Facile Synthesis of the Naturally Cytotoxic Triterpenoid Saponin Patrinia-Glycoside B-II and Its Conformer


Abstract
:1. Introduction
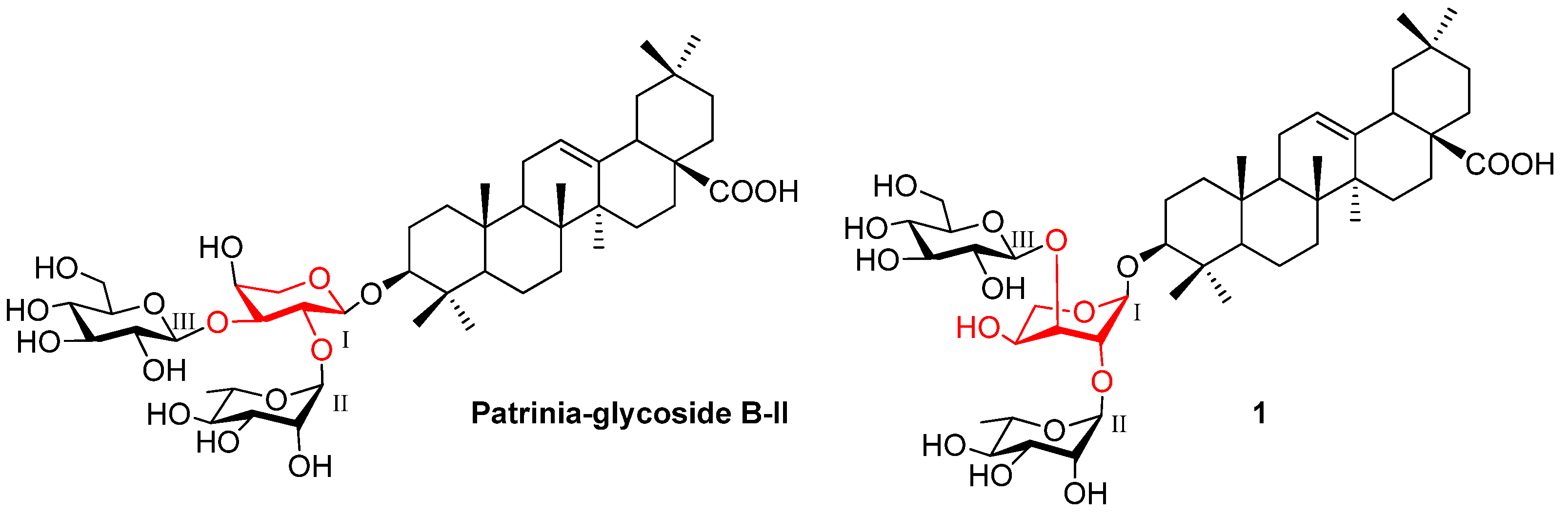
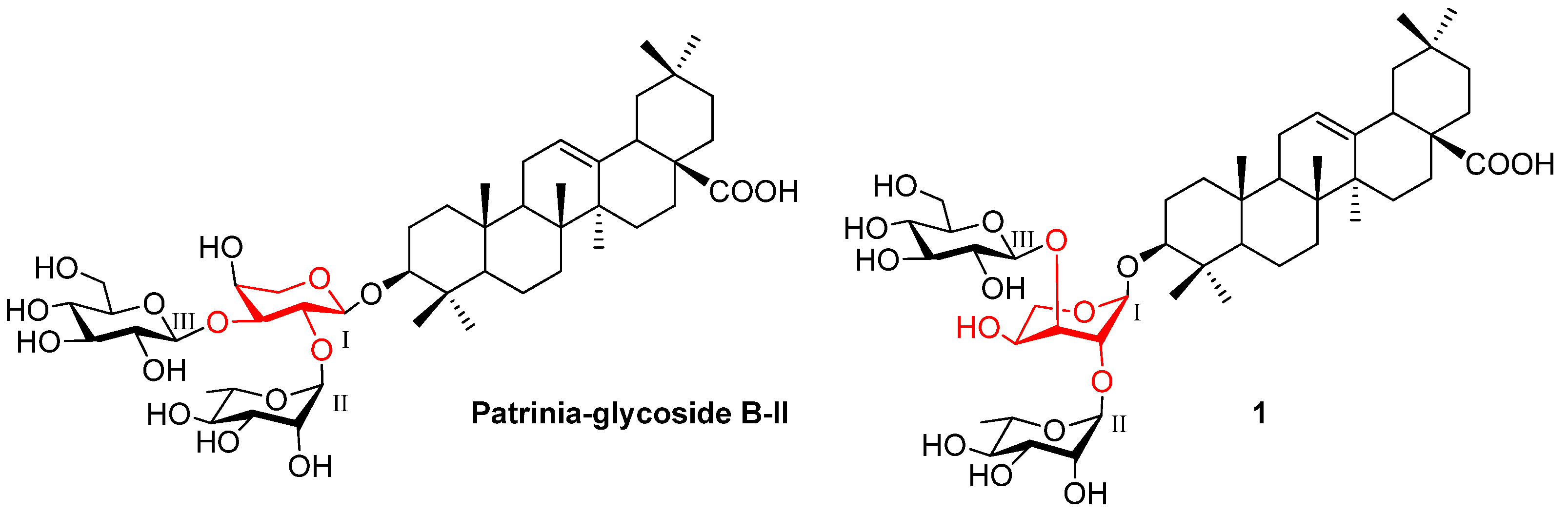
2. Results and Discussion
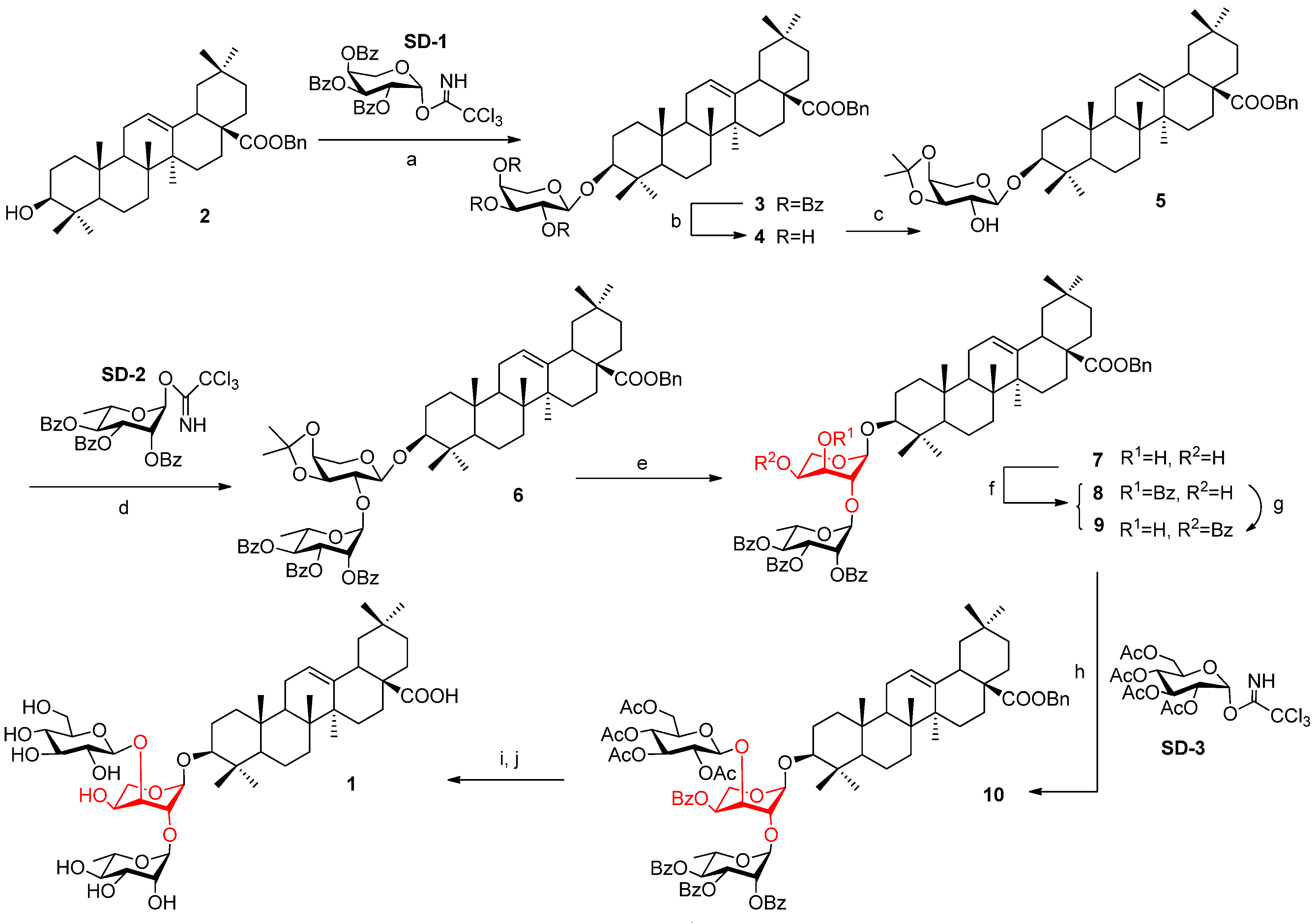
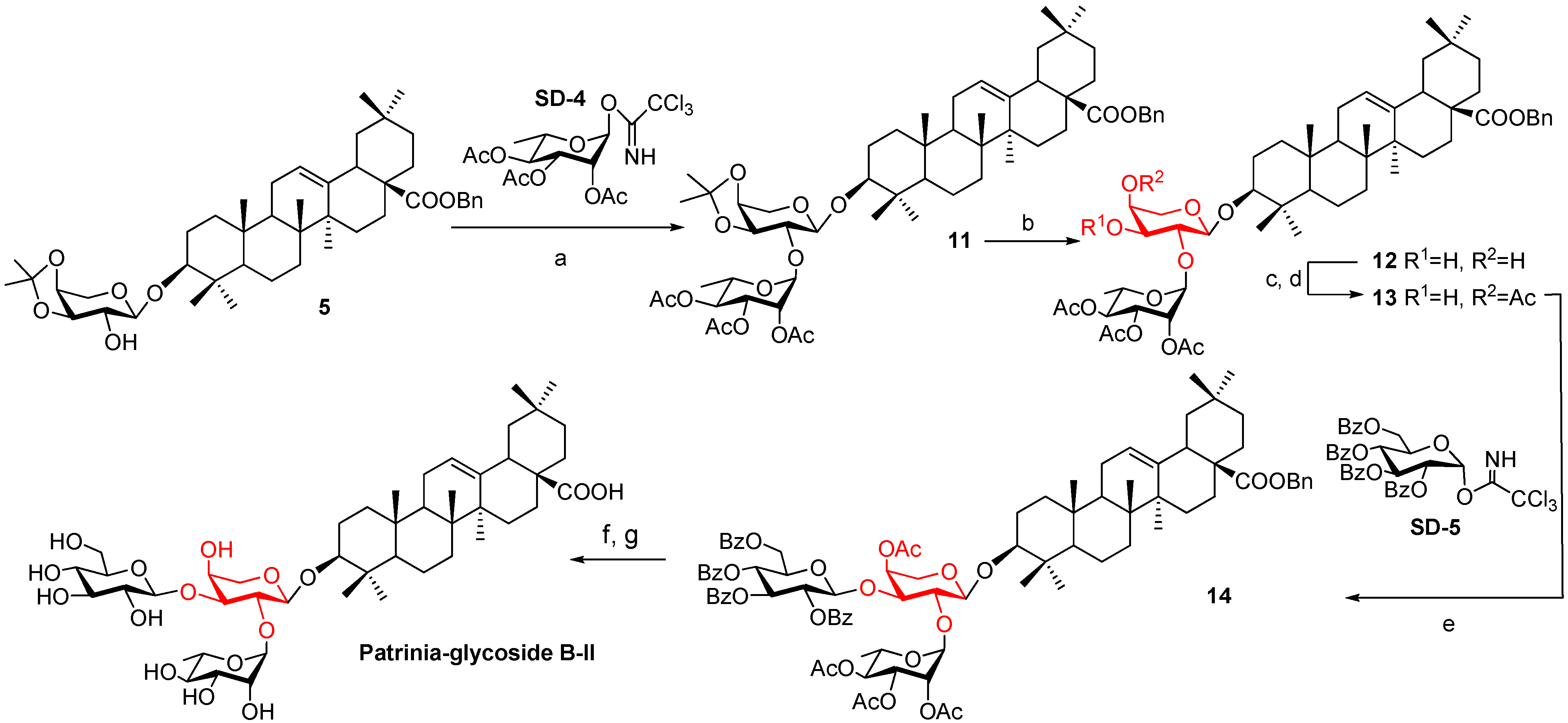
2.1. Synthesis
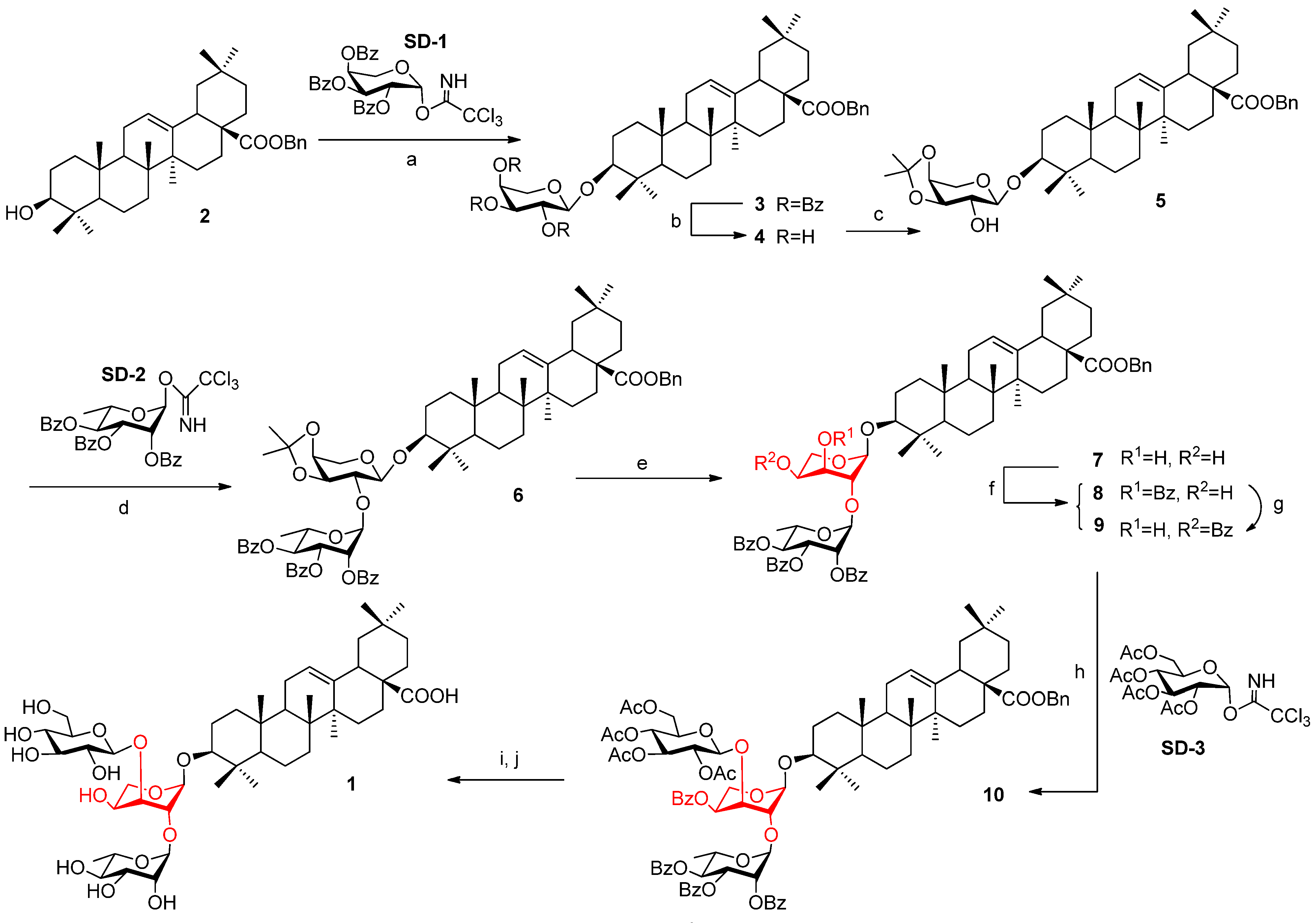
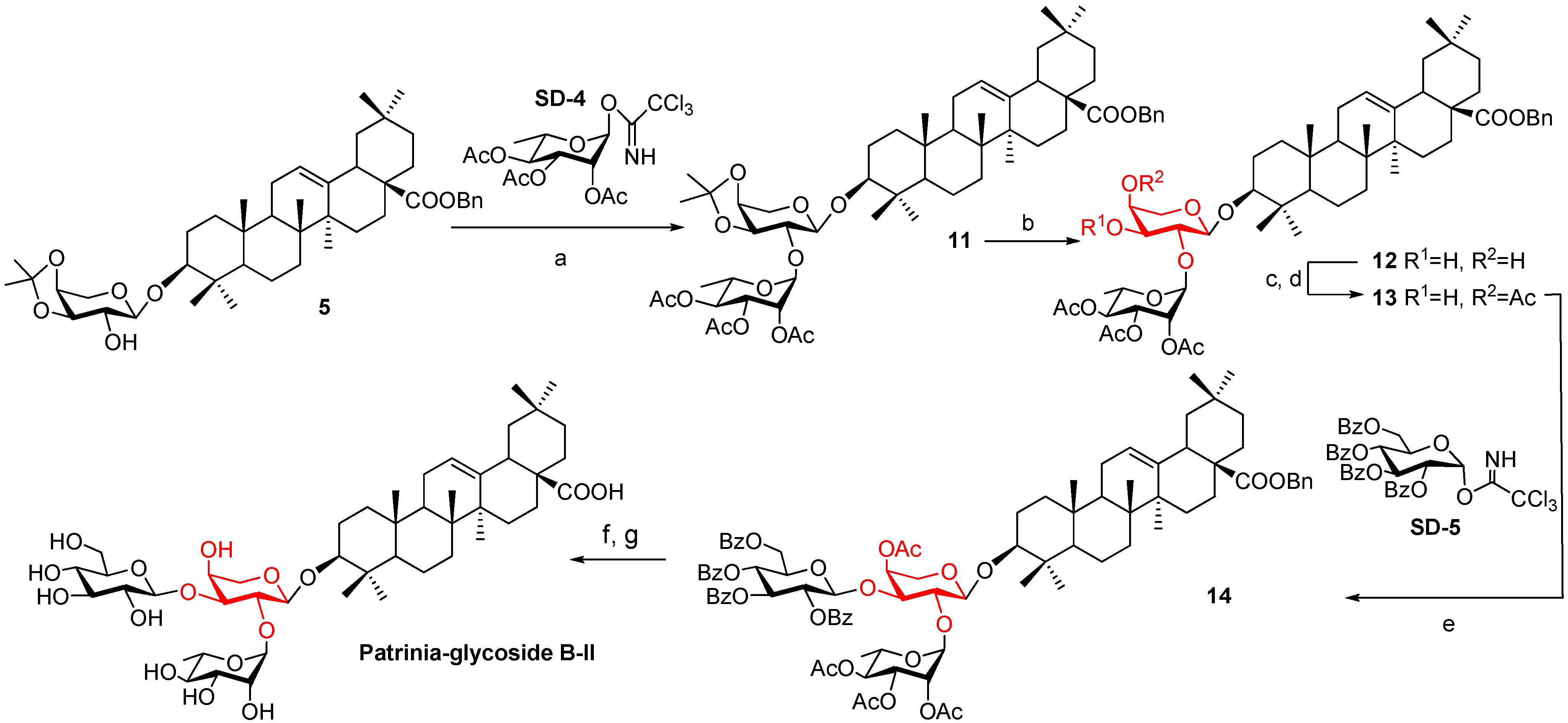
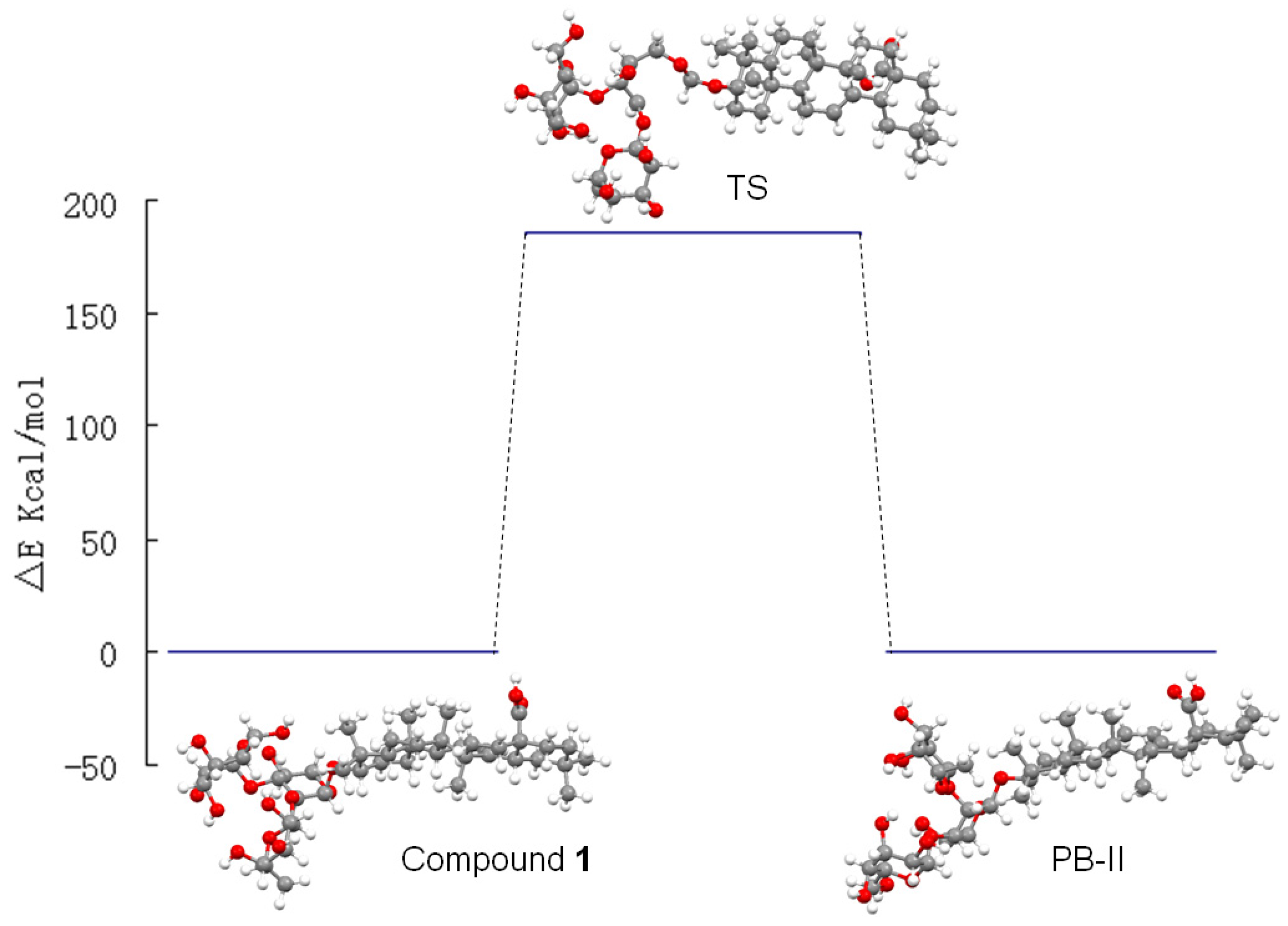
2.2. Computational Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Patrinia-Glycoside B-II | Transition State | |
|---|---|---|---|
| Energy(Kcal/mol) | −19,986.949 | −19,987.263 | −19,801.469 |
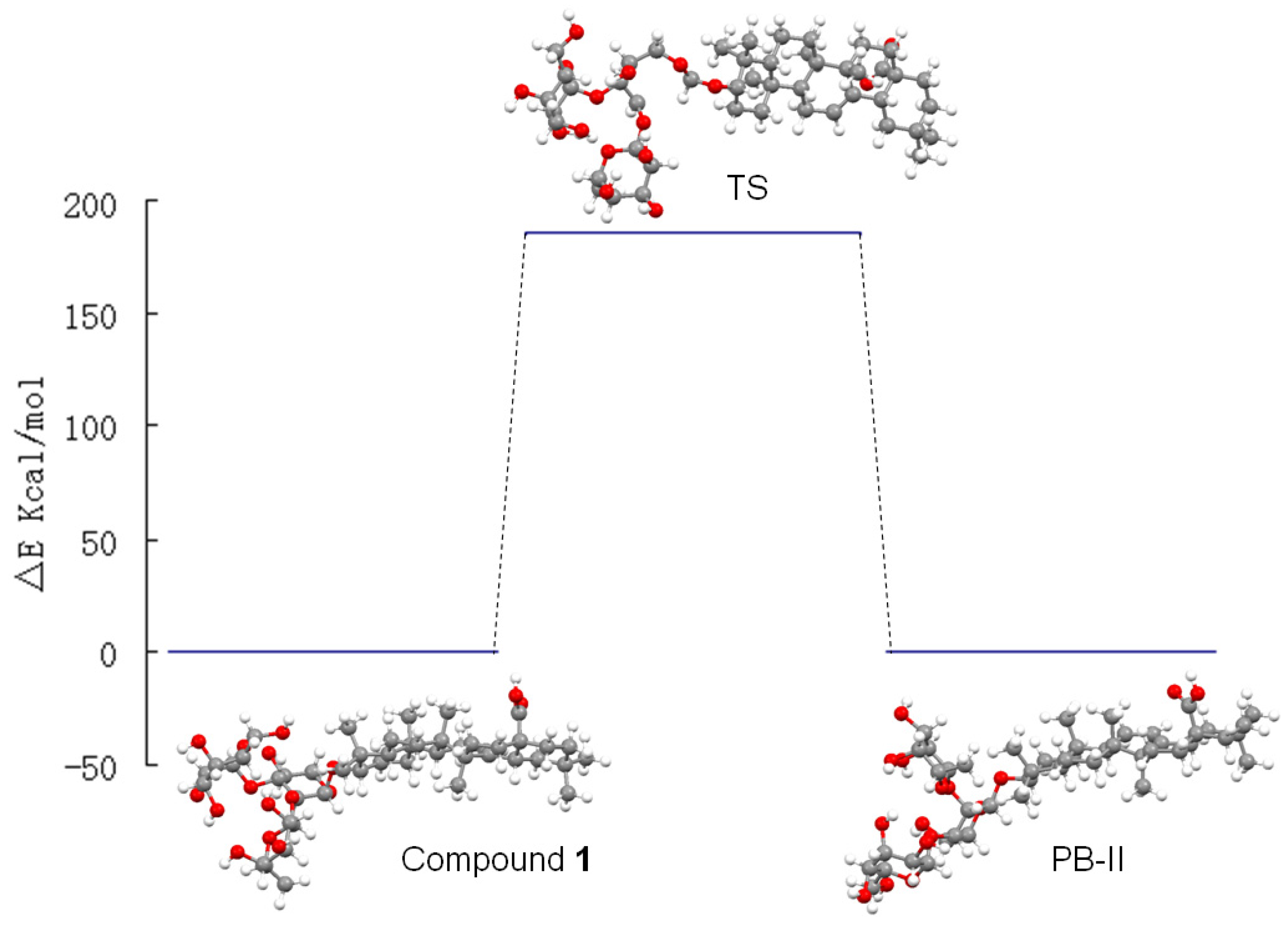

2.3. Biological Evaluation
| Compd | HeLa | HepG2 | HT1080 | A549 | A375-S2 | K562 | HL60 | U937 |
|---|---|---|---|---|---|---|---|---|
| PB-II | 5.4 ± 0.2 | 4.2 ± 0.8 | 18.0 ± 0.5 | 27.9 ± 0.8 | 15.8 ± 0.1 | 6.2 ± 0.6 | 6.6 ± 0.3 | 5.5 ± 0.3 |
| Compound 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 5-FU | >50 | >50 | 15.2 | >50 | 25.31.6 | 36.4 ± 3.1 | 9.6 ± 0.9 | 18.2 ± 2.0 |
3. Experimental
3.1. General
3.2. Benzyl Oleanolate 3-O-3,4-O-isopropylidene-α-l-arabinopyranoside (5)
 +45.0 (c 1.60, CHCl3); Mp 76.8–79.2 °C; 1H-NMR (600 MHz, CDCl3): δ 7.34 (m, 5H, Ar-H), 5.28 (t, J = 3.0 Hz, 1H, H-12), 5.07 (dd, J = 18.7, 12.6 Hz, 2H, PhCH2), 4.22–4.17 (m, 3H, H-1', H-4', H-5'-1), 4.06 (dd, J = 7.8, 6.1 Hz, 1H, H-3'), 3.75 (dd, J = 13.9, 3.5 Hz, 1H, H-5'-2), 3.63 (d, J = 7.8 Hz, 1H, H-2'), 3.12 (dd, J = 11.5, 4.6 Hz, 1H, H-3), 2.91 (dd, J = 13.8, 3.3 Hz, 1H, H-18), 2.30 (br s, 1H, OH), 1.54, 1.36 (s each, 3H each, O-(CH3)2C-O), 1.11, 0.98, 0.92, 0.89, 0.88, 0.82, 0.60 (s each, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.4, 143.6, 136.3, 128.3, 127.9, 127.8, 122.4, 110.0, 104.3, 88.9, 78.1, 74.2, 73.2, 65.9, 63.0, 55.4, 47.5, 46.6, 45.8, 41.6, 41.3, 39.2, 39.0, 38.4, 36.6, 33.8, 33.1, 32.6, 32.3, 30.6, 28.2, 28.0, 27.5, 26.0, 25.8, 23.6, 23.3, 22.9, 18.1, 16.8, 16.6, 15.2; HRMS: calcd for C38H59O7 (M-Bn)−: 627.4261; Found: m/z 627.4255.
+45.0 (c 1.60, CHCl3); Mp 76.8–79.2 °C; 1H-NMR (600 MHz, CDCl3): δ 7.34 (m, 5H, Ar-H), 5.28 (t, J = 3.0 Hz, 1H, H-12), 5.07 (dd, J = 18.7, 12.6 Hz, 2H, PhCH2), 4.22–4.17 (m, 3H, H-1', H-4', H-5'-1), 4.06 (dd, J = 7.8, 6.1 Hz, 1H, H-3'), 3.75 (dd, J = 13.9, 3.5 Hz, 1H, H-5'-2), 3.63 (d, J = 7.8 Hz, 1H, H-2'), 3.12 (dd, J = 11.5, 4.6 Hz, 1H, H-3), 2.91 (dd, J = 13.8, 3.3 Hz, 1H, H-18), 2.30 (br s, 1H, OH), 1.54, 1.36 (s each, 3H each, O-(CH3)2C-O), 1.11, 0.98, 0.92, 0.89, 0.88, 0.82, 0.60 (s each, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.4, 143.6, 136.3, 128.3, 127.9, 127.8, 122.4, 110.0, 104.3, 88.9, 78.1, 74.2, 73.2, 65.9, 63.0, 55.4, 47.5, 46.6, 45.8, 41.6, 41.3, 39.2, 39.0, 38.4, 36.6, 33.8, 33.1, 32.6, 32.3, 30.6, 28.2, 28.0, 27.5, 26.0, 25.8, 23.6, 23.3, 22.9, 18.1, 16.8, 16.6, 15.2; HRMS: calcd for C38H59O7 (M-Bn)−: 627.4261; Found: m/z 627.4255.3.3. Benzyl Oleanolate 3-O-2,3,4-tri-O-benzoyl-α-l-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-l-arabinopyranoside (6)
+96.7 (c 2.58, CHCl3); Mp 122.5–124.2 °C; 1H-NMR (600 MHz, CDCl3): δ 8.12–7.21 (m, 20H, Ar-H), 5.87 (dd, J = 10.2, 3.3 Hz, 1H, H-3''), 5.76 (brs, 1H, H-1''), 5.65 (m, 2H, H-2'', H-4''), 5.30 (brs, H-12), 5.07 (dd, J = 22.4, 12.6 Hz, 2H, PhCH2), 4.53 (m, 1H, H-5''), 4.47 (d, J = 7.8 Hz, 1H, H-1'), 4.25 (m, 2H, H-3', H-4'), 4.17 (m, 1H, H-5'-1), 3.90 (d, J = 7.8 Hz, 1H, H-2'), 3.79 (m, 1H, H-5'-2), 3.17 (dd, J = 11.3, 4.1 Hz, 1H, H-3), 2.92 (m, 1H, H-18), 1.55, 1.35 (s each, 3H each, O-(CH3)2C-O), 1.34 (d, J = 6.1 Hz, 3H, H-6''), 1.14, 0.95, 0.93, 0.92, 0.90, 0.89, 0.64 (s each, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.4, 165.7, 165.5, 165.4, 143.7, 136.4, 133.3, 133.2, 132.9, 130.0, 129.7, 129.6, 129.5, 129.4, 129.3, 128.5, 128.4, 128.3, 128.2, 127.9, 122.5, 110.4, 103.5, 95.4, 89.4, 79.4, 75.5, 73.6, 72.2, 70.8, 70.1, 66.7, 66.1, 62.9, 56.0, 47.6, 46.7, 45.8, 41.7, 41.4, 39.3, 39.2, 38.7, 36.7, 33.8, 33.1, 32.7, 32.4, 30.7, 28.2, 27.8, 27.6, 26.1, 25.9, 23.6, 23.4, 23.0, 18.1, 17.5, 16.9, 16.7, 15.3; HRMS (ESI): Calcd for C65H81O14 (M-Bn)−: 1085.5626; Found: m/z 1085.5619.3.4. Benzyl Oleanolate 3-O-2,3,4-tri-O-benzoyl-α-l-rhamnopyranosyl-(1→2)-α-l-arabinopyranoside (7)
+77.3 (c 2.27, CHCl3); Mp 143.7–147.2 °C; 1H-NMR (600 MHz, CDCl3): δ 8.10–7.23 (m, 20H, Ar-H), 5.82 (dd, J = 10.2, 3.1 Hz, 1H, H-3"), 5.69 (m, 2H, H-2'', H-4''), 5.34 (s, 1H, H-1''), 5.29 (br s, 1H, H-12), 5.07 (dd, J = 18.7, 12.6 Hz, 2H, PhCH2), 4.81 (d, J = 3.0 Hz, 1H, H-1'), 4.34 (m, 1H, H-5''), 4.11–3.98 (m, 3H, H-2', H-4', OH), 3.82 (m, 1H, H-5'-1), 3.67 (m, 1H, H-5'-2), 3.45 (d, J = 7.9 Hz, 1H, H-3'), 3.18 (dd, J = 11.0, 3.2 Hz, 1H, H-3), 2.91 (m, 1H, H-18), 2.52 (br s, 1H, OH), 1.34 (d, J = 6.1 Hz, 3H, H-6''), 1.12, 1.05, 0.92, 0.89, 0.88, 0.84, 0.61 (s each, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.4, 165.7, 165.5, 143.6, 136.4, 133.5, 133.3, 133.1, 129.9, 129.7, 129.6, 129.2, 129.2, 129.1, 128.5, 128.4, 128.2, 127.9, 127.8, 122.4, 102.0, 98.2, 90.3, 76.2, 71.5, 70.7, 70.7, 69.7, 67.3, 65.9, 65.5, 61.1, 55.4, 47.6, 46.7, 45.8, 41.6, 41.3, 39.2, 39.1, 38.5, 36.7, 33.8, 33.1, 32.6, 32.3, 30.6, 28.1, 27.6, 25.8, 25.7, 23.6, 23.4, 23.0, 18.2, 17.5, 16.8, 16.4, 15.3; HRMS: calcd for C62H77O14 (M-Bn)−: 1045.5313; found: m/z 1045.5307.3.5. Benzyl Oleanolate 3-O-2,3,4-tri-O-benzoyl-α-l-rhamnopyranosyl-(1→2)-3-O- benzoyl-a-l-arabinopyranoside (8) and benzyl oleanolate 3-O-2,3,4-tri- O-benzoyl-α-l-rhamnopyranosyl-(1→2)-4-O-benzoyl-α-l-arabinopyranoside (9)
3.6. Benzyl Oleanolate 3-O-2,3,4-tri-O-benzoyl-α-l-rhamnopyranosyl-(1→2)- [2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl-(1→3)]-3-O-benzoyl-α-l-arabinopyranoside (10)
+32.6 (c 1.75, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 8.12–7.25 (m, 25H, Ar-H); 5.77 (dd, J = 10.3, 3.3 Hz, 1H, H-2''), 5.73 (m, 1H, H-3''), 5.69 (t, J = 10.0 Hz, 1H, H-4''), 5.39 (s, 1H, H-1''), 5.31 (s, 1H, H-1'), 5.30 (br s, 1H, H-12), 5.28 (dd, J = 9.9, 9.6 Hz, 1H, H-3'''), 5.22 (d, J = 3.0 Hz, 1H, H-3'), 5.13 (dd, J = 9.4, 10.4 Hz, 1H, H-4'''), 5.11-5.03 (m, 3H, H-2''', PhCH2), 4.74 (d, J = 8.2 Hz, 1H, H-1'''), 4.48 (m, 1H, H-4'), 4.39 (br s, 1H, H-2'), 4.29 (dd, J = 12.4, 4.1 Hz, 1H, H-6'''-1), 4.23 (dd, J = 10.0, 5.9 Hz, 1H, H-5'-1), 4.19-4.17 (m, 2H, H-5'', H-6'''-2), 3.81–3.78 (m, 2H, H-5'-2, H-5'''), 3.16 (dd, J = 11.3, 4.4 Hz, 1H, H-3), 2.91 (dd, J = 13.7, 3.4 Hz, 1H, H-18), 2.07, 2.01, 1.99, 1.96 (s, 3H each, 4 × CH3CO), 1.35 (d, J = 6.1 Hz, 3H, H-6''), 1.13, 0.95, 0.92, 0.90, 0.90, 0.76, 0.61 (s, 3H each, 7 × Me). HRMS: calcd for C82H97O24 (M-Bn)−: 1465.6370; found: m/z 1465.6364.3.7. Oleanolic Acid 3-O-α-l-rhamnopyranosyl-(1→2)-[β-D-glucopyranosyl-(1→3)]-α-l-arabino-pyranoside (1)
−38.5 (c 0.52, MeOH); Mp 210.4–213.8 °C; 1H-NMR (600 MHz, pyridine-d5): δ 5.87 (s, 1H, H-1''), 5.51 (s, 1H, H-1'), 5.48 (br s, 1H, H-12), 4.98 (d, J = 7.8 Hz, 1H, H-1'''), 4.92–4.90 (dd, J = 1.8 Hz, 1H, H-2'), 4.75–4.72 (m, 1H, H-4''), 4.70–4.68 (m, 1H, H-3'), 4.57–4.55 (m, 1H, H-2''), 4.53–4.49 (m, 3H, H-5'-1, H-3'', H-6'''-1), 4.42-4.20 (m, 6H, H-4', H-6'''-2, H-5'', H-5'-2, H-3''', H-4'''), 4.06 (m, 1H, H-2'''), 3.94 (m, 1H, H-5'''), 3.29 (dd, J = 13.8, 3.8 Hz, 1H, H-18), 3.18 (dd, J = 11.7 Hz, 4.3, 1H, H-3), 1.69 (d, J = 6.2 Hz, 3H, H-6''), 1.27, 1.11, 1.00, 0.98, 0.94, 0.90, 0.80 (s, 3H each, 7 × Me); 13C-NMR (150 MHz, pyridine-d5): δ 180.5, 145.1, 122.9, 109.6, 105.4, 101.5, 88.6, 87.7, 82.3, 78.9, 78.8, 78.4, 75.4, 74.3, 72.9, 72.8, 71.9, 70.6, 70.4, 63.0, 56.0, 48.3, 47.0, 46.8, 42.5, 42.3, 40.0, 39.4, 38.9, 37.3, 34.6, 33.6, 33.6, 33.5, 31.3, 28.7, 28.6, 26.5, 26.5, 24.1, 24.1, 24.0, 18.9, 18.9, 17.7, 17.3, 15.8; HRMS (m/z): calcd for C47H76NaO16 (M+Na)+: 919.5026; found: 919.5030.3.8. Benzyl Oleanolate 3-O-2,3,4-tri-O-acetyl-α-l-rhamnopyranosyl-(1→2)-3,4-O- isopropylidene-α-l-arabinopyranoside (11)
+6.8 (c 0.62, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 7.35–7.34 (m, 5H, Ar-H), 5.34 (brs, 1H, H-1''), 5.33-5.32 (m, 2H, H-2'', H-3''), 5.28 (brs, 1H, H-12), 5.08 (dd, J = 20.1, 12.6 Hz, 2H, PhCH2), 4.36 (d, J = 7.2 Hz, 1H, H-1'), 4.21 (m,1H, H-3'), 4.20 (m, 1H, H-5'') 4.19 (m, 1H, H-4'), 4.17 (t, J = 7.2 Hz, 1H, H-2'), 3.77 (dd, J = 13.8, 5.4 Hz, 1H, H-5'-1), 3.75 (dd, J = 13.2, 3.6 Hz, 1H, H-5'-2), 3.09 (dd, J = 11.3, 4.2 Hz, 1H, H-3), 2.91 (dd, J = 10.2, 2.8 Hz, 1H, H-18), 2.15, 2.02, 1.97 (s, 3H each, 3 × CH3CO), 1.53, 1.34 (s, 3H each, O-(CH3)2C-O), 1.20 (d, J = 6.0 Hz, 3H, H-6''), 1.12, 1.03, 0.92, 0.89, 0.89, 0.82, 0.61 (s each, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.6, 170.4, 170.2, 143.9, 136.6, 128.6, 128.2, 122.7, 110.6, 103.4, 95.4, 89.3, 79.4, 75.3, 73.6, 71.4, 69.8, 69.3, 66.4, 66.1, 62.8, 56.1, 47.8, 46.9, 46.1, 41.6, 39.3, 38.9, 36.9, 34.1, 33.3, 32.9, 32.6, 30.9, 29.9, 28.3, 27.9, 27.8, 26.3, 26.1, 23.8, 23.6, 21.2, 21.0, 20.9, 18.4, 17.6, 17.1, 16.6, 15.6; HRMS (m/z): calcd for C58H84NaO13 (M+Na)+: 1013.5610; found: 1013.5615.3.9. Benzyl Oleanolate 3-O-2,3,4-tri-O-acetyl-α-l-rhamnopyranosyl-(1→2)-α-l- arabinopyranoside (12)
+41.0 (c 0.19, CHCl3); Mp 142.7–149.6 °C; 1H-NMR (CDCl3): δ 7.36–7.31 (m, 5H, Ar-H), 5.29–5.26 (m, 3H, H-3'', H-4'', H-12), 5.10–5.05 (m, 3H, H-2'', PhCH2), 5.03 (s, 1H, H-1''), 4.71 (d, J = 5.0 Hz, 1H, H-1'), 4.00 (m, 1H, H-5''), 3.93 (m, 1H, H-4'), 3.89-3.85 (m, 2H, H-2', H-3'), 3.74 (dd, J = 11.4, 4.2 Hz, 1H, H-5'-1), 3.61 (dd, J = 11.4, 4.2 Hz, 1H, H-5'-2), 3.13 (dd, J = 11.4, 4.2 Hz, 1H, H-3), 2.91 (dd, J = 13.8, 3.6 Hz, 1H, H-18), 2.15, 2.05, 2.00 (s, 3H each, 3 × CH3CO), 1.20 (d, J = 6.0 Hz, 3H, H-6''), 1.12, 0.97, 0.93, 0.90, 0.89, 0.80, 0.60 (s, 3H each, 7 × Me); 13C-NMR (CDCl3): δ 128.6, 128.2, 128.1, 122.6, 102.0, 98.4, 76.1, 71.1, 69.9, 69.1, 67.2, 66.2, 47.8, 46.9, 46.1, 41.9, 41.6, 39.5, 39.3, 38.7, 36.9, 33.3, 32.9, 32.6, 30.9, 28.3, 27.8, 26.1, 25.9, 23.9, 23.6, 23.3, 21.1, 21.0, 18.5, 17.6, 17.1, 16.6, 15.5; HRMS (m/z): calcd for C55H80NaO13 (M+Na)+: 973.5278; found: 973.5282.3.10. Benzyl Oleanolate 3-O-2,3,4-tri-O-acetyl-α-l-rhamnopyranosyl-(1→2)-4-O- acetyl-α-l-arabino-pyranoside (13)
+91.0 (c 0.10, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 7.34–7.27 (m, 5H, Ar-H), 5.32–5.31 (m, 3H, H-4', H-3'', H-2''), 5.13 (brs, 1H, H-1''), 5.11–5.04 (m, 4H, PhCH2, H-12, H-2'), 4.09 (d, J = 6.6 Hz, 1H, H-1'), 3.98–3.95 (m, 1H, H-4''), 3.94–3.89 (m, 1H, H-3'), 3.86–3.84 (m, 1H, H-5'-1), 3.64 (dd, J = 3. 6, 12.0 Hz, 1H, H-5'-2), 3.13 (dd, J = 4.2, 1.2 Hz, 1H, H-3), 2.91(dd, 1H, J = 3.6, 10.2 Hz, H-18), 2.15, 2.13, 2.05 (s, 3H each, 3 × CH3CO), 1.22 (d, J = 6.0 Hz, 3H, H-6''), 1.12, 1.00, 0.99, 0.93, 0.90, 0.81, 0.60 (s, 3H each, 7 × Me); 13C-NMR (150 MHz, CDCl3): δ 177.7, 170.87, 170.2, 143.9, 136.7, 129.1, 128.6, 128.2, 128.1, 122.7, 102.9, 98.2, 90.6, 77.4, 77.2, 77.0, 76.1, 71.2, 69.9, 69.1, 67.2, 66.2, 55.8, 47.8, 47.0, 46.1, 41.9, 41.6, 39.5, 39.3, 38.8, 36.9, 34.1, 33.3, 32.9, 32.6, 30.9, 28.3, 27.8, 26.1, 23.9, 23.6, 23.3, 21.3, 21.1, 21.0, 20.9, 19.4, 18.5, 17.6, 17.1, 16.6; HRMS (m/z): calcd for C57H82NaO14 (M+Na)+: 1015.5480; found: 1015.5484.3.11. Oleanolic Acid 3-O-α-l-rhamnopyranosyl-(1→2)-[β-d-glucopyranosyl-(1→3)]-α-l-arabino-pyranoside (Patrinia-glycoside B-II)
−27.5 (c 0.49, MeOH); 1H-NMR (600 MHz, pyridine-d5): δ 6.20 (br s, 1H, H-1''), 5.47 (br s, 1H, H-12), 5.13 (d, 1H, J = 7.8 Hz, H-1'''), 4.87 (d, J = 5.4 Hz, 1H, H-1'), 4.77 (br s, 1H, H-2''), 4.68 (d, 1H, J = 7.8 Hz, H-2'), 4.66–4.50 (m, 4H, H-5'', H-3'', H-4', H-6'''-1), 4.41–4.18 (m, 7H, H-5'-1, H-5'-2, H-6'''-2, H-3', H-3''', H-4''', H-4''), 3.97-3.94 (m, 2H, H-2''', H-5'''), 3.40 (d, J = 14.4 Hz, 1H, H-18), 3.18 (d, J = 10.8 Hz, 1H, H-3), 1.55 (d, J = 6.0 Hz, 3H, H-6''), 1.26, 1.18, 1.08, 0.97, 0.92, 0.87, 0.80 (s, 3H each, 7 × Me); 13C-NMR (150 MHz, pyridine-d5): δ 179.9, 144.5, 122.2, 104.5, 104.5, 101.7, 87.9, 82.0, 78.3, 78.0, 74.7, 74.5, 73.7, 72.3, 72.3, 71.2, 69.8, 67.9, 64.8, 62.3, 55.7, 47.8, 46.4, 46.2, 46.0, 41.9, 41.7, 39.5, 39.3, 38.7, 36.8, 34.0, 33.0, 33.0, 31.9, 30.7, 29.8, 29.4, 28.1, 27.8, 26.4, 28.1, 27.8, 26.4, 25.9, 23.5, 23.4, 18.4, 18.2, 17.1, 16.8, 15.3; HRMS (m/z): calcd for C47H76NaO16 (M+Na)+: 919.5026; found: 919.5030.3.12. Computational Methods
3.13. Cell Culture
3.14. Cell Viability Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Parente, J.P.; da Silva, B.P. Bioactive complex triterpenoid saponins from the Leguminosae family. Nat. Prod. Commun. 2009, 4, 143–155. [Google Scholar]
- Luo, J.; Ma, L.; Kong, L. New triterpenoid saponins with strong alpha-glucosidase inhibitory activity from the roots of Gypsophila oldhamiana. Bioorg. Med. Chem. 2008, 16, 2912–2920. [Google Scholar] [CrossRef]
- Lanzotti, V. Bioactive saponins from allium and aster plants. Phytochem. Rev. 2005, 4, 95–110. [Google Scholar] [CrossRef]
- Ekabo, O.A.; Farnsworth, N.R. Antifungal and molluscicidal saponins from Serjania salzmanniana. J. Nat. Prod. 1996, 59, 431–435. [Google Scholar] [CrossRef]
- Mimaki, Y.; Kuroda, M.; Asano, T.; Sashida, Y. Triterpene saponins and lignans from the roots of Pulsatilla chinensis and their cytotoxic activity against HL-60 cells. J. Nat. Prod. 1999, 62, 1279–1283. [Google Scholar] [CrossRef]
- Barthomeuf, C.; Debiton, E.; Mshvidadze, V.; Kemertelidze, E.; Balansard, G. In vitro activity of hederacolchisid A1 compared with other saponins from Hedera colchica against proliferation of human carcinoma and melanoma cells. Planta Med. 2002, 68, 672–675. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, C.O.; Lee, K.T.; Choi, J.; Park, H.J. Structure-activity relationship of oleanane disaccharides isolated from Akebia quinata versus cytotoxicity against cancer cells and NO inhibition. Biol. Pharm. Bull. 2004, 27, 744–747. [Google Scholar] [CrossRef]
- Park, H.J.; Kwon, S.H.; Lee, J.H.; Lee, K.H.; Miyamoto, K.; Lee, K.T. Kalopanaxsaponin A is a basic saponin structure for the anti-tumor activity of hederagenin monodesmosides. Planta Med. 2001, 67, 118–121. [Google Scholar] [CrossRef]
- Cheng, M.S.; Yan, M.C.; Liu, Y.; Zheng, L.G.; Liu, J. Synthesis of β-hederin and Hederacolchiside A1: Triterpenoid saponins bearing a unique cytotoxicity-inducing disaccharide moiety. Carbohydr. Res. 2006, 341, 60–67. [Google Scholar] [CrossRef]
- Yan, M.C.; Liu, Y.; Lu, W.X.; Wang, H.; Sha, Y.; Cheng, M.S. Facile synthesis and cytotoxicity of triterpenoid saponins bearing a unique disaccharide moiety: Hederacolchiside A1 and its analogues. Carbohydr. Res. 2008, 343, 780–784. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tanaka, K.; Murata, H.; Somekawa, M.; Inada, A. Phytochemical studies of seeds of medicinal plants. III. Ursolic acid and oleanolic acid glycosides from seeds of Patrinia scabiosaefolia Fischer. Chem. Pharm. Bull. 1993, 41, 183–186. [Google Scholar] [CrossRef]
- Schmidt, R.R. New methods for the synthesis of glycosides and oligosaccharides—Are there alternatives to the koenigs-knorr method? Angew. Chem. Int. Ed. Engl. 1986, 25, 212–235. [Google Scholar] [CrossRef]
- Yu, B.; Yu, H.; Hui, Y.; Han, X. Trichloroacetimidate as an efficient protective group for alcohols. Synlett 1999, 6, 753–755. [Google Scholar]
- David, S.; Hanessian, S. Regioselective manipulation of hydroxyl groups via organotin derivatives. Tetrahedron 1985, 41, 643–663. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.; Zang, J.; Song, N.; Zhang, X.; Li, Y.X. Synthesis of two bidesmosidic ursolic acid saponins bearing a 2,3-branched trisaccharide residue. Carbohydr. Res. 2005, 340, 2086–2096. [Google Scholar] [CrossRef]
- Gu, G.; Du, Y.; Linhardt, R.J. Facile synthesis of saponins containing 2,3-branched oligosaccharides by using partially protected glycosyl donors. J. Org. Chem. 2004, 69, 5497–5500. [Google Scholar] [CrossRef]
- Yamada, H.; Nakatani, M.; Ikeda, T.; Marumoto, Y. Stable axial-rich conformation of pyranoses derived from L-rhamnose and d-mannose. Tetrahedron Lett. 1999, 40, 5573–5576. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, W.X.; Yan, M.C.; Yu, Y.; Ikejima, T.; Cheng, M.S. Synthesis and tumor cytotoxicity of novel amide derivatives of β-Hederin. Molecules 2010, 15, 7871–7883. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; de Ninno, M.P.; Chen, S.H. Stereoselective total syntheses of the naturally occurring enantiomers of N-acetylneuraminic acid and 3-deoxy-d-manno-2-octulosonic acid. A new and stereospecific approach to sialo and 3-deoxy-d-manno-2-octulosonic acid conjugates. J. Am. Chem. Soc. 1988, 110, 3929–3940. [Google Scholar] [CrossRef]
- Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, F.C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ren, L.; Liu, Y.-X.; Lv, D.; Yan, M.-C.; Nie, H.; Liu, Y.; Cheng, M.-S. Facile Synthesis of the Naturally Cytotoxic Triterpenoid Saponin Patrinia-Glycoside B-II and Its Conformer. Molecules 2013, 18, 15193-15206. https://doi.org/10.3390/molecules181215193
Ren L, Liu Y-X, Lv D, Yan M-C, Nie H, Liu Y, Cheng M-S. Facile Synthesis of the Naturally Cytotoxic Triterpenoid Saponin Patrinia-Glycoside B-II and Its Conformer. Molecules. 2013; 18(12):15193-15206. https://doi.org/10.3390/molecules181215193
Chicago/Turabian StyleRen, Li, Yong-Xiang Liu, Dan Lv, Mao-Cai Yan, Han Nie, Yang Liu, and Mao-Sheng Cheng. 2013. "Facile Synthesis of the Naturally Cytotoxic Triterpenoid Saponin Patrinia-Glycoside B-II and Its Conformer" Molecules 18, no. 12: 15193-15206. https://doi.org/10.3390/molecules181215193
APA StyleRen, L., Liu, Y.-X., Lv, D., Yan, M.-C., Nie, H., Liu, Y., & Cheng, M.-S. (2013). Facile Synthesis of the Naturally Cytotoxic Triterpenoid Saponin Patrinia-Glycoside B-II and Its Conformer. Molecules, 18(12), 15193-15206. https://doi.org/10.3390/molecules181215193