Chemical Genetics: Budding Yeast as a Platform for Drug Discovery and Mapping of Genetic Pathways

{kind=link}
Abstract
:1. Introduction
2. History of Budding Yeast as a Model Organism
3. Classical Genetics
4. Genetics in the Post-Genomic Era
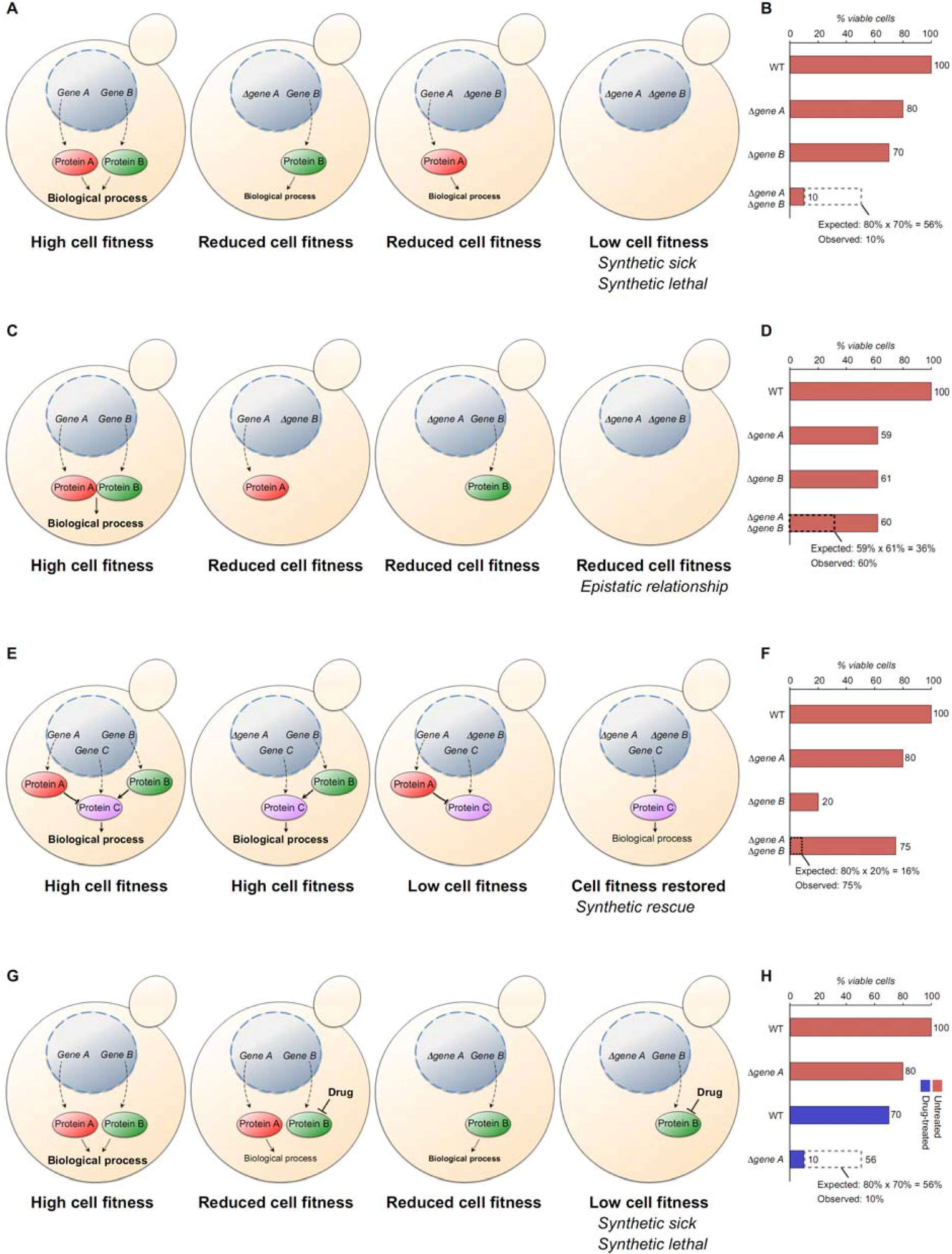
4.1. Defining Genetic Interactions
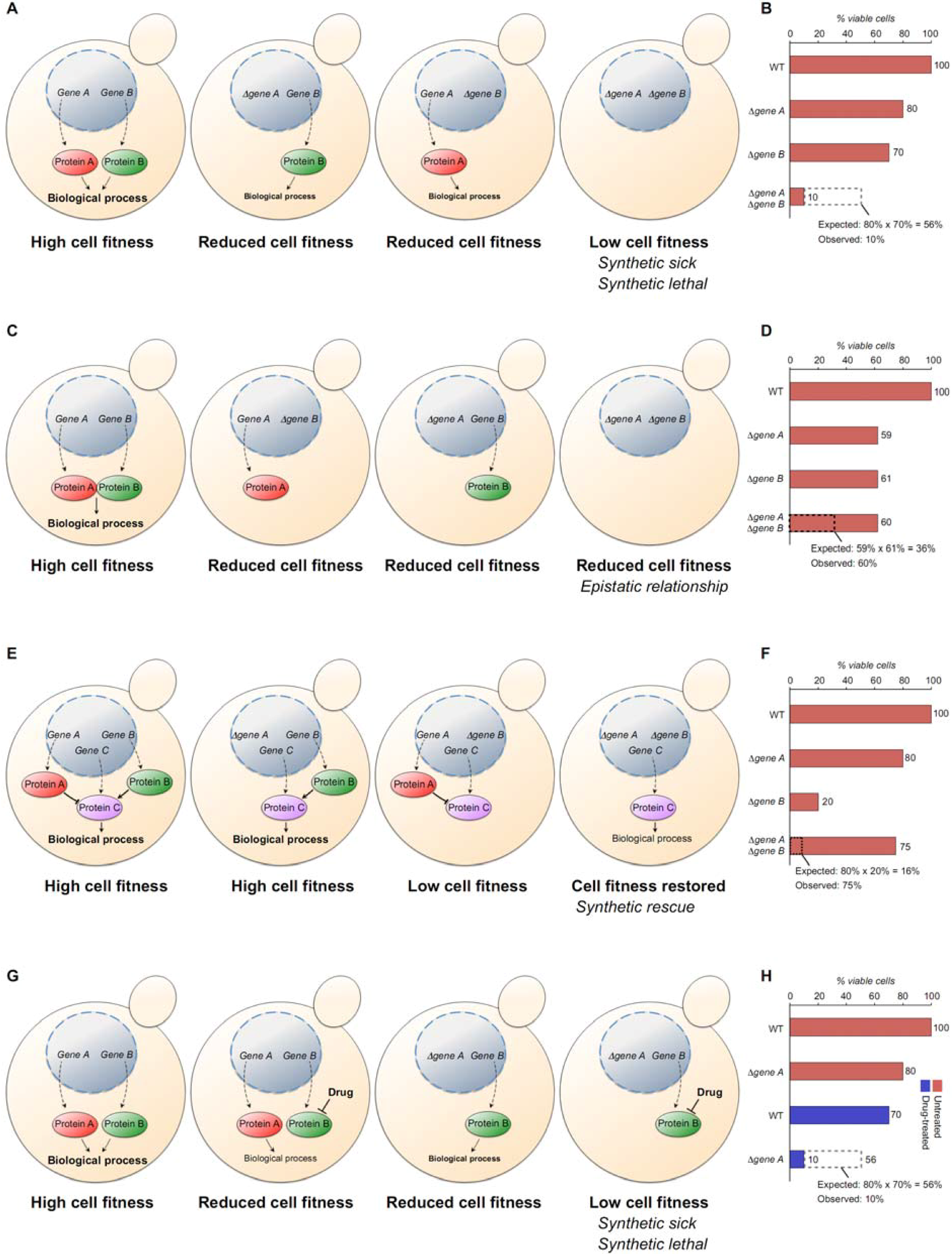
4.2. High-Throughput Systematic Screens
4.3. High-Throughput Chemical-Genetic Screens
5. Drug Target Identification and Chemical Profiling of the Yeast Genome
6. Conclusions and Future Directions
Acknowledgments
References
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 563–567. [Google Scholar]
- Morgan, T.H. The Origin of Five Mutations in Eye Color in Drosophila and Their Modes of Inheritance. Science 1911, 33, 534–537. [Google Scholar]
- Morgan, T.H. Sex Limited Inheritance in Drosophila. Science 1910, 32, 120–122. [Google Scholar]
- Morgan, T.H. Chromosomes and Associative Inheritance. Science 1911, 34, 636–638. [Google Scholar]
- Pena-Castillo, L.; Hughes, T.R. Why are there still over 1,000 uncharacterized yeast genes? Genetics 2007, 176, 7–14. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and genetic assimilation of acquired characters. Nature 1959, 183, 1654–1655. [Google Scholar] [CrossRef]
- Hartman, J.L., IV; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar]
- Tishkoff, D.X.; Filosi, N.; Gaida, G.M.; Kolodner, R.D. A novel mutation avoidance mechanism dependent on S.cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 1997, 88, 253–263. [Google Scholar] [CrossRef]
- D’Amours, D.; Jackson, S.P. The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev. 2001, 15, 2238–2249. [Google Scholar] [CrossRef]
- Zhao, X.; Muller, E.G.; Rothstein, R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 1998, 2, 329–340. [Google Scholar] [CrossRef]
- Zhao, X.; Chabes, A.; Domkin, V.; Thelander, L.; Rothstein, R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 2001, 20, 3544–3553. [Google Scholar]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef]
- Dolinski, K.; Botstein, D. Changing perspectives in yeast research nearly a decade after the genome sequence. Genome Res. 2005, 15, 1611–1619. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar]
- Tong, A.H.; Lesage, G.; Bader, G.D.; Ding, H.; Xu, H.; Xin, X.; Young, J.; Berriz, G.F.; Brost, R.L.; Chang, M.; et al. Global mapping of the yeast genetic interaction network. Science 2004, 303, 808–813. [Google Scholar]
- Tong, A.H.; Evangelista, M.; Parsons, A.B.; Xu, H.; Bader, G.D.; Page, N.; Robinson, M.; Raghibizadeh, S.; Hogue, C.W.; Bussey, H.; et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001, 294, 2364–2368. [Google Scholar]
- Dixon, S.J.; Costanzo, M.; Baryshnikova, A.; Andrews, B.; Boone, C. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 2009, 43, 601–625. [Google Scholar] [CrossRef]
- Ooi, S.L.; Shoemaker, D.D.; Boeke, J.D. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 2003, 35, 277–286. [Google Scholar]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar]
- Davierwala, A.P.; Haynes, J.; Li, Z.; Brost, R.L.; Robinson, M.D.; Yu, L.; Mnaimneh, S.; Ding, H.; Zhu, H.; Chen, Y.; et al. The synthetic genetic interaction spectrum of essential genes. Nat. Genet. 2005, 37, 1147–1152. [Google Scholar]
- Mnaimneh, S.; Davierwala, A.P.; Haynes, J.; Moffat, J.; Peng, W.T.; Zhang, W.; Yang, X.; Pootoolal, J.; Chua, G.; Lopez, A.; et al. Exploration of essential gene functions via titratable promoter alleles. Cell 2004, 118, 31–44. [Google Scholar] [CrossRef]
- Li, Z.; Vizeacoumar, F.J.; Bahr, S.; Li, J.; Warringer, J.; Vizeacoumar, F.S.; Min, R.; Vandersluis, B.; Bellay, J.; Devit, M.; et al. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 2011, 29, 361–367. [Google Scholar]
- Ben-Aroya, S.; Coombes, C.; Kwok, T.; O’Donnell, K.A.; Boeke, J.D.; Hieter, P. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell 2008, 30, 248–258. [Google Scholar] [CrossRef]
- Enserink, J.M.; Kolodner, R.D. An overview of Cdk1-controlled targets and processes. Cell Div. 2010, 5, 11. [Google Scholar] [CrossRef]
- Rodriguez, J.; Crespo, P. Working without kinase activity: Phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci. Signal. 2011, 4, re3. [Google Scholar] [CrossRef]
- Toya, M.; Terasawa, M.; Nagata, K.; Iida, Y.; Sugimoto, A. A kinase-independent role for Aurora A in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat. Cell Biol. 2011, 13, 708–714. [Google Scholar]
- Au-Yeung, B.B.; Levin, S.E.; Zhang, C.; Hsu, L.Y.; Cheng, D.A.; Killeen, N.; Shokat, K.M.; Weiss, A. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat. Immunol. 2010, 11, 1085–1092. [Google Scholar] [CrossRef]
- Zhao, X.; Peng, X.; Sun, S.; Park, A.Y.; Guan, J.L. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J. Cell Biol. 2010, 189, 955–965. [Google Scholar] [CrossRef]
- Yu, V.P.; Baskerville, C.; Grunenfelder, B.; Reed, S.I. A kinase-independent function of Cks1 and Cdk1 in regulation of transcription. Mol. Cell 2005, 17, 145–151. [Google Scholar] [CrossRef]
- Bishop, A.C.; Ubersax, J.A.; Petsch, D.T.; Matheos, D.P.; Gray, N.S.; Blethrow, J.; Shimizu, E.; Tsien, J.Z.; Schultz, P.G.; Rose, M.D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. [Google Scholar]
- Zimmermann, C.; Chymkowitch, P.; Eldholm, V.; Putnam, C.D.; Lindvall, J.M.; Omerzu, M.; Bjoras, M.; Kolodner, R.D.; Enserink, J.M. A chemical-genetic screen to unravel the genetic network of CDC28/CDK1 links ubiquitin and Rad6-Bre1 to cell cycle progression. Proc. Natl. Acad. Sci. USA 2011, 108, 18748–18753. [Google Scholar]
- Chymkowitch, P.; Eldholm, V.; Lorenz, S.; Zimmermann, C.; Lindvall, J.M.; Bjoras, M.; Meza-Zepeda, L.A.; Enserink, J.M. Cdc28 kinase activity regulates the basal transcription machinery at a subset of genes. Proc. Natl. Acad. Sci. USA 2012, 109, 10450–10455. [Google Scholar]
- Enserink, J.M.; Kolodner, R.D. What makes the engine hum: Rad6, a cell cycle supercharger. Cell Cycle 2012, 11, 249–252. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Woodbury, E.L.; Quang, P.N.; Paraz, M.; Blethrow, J.D.; Shah, K.; Shokat, K.M.; Morgan, D.O. Targets of the cyclin-dependent kinase Cdk1. Nature 2003, 425, 859–864. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villen, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef]
- Enserink, J.M.; Hombauer, H.; Huang, M.E.; Kolodner, R.D. Cdc28/Cdk1 positively and negatively affects genome stability in S. cerevisiae. J. Cell Biol. 2009, 185, 423–437. [Google Scholar] [CrossRef]
- Granata, M.; Lazzaro, F.; Novarina, D.; Panigada, D.; Puddu, F.; Abreu, C.M.; Kumar, R.; Grenon, M.; Lowndes, N.F.; Plevani, P.; et al. Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle-regulated by CDK1 activity. PLoS Genet. 2010, 6, e1001047. [Google Scholar] [CrossRef]
- Buratowski, S. Progression through the RNA polymerase II CTD cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef]
- Cismowski, M.J.; Laff, G.M.; Solomon, M.J.; Reed, S.I. KIN28 encodes a C-terminal domain kinase that controls mRNA transcription in Saccharomyces cerevisiae but lacks cyclin-dependent kinase-activating kinase (CAK) activity. Mol. Cell Biol. 1995, 15, 2983–2992. [Google Scholar]
- Valay, J.G.; Simon, M.; Dubois, M.F.; Bensaude, O.; Facca, C.; Faye, G. The KIN28 gene is required both for RNA polymerase II mediated transcription and phosphorylation of the Rpb1p CTD. J. Mol. Biol. 1995, 249, 535–544. [Google Scholar] [CrossRef]
- Qiu, H.; Hu, C.; Hinnebusch, A.G. Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol. Cell 2009, 33, 752–762. [Google Scholar] [CrossRef]
- Kanin, E.I.; Kipp, R.T.; Kung, C.; Slattery, M.; Viale, A.; Hahn, S.; Shokat, K.M.; Ansari, A.Z. Chemical inhibition of the TFIIH-associated kinase Cdk7/Kin28 does not impair global mRNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 5812–5817. [Google Scholar]
- Hong, S.W.; Hong, S.M.; Yoo, J.W.; Lee, Y.C.; Kim, S.; Lis, J.T.; Lee, D.K. Phosphorylation of the RNA polymerase II C-terminal domain by TFIIH kinase is not essential for transcription of Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. USA 2009, 106, 14276–14280. [Google Scholar]
- Viladevall, L.; St Amour, C.V.; Rosebrock, A.; Schneider, S.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Schwer, B.; Leatherwood, J.K.; Fisher, R.P. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol. Cell 2009, 33, 738–751. [Google Scholar] [CrossRef]
- Tanaka, S.; Araki, H. Regulation of the initiation step of DNA replication by cyclin-dependent kinases. Chromosoma 2010, 119, 565–574. [Google Scholar] [CrossRef]
- Lo, H.C.; Wan, L.; Rosebrock, A.; Futcher, B.; Hollingsworth, N.M. Cdc7-Dbf4 regulates NDT80 transcription as well as reductional segregation during budding yeast meiosis. Mol. Biol. Cell 2008, 19, 4956–4967. [Google Scholar] [CrossRef]
- Shi, Y.; Stefan, C.J.; Rue, S.M.; Teis, D.; Emr, S.D. Two novel WD40 domain-containing proteins, Ere1 and Ere2, function in the retromer-mediated endosomal recycling pathway. Mol. Biol. Cell 2011, 22, 4093–4107. [Google Scholar] [CrossRef]
- Andrusiak, K.; Piotrowski, J.S.; Boone, C. Chemical-genomic profiling: Systematic analysis of the cellular targets of bioactive molecules. Bioorg. Med. Chem. 2012, 20, 1952–1960. [Google Scholar] [CrossRef]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Loewith, R. A brief history of TOR. Biochem. Soc. Trans. 2011, 39, 437–442. [Google Scholar] [CrossRef]
- Yan, Z.; Costanzo, M.; Heisler, L.E.; Paw, J.; Kaper, F.; Andrews, B.J.; Boone, C.; Giaever, G.; Nislow, C. Yeast Barcoders: A chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat. Methods 2008, 5, 719–725. [Google Scholar]
- Giaever, G.; Shoemaker, D.D.; Jones, T.W.; Liang, H.; Winzeler, E.A.; Astromoff, A.; Davis, R.W. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 1999, 21, 278–283. [Google Scholar] [CrossRef]
- Sopko, R.; Huang, D.; Preston, N.; Chua, G.; Papp, B.; Kafadar, K.; Snyder, M.; Oliver, S.G.; Cyert, M.; Hughes, T.R.; et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell 2006, 21, 319–330. [Google Scholar] [CrossRef]
- Chang, M.; Bellaoui, M.; Boone, C.; Brown, G.W. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc. Natl. Acad. Sci. USA 2002, 99, 16934–16939. [Google Scholar]
- Hanway, D.; Chin, J.K.; Xia, G.; Oshiro, G.; Winzeler, E.A.; Romesberg, F.E. Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 10605–10610. [Google Scholar] [CrossRef]
- Hillenmeyer, M.E.; Fung, E.; Wildenhain, J.; Pierce, S.E.; Hoon, S.; Lee, W.; Proctor, M.; St.Onge, R.P.; Tyers, M.; Koller, D.; et al. The chemical genomic portrait of yeast: Uncovering a phenotype for all genes. Science 2008, 320, 362–365. [Google Scholar]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.W.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2004, 22, 62–69. [Google Scholar] [CrossRef]
- Hughes, T.R.; Marton, M.J.; Jones, A.R.; Roberts, C.J.; Stoughton, R.; Armour, C.D.; Bennett, H.A.; Coffey, E.; Dai, H.; He, Y.D.; et al. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–126. [Google Scholar] [CrossRef]
- Roemer, T.; Davies, J.; Giaever, G.; Nislow, C. Bugs, drugs and chemical genomics. Nat. Chem. Biol. 2012, 8, 46–56. [Google Scholar]
- Marton, M.J.; DeRisi, J.L.; Bennett, H.A.; Iyer, V.R.; Meyer, M.R.; Roberts, C.J.; Stoughton, R.; Burchard, J.; Slade, D.; Dai, H.; et al. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998, 4, 1293–1301. [Google Scholar] [CrossRef]
- Dos Santos, S.C.; Teixeira, M.C.; Cabrito, T.R.; Sa-Correia, I. Yeast toxicogenomics: Genome-wide responses to chemical stresses with impact in environmental health, pharmacology, and biotechnology. Front. Genet. 2012, 3, 63. [Google Scholar]
- Venancio, T.M.; Bellieny-Rabelo, D.; Aravind, L. Evolutionary and Biochemical Aspects of Chemical Stress Resistance in Saccharomyces cerevisiae. Front. Genet. 2012, 3, 47. [Google Scholar]
- Venancio, T.M.; Balaji, S.; Geetha, S.; Aravind, L. Robustness and evolvability in natural chemical resistance: Identification of novel systems properties, biochemical mechanisms and regulatory interactions. Mol. Biosyst. 2010, 6, 1475–1491. [Google Scholar] [CrossRef]
- Venancio, T.M.; Balaji, S.; Aravind, L. High-confidence mapping of chemical compounds and protein complexes reveals novel aspects of chemical stress response in yeast. Mol. Biosyst. 2010, 6, 175–181. [Google Scholar] [CrossRef]
- Hoon, S.; St. Onge, R.P.; Giaever, G.; Nislow, C. Yeast chemical genomics and drug discovery: An update. Trends Pharmacol. Sci. 2008, 29, 499–504. [Google Scholar] [CrossRef]
- Lehar, J.; Stockwell, B.R.; Giaever, G.; Nislow, C. Combination chemical genetics. Nat. Chem. Biol. 2008, 4, 674–681. [Google Scholar] [CrossRef]
- Lopez, A.; Parsons, A.B.; Nislow, C.; Giaever, G.; Boone, C. Chemical-genetic approaches for exploring the mode of action of natural products. Fortschr. Arzneimittelforsch. 2008, 237, 239–271. [Google Scholar]
- Smith, A.M.; Ammar, R.; Nislow, C.; Giaever, G. A survey of yeast genomic assays for drug and target discovery. Pharmacol. Ther. 2010, 127, 156–164. [Google Scholar] [CrossRef]
- McClellan, A.J.; Xia, Y.; Deutschbauer, A.M.; Davis, R.W.; Gerstein, M.; Frydman, J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 2007, 131, 121–135. [Google Scholar] [CrossRef]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; Macbeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef]
- Janes, K.A.; Albeck, J.G.; Gaudet, S.; Sorger, P.K.; Lauffenburger, D.A.; Yaffe, M.B. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 2005, 310, 1646–1653. [Google Scholar]
- Janes, K.A.; Reinhardt, H.C.; Yaffe, M.B. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell 2008, 135, 343–354. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Enserink, J.M. Chemical Genetics: Budding Yeast as a Platform for Drug Discovery and Mapping of Genetic Pathways. Molecules 2012, 17, 9258-9273. https://doi.org/10.3390/molecules17089258
Enserink JM. Chemical Genetics: Budding Yeast as a Platform for Drug Discovery and Mapping of Genetic Pathways. Molecules. 2012; 17(8):9258-9273. https://doi.org/10.3390/molecules17089258
Chicago/Turabian StyleEnserink, Jorrit M. 2012. "Chemical Genetics: Budding Yeast as a Platform for Drug Discovery and Mapping of Genetic Pathways" Molecules 17, no. 8: 9258-9273. https://doi.org/10.3390/molecules17089258