans and cyclopentanones. Org. Lett. 2007, 9, 541–544. [Google Scholar] [CrossRef]
- Bernard, A.M.; Frongia, A.; Secci, F.; Piras, P.P. 2,2-Dimethyl cyclopentanones by acid catalyzed ring expansion of isopropenylcyclobutanols. A short synthesis of (±)-α-cuparenone and (±)-herbertene. Chem. Commun. 2005, 3853–3855. [Google Scholar]
- Natarajan, A.; Ng, D.; Yang, Z.; Garcia-Garibay, M.A. Parallel syntheses of (+)-and (−)-α-cuparenone by radical combination in crystalline solids. Angew. Chem. Int. Ed. 2007, 46, 6485–6487. [Google Scholar] [CrossRef]
- Shen, Y.-M.; Wang, B.; Shi, Y. Enantioselective synthesis of 2-Aryl cyclopentanones by asymmetric epoxidation and epoxide rearrangement. Angew. Chem. Int. Ed. 2006, 45, 1429–1432. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. An efficient catalytic asymmetric epoxidation method. J. Am. Chem. Soc. 1997, 119, 11224–11235. [Google Scholar] [CrossRef]
- Tian, H.; She, X.; Shu, L.; Yu, H.; Shi, Y. Highly enantioselective epoxidation of cis-olefins by chiral dioxirane. J. Am. Chem. Soc. 2000, 122, 11551–11552. [Google Scholar] [CrossRef]
- Wang, B.; Wong, O.A.; Zhao, M.-X.; Shi, Y. Asymmetric epoxidation of 1,1-disubstituted terminal olefins by chiral dioxirane via a planar-like transition state. J. Org. Chem. 2008, 73, 9539–9543. [Google Scholar] [CrossRef]
- Bernard, A.M.; Frongia, A.; Piras, P.P.; Secci, F. Unexpected stereochemistry in the lithium salt catalyzed ring expansion of nonracemic oxaspiropentanes. Formal syntheses of (−)-(4R,5R)-muricatacin and the pheromone (R)-japonilure. Org. Lett. 2003, 5, 2923–2926. [Google Scholar] [CrossRef]
- Zhang, E.; Fan, C.-A.; Tu, Y.-Q.; Zhang, F.-M.; Song, Y.-L. Organocatalytic asymmetric vinylogous α-ketol rearrangement: Enantioselective construction of chiral all-Carbon quaternary stereocenters in spirocyclic diketones via semipinacol-type 1,2-carbon migration. J. Am. Chem. Soc. 2009, 131, 14626–14627. [Google Scholar] [CrossRef]
- Li, B.-S.; Zhang, E.; Zhang, Q.-W.; Zhang, F.-M.; Tu, Y.-Q.; Cao, X.-P. One-pot construction of multi-substituted spiro-cycloalkanediones by an organocatalytic asymmetric epoxidation/semipinacol rearrangement. Chem.-Asian J. 2011, 6, 2269–2272. [Google Scholar] [CrossRef]
- Zhang, Q.-W.; Fan, C.-A.; Zhang, H.-J.; Tu, Y.-Q.; Zhao, Y.-M.; Gu, P.; Chen, Z.-M. Brønsted acid catalyzed enantioselective semipinacol rearrangement for the synthesis of chiral spiroethers. Angew. Chem. Int. Ed. 2009, 48, 8572–8574. [Google Scholar] [CrossRef]
- Hurley, P.B.; Dake, G.R. Synthetic studies toward halichlorine: Complex azaspirocycle formation with use of an NBS-promoted semipinacol reaction. J. Org. Chem. 2008, 73, 4131–4138. [Google Scholar] [CrossRef]
- Paquette, L.A.; Lanter, J.C.J.; Johnston, N. Single stereodifferentiation associated with carbon atom insertion during the oxonium ion-initiated pinacol rearrangement of dihydrofuranyl and dihydropyranyl carbinols. J. Org. Chem. 1997, 62, 1702–1712. [Google Scholar] [CrossRef]
- Paquette, L.A.; Owen, D.R.; Bibart, R.T.; Seekamp, C.K.; Kahane, A.L.; Lanter, J.C.; Corral, M.A. 1-Oxaspiro[4.4]nonan-6-ones. Synthetic access via oxonium ion technology, optical resolution, and conversion into enantiopure spirocyclic α,β-butenolides. J. Org. Chem. 2001, 66, 2828–2834. [Google Scholar] [CrossRef]
- Romanov-Michailidis, F.; Guénée, L.; Alexakis, A. Enantioselective organocatalytic fluorination-induced Wagner-Meerwein rearrangement. Angew. Chem. Int. Ed. 2013, 52, 9266–9270. [Google Scholar] [CrossRef]
- Bernard, A.M.; Frongia, A.; Ollivier, J.; Piras, P.P.; Secci, F.; Spiga, M. A highly stereocontrolled formal total synthesis of (±)- and of (−)-grandisol by 1,4-conjugated addition of organocopper reagents to cyclobutylidene derivatives. Tetrahedron 2007, 63, 4968–4974. [Google Scholar] [CrossRef]
- Quinn, K.J.; Isaacs, A.K.; Arvary, R.A. Concise total synthesis of (−)-muricatacin by tandem ring-closing/cross metathesis. Org. Lett. 2004, 6, 4143–4145. [Google Scholar] [CrossRef]
- Malkow, A.V.; Friscourt, F.; Bell, M.; Swarbrick, M.E.; Kocovsky, P. Enantioselective Baeyer–Villiger oxidation catalyzed by Palladium (II) complexes with chiral P,N-Ligands. J. Org. Chem. 2008, 73, 3996–4003. [Google Scholar] [CrossRef]
- Noyori, R.; Aoki, M.; Sato, K. Green oxidation with aqueous hydrogen peroxide. Chem. Commun. 2003, 2003, 1977–1986. [Google Scholar] [CrossRef]
- Piera, J.; Backvall, J.-E. Catalytic oxidation of organic substrates by molecular oxygen and hydrogen peroxide by multistep electron transfer. A biomimetic approach. Angew. Chem. Int. Ed. 2008, 47, 3506–3523. [Google Scholar] [CrossRef]
- Michelin, R.A.; Sgarbossa, P.; Scarso, A.; Strukul, G. The Baeyer–Villiger oxidation of ketones: A paradigm for the role of soft Lewis acidity in homogeneous catalysis. Coord. Chem. Rev. 2010, 254, 646–660. [Google Scholar] [CrossRef]
- Renz, M.; Meunier, B. 100 years of baeyer–villiger oxidations. Eur. J. Org. Chem. 1999, 1999, 737–750. [Google Scholar] [CrossRef]
- Imada, Y.; Naota, T. Flavins as organocatalysts for environmentally benign molecular transformations. Chem. Rec. 2007, 7, 354–361. [Google Scholar] [CrossRef]
- Sasakura, N.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. A new environmentally friendly method for the Bayer-Villiger oxidation of cyclobutanones catalyzed by thioureas using H2O2 as an oxidant. RSC Adv. 2012, 2, 6135–6139. [Google Scholar] [CrossRef]
- Imada, Y.; Iida, H.; Marahashi, S.-I.; Naota, T. An Aerobic, organocatalytic, and chemoselective method for Baeyer-Villiger oxidation. Angew. Chem. Int. Ed. 2005, 44, 1704–1706. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Ono, S.; Imada, Y. Asymmetric Baeyer–Villiger reaction with hydrogen peroxide catalyzed by a novel planar-chiral bisflavin. Angew. Chem. Int. Ed. 2002, 41, 2366–2368. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Z.; Zhang, X.; Zhang, X.; Ding, K. Chiral Brønsted acid catalyzed asymmetric Baeyer–Villiger reaction of 3-substituted cyclobutanones by using aqueous H2O2. Angew. Chem. Int. Ed. 2008, 47, 2840–2843. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Z.; Li, Y.; Zhang, X.; Zhang, X.; Wang, H.; Ding, K. Mechanistic investigation of chiral phosphoric acid catalyzed asymmetric Baeyer–Villiger reaction of 3-substituted cyclobutanones with H2O2 as the oxidant. Chem. Eur. J. 2010, 16, 3021–3035. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.; Ji, J.; Zhang, Y.; Hu, X.; Lin, L.; Feng, X. Enantioselective Baeyer–Villiger oxidation: Desymmetrization of meso cyclic ketones and kinetic resolution of racemic 2-arylcyclohexanones. J. Am. Chem. Soc. 2012, 134, 17023–17026. [Google Scholar] [CrossRef]
- Mihovilovich, M.D.; Kapitàn, P. Regiodivergent Baeyer–Villiger oxidation of fused ketone substrates by recombinant whole-cells expressing two monooxygenases from Brevibacterium. Tetrahedron Lett. 2004, 45, 2751–2754. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wu, S. Laboratory evolution of robust and enantioselective Baeyer-Villiger monooxygenases for asymmetric catalysis. J. Am. Chem. Soc. 2009, 131, 15424–15432. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Secci, F.; Frongia, A.; Piras, P.P. Stereocontrolled Synthesis and Functionalization of Cyclobutanes and Cyclobutanones. Molecules 2013, 18, 15541-15572. https://doi.org/10.3390/molecules181215541
Secci F, Frongia A, Piras PP. Stereocontrolled Synthesis and Functionalization of Cyclobutanes and Cyclobutanones. Molecules. 2013; 18(12):15541-15572. https://doi.org/10.3390/molecules181215541
Chicago/Turabian StyleSecci, Francesco, Angelo Frongia, and Pier Paolo Piras. 2013. "Stereocontrolled Synthesis and Functionalization of Cyclobutanes and Cyclobutanones" Molecules 18, no. 12: 15541-15572. https://doi.org/10.3390/molecules181215541