3-Phenylcoumarins as Inhibitors of HIV-1 Replication

 , and
, and
Abstract
:1. Introduction
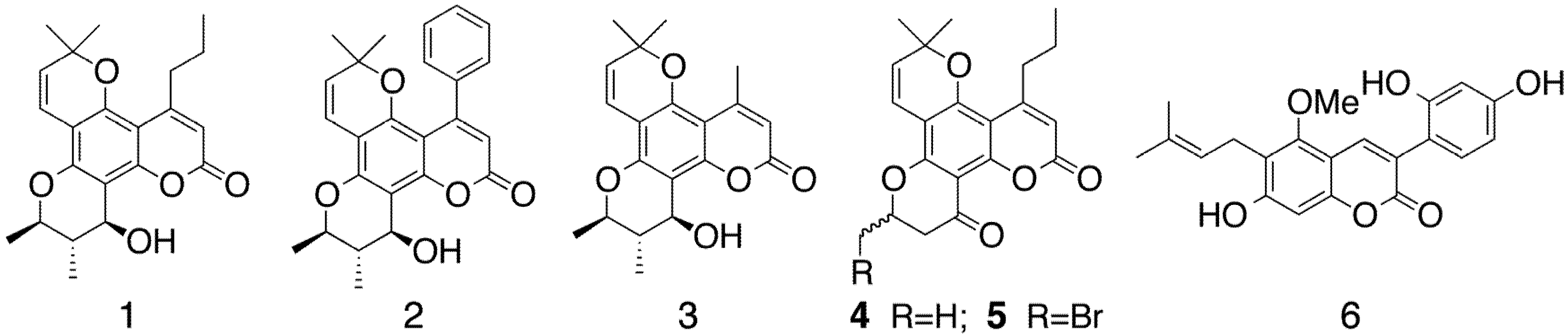
2. Results and Discussion
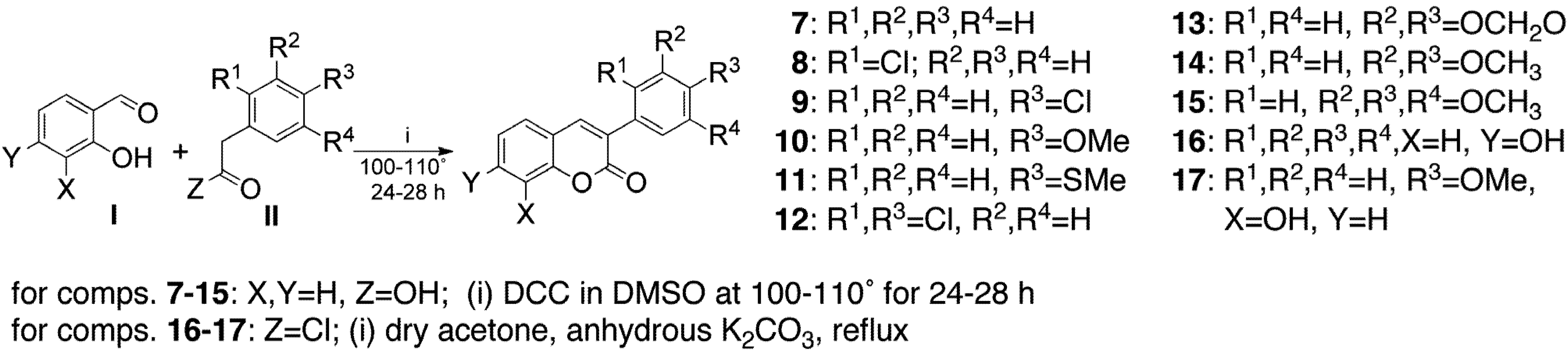
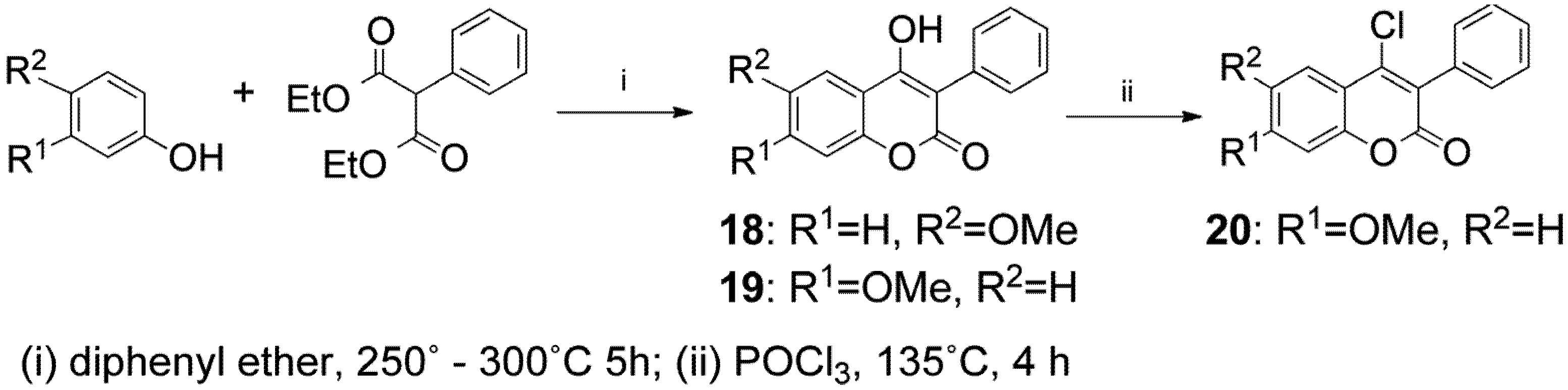

{kind=link}
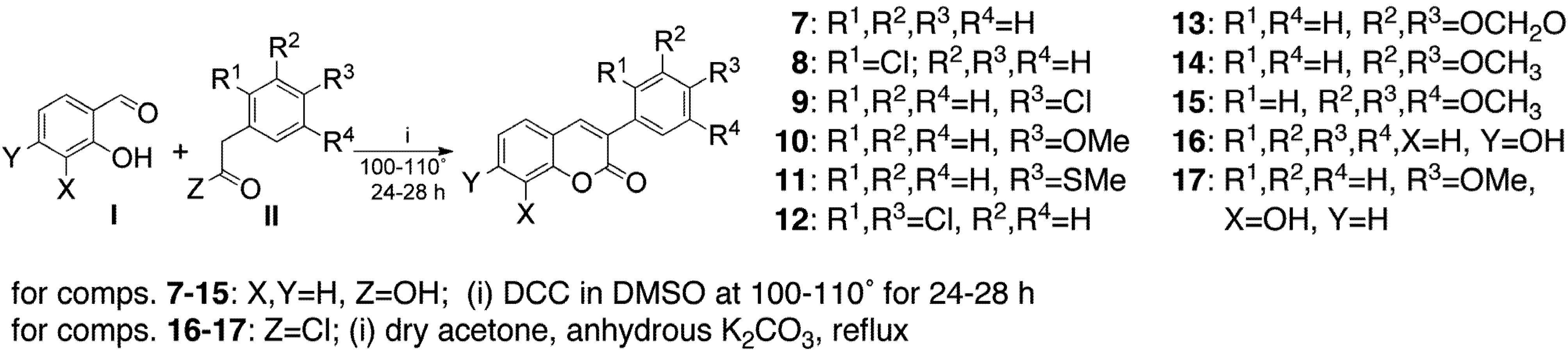{kind=link}
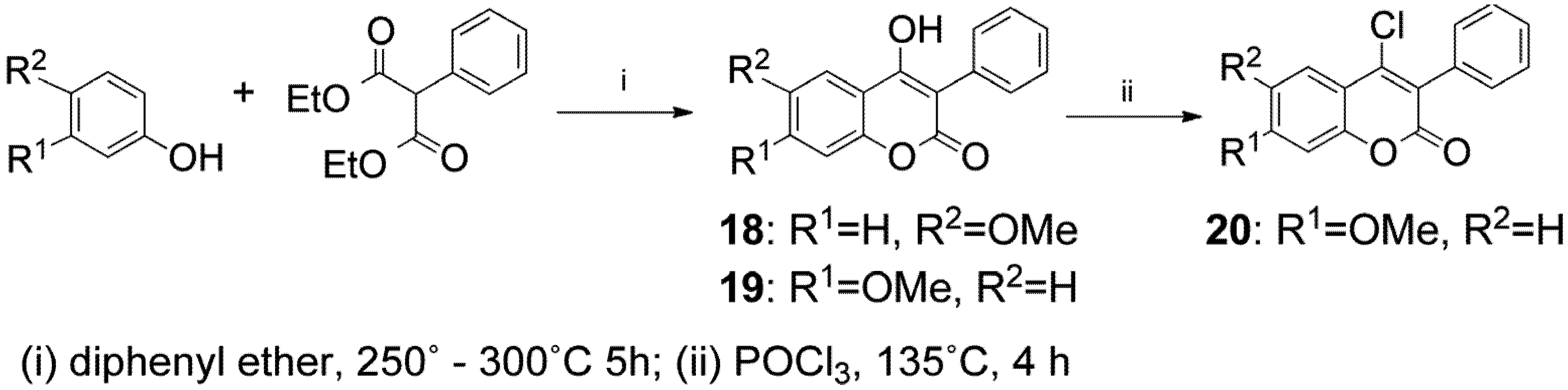{kind=link}
| Compound | Antiviral activity | ||||||
|---|---|---|---|---|---|---|---|
| 5.1 + TNFα | Hela-Tat-luc | HeLa-Tet-ON | RV (VIH) | Toxicity (%) | |||
| 25 µM | 50 µM | 25 µM | 50 µM | 50 µM | IC50, µM | CC50 | |
| 7 | nt | 21.7 ± 3 | 26.8 ± 2.9 | 33.7 ± 1.0 | S | 130 ± 11.4 | >50 |
| 8 | nt | −28.7 ± 4 | nt | 17.6 ± 2.1 | S | 20.7 ± 1.9 | >50 |
| 9 | nt | 33.0 ± 2.33 | nt | −4.9 ± 0.8 | nt | >195 | >50 |
| 10 | nt | 61.2 ± 2.4 | 73.5 ± 6.7 | 66.6 ± 3.4 | U | 73 ± 8.6 | >50 |
| 11 | 84.3 ± 3 | 81.6 ± 5.6 | 77.5 ± 8.5 | 80.1 ± 5.6 | U | 18.2 ± 0.2 | >50 |
| 12 | nt | −40.9 ± 2.3 | nt | 14.1 ± 0.6 | S | >172 | >50 |
| 13 | nt | 68.5 ± 3.4 | 32.8 ± 4.3 | 39.5 ± 2.3 | S | 138 ± 22,3 | 16.7 ± 3.4 |
| 14 | nt | −23.0 ± 2.9 | nt | −25.1 ± 5.3 | nt | >177 | >50 |
| 15 | nt | 49.7 ± 7.0 | 47.6 ± 6.2 | 35.1 ± 3.9 | U | 62.7 ± 13.1 | 42.8 ± 16.2 |
| 16 | nt | 59.0 ± 8.2 | nt | 17.2 ± 0.5 | S | 81.4 ± 14.0 | >50 |
| 17 | 82.2 ± 3.1 | 87.8 ± 9.8 | 85.7 ± 3.4 | 88.3 ± 5.5 | U | 84.6 ± 12.1 | >50 |
| 18 | nt | −3.5 ± 0.9 | nt | 22.8 ± 3.4 | S | 186 ± 10.8 | >50 |
| 19 | −34.26 ± 5 | −4.7 ± 0.3 | 48.9 ± 3.4 | 55.7 ± 4.9 | S | 23.8 ± 1.7 | >50 |
| 20 | nt | −21.2 ± 7.0 | nt | −64.4 ± 10 | nt | >209 | 18.4 ± 8.2 |
| Mesuol | 71.0 ± 4.8 | 77.9 ± 4.9 | nt | 71.3 ± 8.9 | S | 2.5 | >4 µM |
| AZT | nt | nt | nt | nt | nt | 0.01 | >1 µM |
3. Experimental
3.1. Material and Reagents
3.2. General Procedure for the Synthesis of Compounds 7–20
3.3. Anti-HIV Bioassay
4. Conclusions
Acknowledgments
References
- Piot, P.; Bartos, M.; Ghys, P.D.; Walker, N.; Schwartlander, B. The global impact of HIV/AIDS. Nature 2001, 410, 968–973. [Google Scholar]
- Center for HIV Information, HIV InSite. Report on the Global HIV/AIDS Epidemic, UNAIDS (2011). Available online: http://hivinsite.ucsf.edu/ (accessed on 25 July 2012).
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Barouch, D.H. Challenges in the development of an HIV-1 vaccine. Nature 2008, 455, 613–619. [Google Scholar] [CrossRef]
- Adamson, C.S.; Freed, E.O. Recent progress in antiretrovirals—Lessons from resistance. Drug Discov. Today 2008, 13, 424–432. [Google Scholar] [CrossRef]
- Rabson, A.B.; Lin, H.C. NF-kappa B and HIV: Linking viral and immune activation. Adv. Pharmacol. 2000, 48, 161–207. [Google Scholar] [CrossRef]
- Alcami, J.; Lain de Lera, T.; Folgueira, L.; Pedraza, M.A.; Jacque, J.M.; Bachelerie, F.; Noriega, A.R.; Hay, R.T.; Harrich, D.; Gaynor, R.B.; et al. Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J. 1995, 14, 1552–1560. [Google Scholar]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Stevenson, M. Tat’s seductive side. Nat. Med. 2003, 9, 163–164. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Musa, M.A.; Cooperwood, J.S.; Khan, M.O. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr. Med. Chem. 2008, 15, 2664–2679. [Google Scholar] [CrossRef]
- Kulkarni, M.V.; Kulkarni, G.M.; Lin, C.H.; Sun, C.M. Recent advances in coumarins and 1-azacoumarins as versatile biodynamic agents. Curr. Med. Chem. 2006, 13, 2795–2818. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Kumar, A.; Chatterjee, M.; Rao, K.B.; Singh, S.; Verma, A.K.; Palit, G. Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents. Bioorg. Med. Chem. Lett. 2011, 21, 1937–1941. [Google Scholar]
- Quezada, E.; Delogu, G.; Picciau, C.; Santana, L.; Podda, G.; Borges, F.; García-Morales, V.; Viña, D.; Orallo, F. Synthesis and vasorelaxant and platelet antiaggregatory activities of a new series of 6-halo-3-phenylcoumarins. Molecules 2010, 15, 270–279. [Google Scholar] [CrossRef]
- Matos, M.J.; Viña, D.; Janeiro, P.; Borges, F.; Santana, L.; Uriarte, E. New halogenated 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5157–5160. [Google Scholar]
- Matos, M.J.; Viña, D.; Quezada, E.; Picciau, C.; Delogu, G.; Orallo, F.; Santana, L.; Uriarte, E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3268–3270. [Google Scholar]
- Matos, M.J.; Vina, D.; Picciau, C.; Orallo, F.; Santana, L.; Uriarte, E. Synthesis and evaluation of 6-methyl-3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5053–5055. [Google Scholar]
- Roussaki, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Hamilakis, S.; Detsi, A. A novel synthesis of 3-aryl coumarins and evaluation of their antioxidant and lipoxygenase inhibitory activity. Bioorg. Med. Chem. Lett. 2010, 20, 3889–3892. [Google Scholar]
- Belluti, F.; Fontana, G.; dal Bo, L.; Carenini, N.; Giommarelli, C.; Zunino, F. Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: Identification of novel proapoptotic agents. Bioorg. Med. Chem. 2010, 18, 3543–3550. [Google Scholar] [CrossRef]
- Riveiro, M.E.; Moglioni, A.; Vazquez, R.; Gomez, N.; Facorro, G.; Piehl, L.; de Celis, E.R.; Shayo, C.; Davio, C. Structural insights into hydroxycoumarin-induced apoptosis in U-937 cells. Bioorg. Med. Chem. 2008, 16, 2665–2675. [Google Scholar]
- Kostova, I.; Raleva, S.; Genova, P.; Argirova, R. Structure-activity relationships of synthetic coumarins as HIV-1 inhibitors. Bioinorg. Chem. Appl. 2006, 2006, 68274:1–68274:9. [Google Scholar]
- Kostova, I. Coumarins as inhibitors of HIV reverse transcriptase. Curr. HIV Res. 2006, 4, 347–363. [Google Scholar]
- Mastrolorenzo, A.; Rusconi, S.; Scozzafava, A.; Barbaro, G.; Supuran, C.T. Inhibitors of HIV-1 protease: Current state of the art 10 years after their introduction. From antiretroviral drugs to antifungal, antibacterial and antitumor agents based on aspartic protease inhibitors. Curr. Med. Chem. 2007, 14, 2734–2748. [Google Scholar] [CrossRef]
- Kashman, Y.; Gustafson, K.R.; Fuller, R.W.; Cardellina, J.H., 2nd; McMahon, J.B.; Currens, M.J.; Buckheit, R.W., Jr.; Hughes, S.H.; Cragg, G.M.; Boyd, M.R. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum. J. Med. Chem. 1992, 35, 2735–2743. [Google Scholar]
- Patil, A.D.; Freyer, A.J.; Eggleston, D.S.; Haltiwanger, R.C.; Bean, M.F.; Taylor, P.B.; Caranfa, M.J.; Breen, A.L.; Bartus, H.R.; Johnson, R.K.; et al. The inophyllums, novel inhibitors of HIV-1 reverse transcriptase isolated from the Malaysian tree, Calophyllum inophyllum L. J. Med. Chem. 1993, 36, 4131–4138. [Google Scholar]
- Al-Mawsawi, L.Q.; Fikkert, V.; Dayam, R.; Witvrouw, M.; Burke, T.R., Jr.; Borchers, C.H.; Neamati, N. Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 10080–10085. [Google Scholar]
- Chiang, C.C.; Mouscadet, J.F.; Tsai, H.J.; Liu, C.T.; Hsu, L.Y. Synthesis and HIV-1 integrase inhibition of novel bis- or tetra-coumarin analogues. Chem. Pharm. Bull. 2007, 55, 1740–1743. [Google Scholar] [CrossRef]
- Uchiumi, F.; Hatano, T.; Ito, H.; Yoshida, T.; Tanuma, S. Transcriptional suppression of the HIV promoter by natural compounds. Antiviral Res. 2003, 58, 89–98. [Google Scholar] [CrossRef]
- Ma, T.; Gao, Q.; Chen, Z.; Wang, L.; Liu, G. Chemical resolution of (±)-calanolide A, (±)-cordatolide A and their 11-demethyl analogues. Bioorg. Med. Chem. Lett. 2008, 18, 1079–1083. [Google Scholar] [CrossRef]
- Kirkiacharian, S.; Thuy, D.T.; Sicsic, S.; Bakhchinian, R.; Kurkjian, R.; Tonnaire, T. Structure-activity relationships of some 3-substituted-4-hydroxycoumarins as HIV-1 protease inhibitors. Farmaco 2002, 57, 703–708. [Google Scholar] [CrossRef]
- Bedoya, L.M.; Beltran, M.; Sancho, R.; Olmedo, D.A.; Sanchez-Palomino, S.; del Olmo, E.; López-Pérez, J.L.; Muñoz, E.; San Feliciano, A.; Alcami, J. 4-Phenylcoumarins as HIV transcription inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4447–4450. [Google Scholar] [CrossRef]
- Márquez, N.; Sancho, R.; Bedoya, L.M.; Alcamí, J.; López-Pérez, J.L.; San Feliciano, A.; Fiebich, B.L.; Muñoz, E. Mesuol, a natural occurring 4-phenylcoumarin, inhibits HIV-1 replication by targeting the NF-kappaB pathway. Antiviral Res. 2005, 66, 137–145. [Google Scholar] [CrossRef]
- López-Pérez, J.L.; Olmedo, D.A.; del Olmo, E.; Vásquez, Y.; Solis, P.N.; Gupta, M.P.; San Feliciano, A. Cytotoxic 4-phenylcoumarins from the leaves of Marila pluricostata. J. Nat. Prod. 2005, 68, 369–373. [Google Scholar] [CrossRef]
- Mohanty, S.; Makrandi, J.K.; Grover, S.K. Phase-transfer catalyzed synthesis of 3-phenylcoumarins. Indian J. Chem. Sec. B 1989, 28, 766–767. [Google Scholar]
- Deschamps-Vallet, C.; Ilotse, J.B.; Meyer-Dayan, M. Transformation of the isoflavylium cation into 3-phenylcoumarins, isoflav-3-enes, and isoflavan. Tetrahedron Lett. 1983, 24, 3993–3996. [Google Scholar] [CrossRef]
- Hans, N.; Singhi, M.; Sharma, V.; Grover, S.K. A novel one-step synthesis of 3-phenyl-, 4-methyl-3-phenyl- and 3-phenyl-4-styrylcoumarins using DCC-DMSO. Indian J. Chem. Sec. B Org. Chem. Incl. Med. Chem. 1996, 35B, 1159–1162. [Google Scholar]
- Neelakantan, S.; Raman, P.V.; Tinabaye, A. A new and convenient synthesis of 4-methyl-3-phenyl-coumarins and 3-phenylcoumarins. Indian J. Chem. Sec. B 1982, 21, 256–257. [Google Scholar]
- Kappe, T.; Brandner, A. A simple synthesis of coumestrol. Z. Naturforsch. 1974, 29, 292–293. [Google Scholar]
- Stadlbauer, W.; Badawey, E.-S.; Hojas, G.; Roschger, P.; Kappe, T. Malonates in cyclocondensation reactions. Molecules 2001, 6, 338–352. [Google Scholar] [CrossRef]
- Sancho, R.; Medarde, M.; Sanchez-Palomino, S.; Madrigal, B.M.; Alcamí, J.; Munoz, E.; San Feliciano, A. Anti-HIV activity of some synthetic lignanolides and intermediates. Bioorg. Med. Chem. Lett. 2004, 14, 4483–4486. [Google Scholar] [CrossRef]
- Israel, N.; Gougerot-Pocidalo, M.A.; Aillet, F.; Virelizier, J.L. Redox status of cells influences constitutive or induced NF-kappa B translocation and HIV long terminal repeat activity in human T and monocytic cell lines. J. Immunol 1992, 149, 3386–3393. [Google Scholar]
- García-Pérez, J.; Sánchez-Palomino, S.; Pérez-Olmeda, M.; Fernández, B.; Alcamí, J. A new strategy based on recombinant viruses as a tool for assessing drug susceptibility of human immunodeficiency virus type 1. J. Med. Virol. 2007, 79, 127–137. [Google Scholar] [CrossRef]
- Mhiri, C.; Ladhar, F.; El Gharbi, R.; Le Bigot, Y. A convenient synthesis of 3-arylcoumarins from arylacetonitriles. Synth. Commun. 1999, 29, 1451–1461. [Google Scholar] [CrossRef]
- Kamat, S.P.; D’Souza, A.M.; Paknikar, S.K.; Beauchamp, P.S. A convenient one-pot synthesis of 4-methyl-3-phenyl-, 3-aryl- and 3-aryl-4-phenylcoumarins. J. Chem. Res. Synop. 2002, 2002, 242–246. [Google Scholar]
- Aparecido de Marchi, A.; Santos Castilho, M.; Barboni Nascimento, P.G.; Costa Archanjo, F.; del Ponte, G.; Oliva, G.; Tallarico Pupo, M. New 3-piperonylcoumarins as inhibitors of glycosomal glyceraldehyde-3-phosphate dehydrogenase (gGAPDH) from Trypanosoma cruzi. Bioorg. Med. Chem. 2004, 12, 4823–4833. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olmedo, D.; Sancho, R.; Bedoya, L.M.; López-Pérez, J.L.; Del Olmo, E.; Muñoz, E.; Alcamí, J.; Gupta, M.P.; San Feliciano, A. 3-Phenylcoumarins as Inhibitors of HIV-1 Replication. Molecules 2012, 17, 9245-9257. https://doi.org/10.3390/molecules17089245
Olmedo D, Sancho R, Bedoya LM, López-Pérez JL, Del Olmo E, Muñoz E, Alcamí J, Gupta MP, San Feliciano A. 3-Phenylcoumarins as Inhibitors of HIV-1 Replication. Molecules. 2012; 17(8):9245-9257. https://doi.org/10.3390/molecules17089245
Chicago/Turabian StyleOlmedo, Dionisio, Rocío Sancho, Luis M. Bedoya, José L. López-Pérez, Esther Del Olmo, Eduardo Muñoz, José Alcamí, Mahabir P. Gupta, and Arturo San Feliciano. 2012. "3-Phenylcoumarins as Inhibitors of HIV-1 Replication" Molecules 17, no. 8: 9245-9257. https://doi.org/10.3390/molecules17089245