Large-Scale Solvent-Free Chlorination of Hydroxy-Pyrimidines, -Pyridines, -Pyrazines and -Amides Using Equimolar POCl3

Abstract
:
1. Introduction
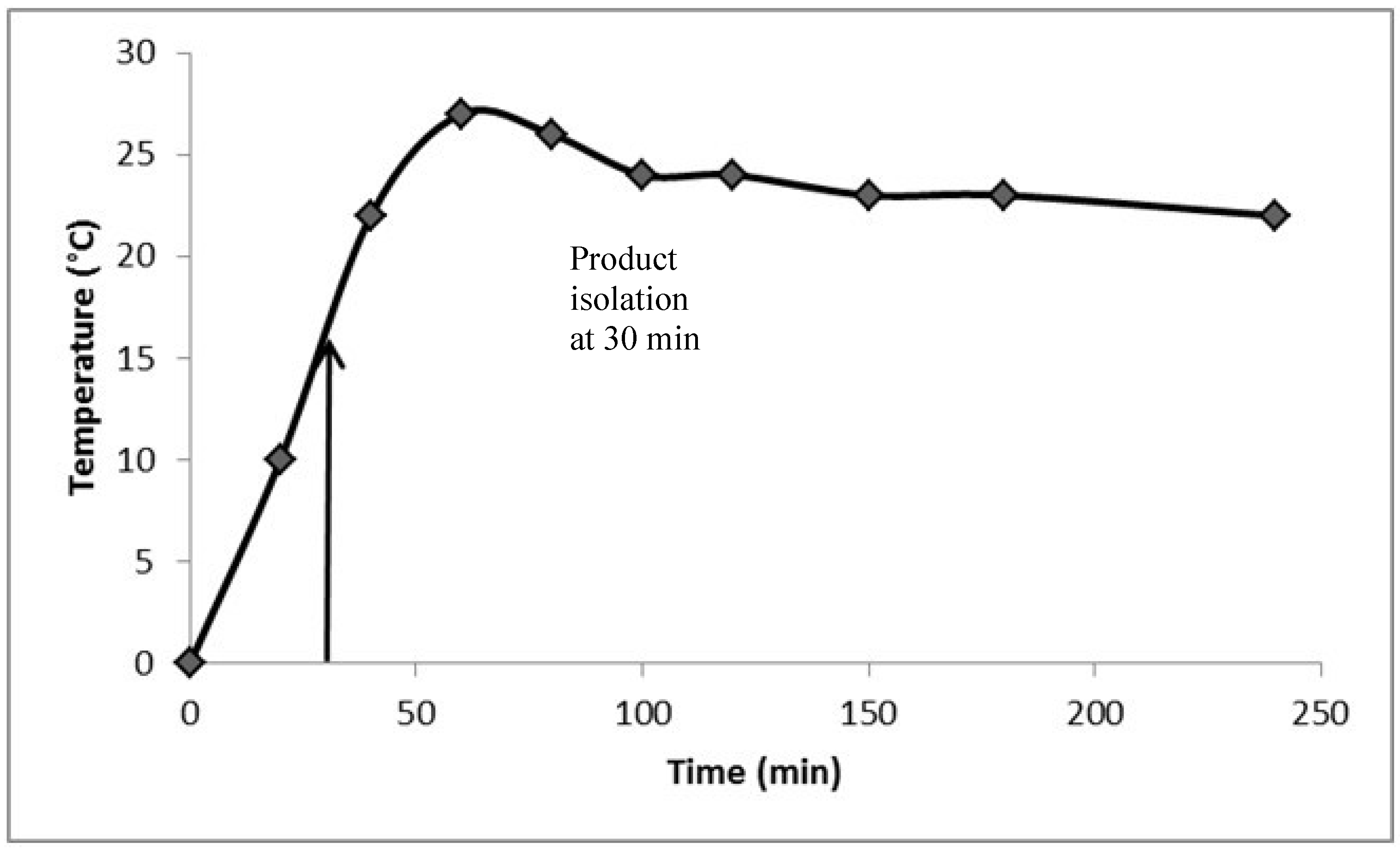
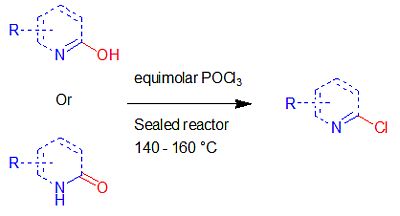
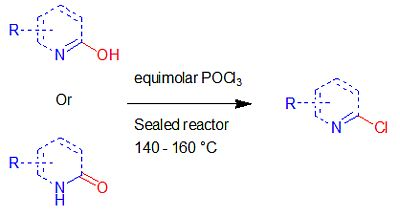
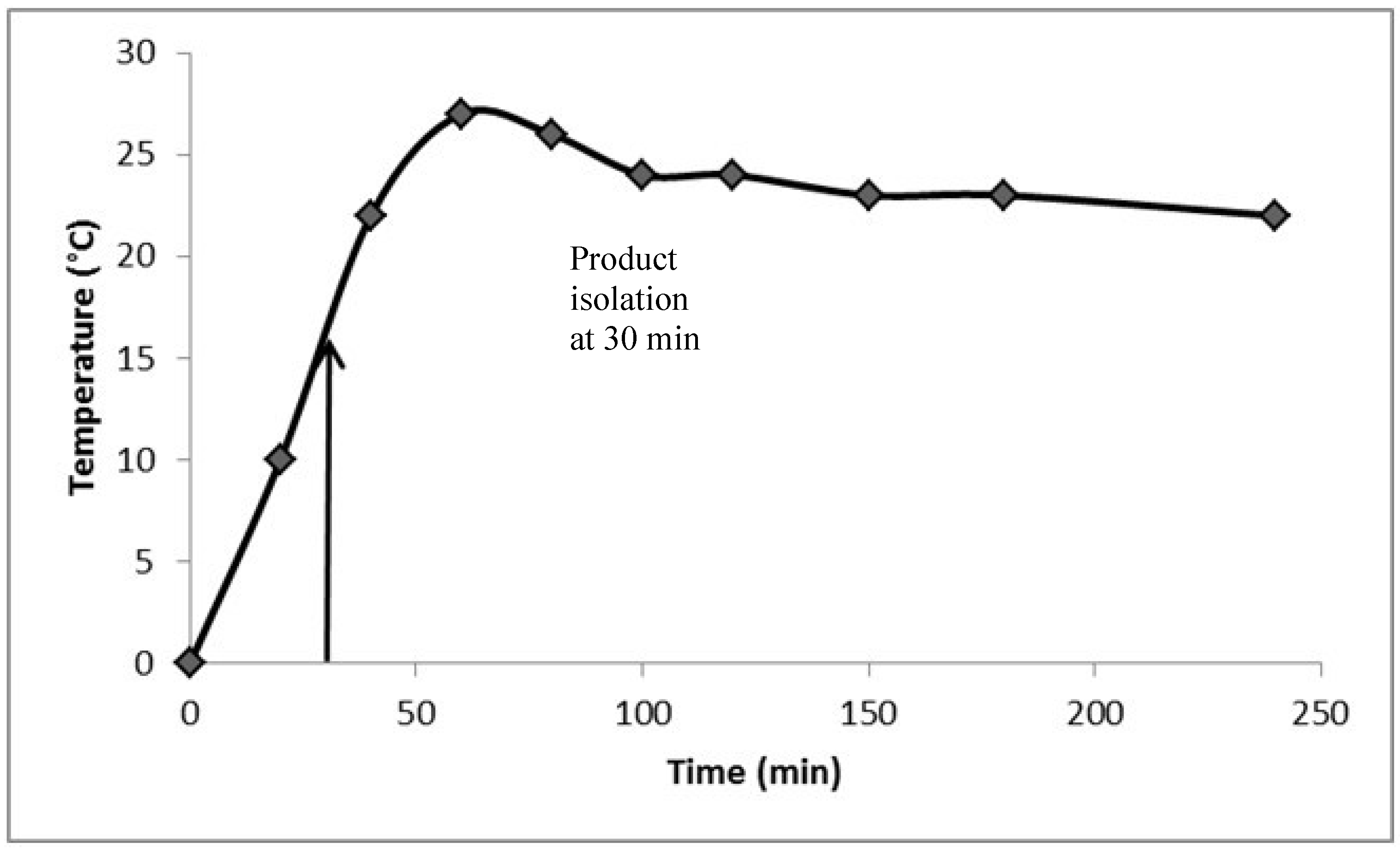
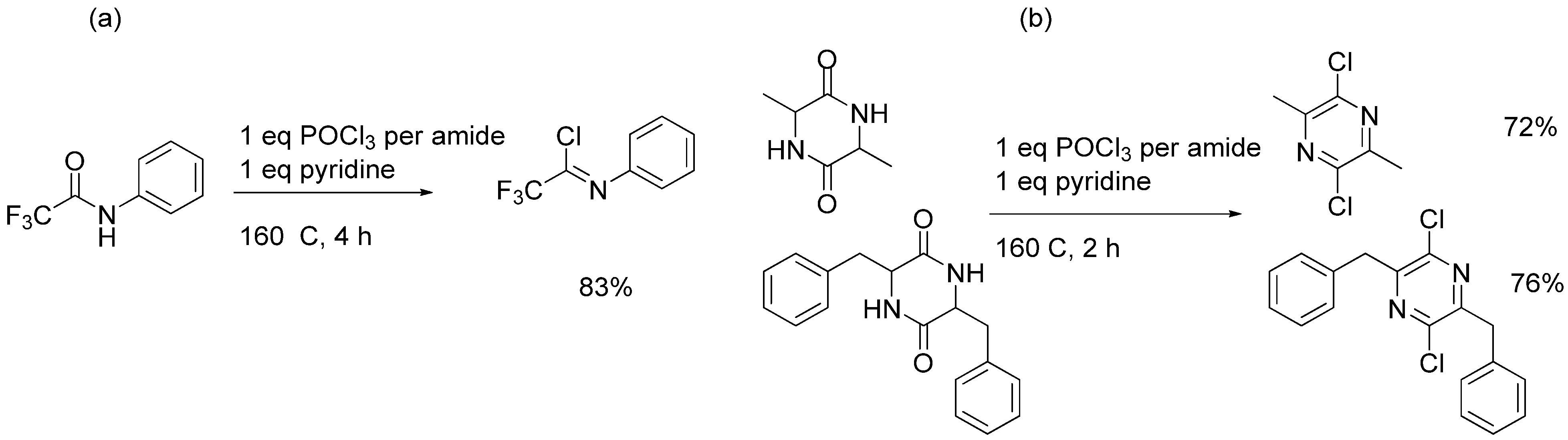
2. Results and Discussion


| Entry | Starting Material | Scale | Product | Yield | Lit. Yield (Scale, Time) |
|---|---|---|---|---|---|
| 1 |  | 57 g |  | 91% | 88% (10 g, 1.7 hours) [8] |
| 2 | 38 g | 88% | 92% (252 g, 20 hours) [9] | ||
| 3 | 44 g | 89% | 69% (25 g, 3 hours) [10] | ||
| 4 | 38 g | 85% | 27% (10 g, 10 hours) [11] | ||
| 5 | 38 g | 95% | 70% (10 g, 3 hours) [12] | ||
| 6 | 33 g | 83% | 59% (alternative reaction) [13] |

| Entry | Starting Material | Scale | Product | Yield | Lit. Yield (Scale, Time) |
|---|---|---|---|---|---|
| 1 |  | 87 g |  | 95% | 49–83% * [14,15] |
| 2 | 127 g | 92% | 66% * [16] | ||
| 3 | 70 g | 93% | 66%, 89% (12 g, 3 hours) [17,18] | ||
| 4 | 87 g | 91% | 52% * [19] | ||
| 5 | 87 g | 90% | 65% * [20] | ||
| 6 | 94 g | 91% | 48–50% * [21,22] | ||
| 7 | 109 g | 93% | 65% (3.1 g, overnight) [23] | ||
| 8 | 81 g | 88% | 94% (2 g, 24 hours) [24] |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Scale | Product | Yield | Lit. Yield (Scale, Time) |
|---|---|---|---|---|---|
| 1 |  | 44 g |  | 94% | 56% (60 g, 2 hours) [25] 88% (15 g, 0.7 hour) [26] |
| 2 | 49 g | 96% | 93% (64 g, 4 hours) [27] | ||
| 3 | 54 g | 86% | 40% (164 g, 0.5 hour) [28] | ||
| 4 | 73 g | 94% | 5% (6 g, 1.5 hours) [29] |

3. Experimental
3.1. General
3.2. Procedures for Chlorination of Pyrimidines, Benzo- and Pyrido-pyrazines, and Amides
3.3. Pyrimidine Products
3.4. Quinoxaline and Pyridopyrazine Products
3.5. Imidoylchloride and Pyrazine Products
3.6. Procedures for Chlorination of 2-Hydroxypyridines and 2,4-Dihydroxyquinoline
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Gabriel, S. Notizen über Brom-dihydrouracil. Chem. Ber. 1905, 38, 1689–1991. [Google Scholar]
- Stucky, G.; Imwinkelried, R. N-(2-amino-4,6-dichloro-pyrimidin-5-yl)formamide and process for its production. EP Patent 0,684,236, 24 June 1998. [Google Scholar]
- Whittaker, N.; Jones, T.S.G. A new synthesis and the chemical properties of 5-aminopyrimidine. J. Chem. Soc. 1951, 1565–1570. [Google Scholar]
- Achmatowicz, M.M.; Thiel, O.R.; Colyer, J.T.; Hu, J.; Elipe, M.V.S.; Tomaskevitch, J.; Tedrow, J.S.; Larsen, R.D. Hydrolysis of phosphoryl trichloride (POCl3): Characterization, in situ detection, and safe quenching of energetic metastable intermediates. Org. Proc. Res. Dev. 2010, 14, 1490–1500. [Google Scholar] [CrossRef]
- Hunds, A. Process for the preparation of chloropyrimidines. U.S. Patent 5,525,724, 11 June 1996. [Google Scholar]
- Leng, R.B.; Maurer, J.L.; Ringer, J.W. Chloropyrimidine Process. U.S. Patent 6,066,734, 23 May 2000. [Google Scholar]
- Sun, Z.; Wang, H.; Wen, K.; Li, Y.; Fan, E. Solvent-free or low-solvent large-scale preparation of chloropyrimidine and analogues. J. Org. Chem. 2011, 76, 4149–4153. [Google Scholar]
- Bader, H.; Downer, J.D.; Gibbons, D.; Mulholland, T.P.C.; Ward, G.; Brown, D.J.; Whittaker, N.; Spivey, A.M.; Osbond, J.M. Notes. J. Chem. Soc. 1953, 1641–1650. [Google Scholar]
- Andres, P.; Marhold, A. A new synthesis of 5-trifluoromethyluracil. J. Fluo. Chem. 1996, 77, 93–95. [Google Scholar]
- Krueger, E.B.; Rawson, T.E.; Burdick, D.J.; Liang, J.; Zhu, B.Y. Pyrimidine kinase inhibitors. WO 2008/079719 A1, 2008. [Google Scholar]
- Smith, V.H.; Christensen, B.E. Pyrimidines. v. dehalogenation and nuclear reduction of certain pyrimidines. J. Org. Chem. 1955, 20, 829–838. [Google Scholar] [CrossRef]
- Barany, F.; Pingle, M.; Bergstrom, D.S.; Giardina, F.; Arnold, L.D. Coferons and methods of making and using them. WO 2011/043817 A1, 2011. [Google Scholar]
- Minakawa, N.; Kojima, N.; Hikishima, S.; Sasaki, T.; Kiyosue, A.; Atsumi, N.; Ueno, Y.; Matsuda, A. New base pairing motifs. The synthesis and thermal stability of oligodeoxynucleotides containing imidazopyridopyrimidine nucleosides with the ability to form four hydrogen bonds. J. Am. Chem. Soc. 2003, 125, 9970–9982. [Google Scholar]
- Birch, M.; Sibley, G.E.M.; Law, D.; Oliver, J.D. Antifungal combination therapy. WO 2009/144473 A1, 2009. [Google Scholar]
- Nara, S.J.; Jha, M.; Brinkhorst, J.; Zemanek, T.J.; Pratt, D.A. A Simple cu-catalyzed coupling approach to substituted 3-pyridinol and 5-pyrimidinol antioxidants. J. Org. Chem. 2008, 73, 9326–9333. [Google Scholar]
- Zhou, Y.; Vourloumis, D.; Gregor, V.; Winters, E.G.; Hermann, T.; Ayida, B.; Sun, Z.; Murphy, D.; Simonsen, K. Antibacterial 3,5-diaminopiperidine-substituted aromatic and heteroaromatic compounds. WO 2005/028467 A1, 2005. [Google Scholar]
- Kokars, V.; Yanishevskii, A.; Kampars, V. Synthesis of 2-azastilbene derivatives with intramolecular charge transfer. Chem. Heterocycl. Compd. 2002, 38, 805–809. [Google Scholar]
- Kijima, A.; Sekiguchi, S. Alkaline hydrolysis of N-methyl-2,4-dinitroacetanilide and N-alkyl-N-(5-nitro-2-pyridyl)acetamides. Bull. Chem. Soc. Jpn. 1987, 60, 3597–3601. [Google Scholar]
- Capet, M.; Danvy, D.; Levoin, N.; Morvan, M.; Berrebi-Bertrand, I.; Calmels, T.; Robert, P.; Schwartz, J.C.; Lecomte, J.M. Arylpiperaszine derivatives, to the process for the production thereof and to the use thereof as therapeutic agents. U.S. Patenet 7,432,269 B2, 2008. [Google Scholar]
- Gros, P.C.; Elaachbouni, F. Bromine-lithium exchange under non-cryogenic conditions: TMSCH2Li-LiDMAE promoted C-2 lithiation of 2,3-dibromopyridine. Chem. Commun. 2008, 4813–4815. [Google Scholar]
- Pierrat, P.; Gros, P.; Fort, Y. Unusual t-buli induced ortholithiation versus halogen-lithium exchange in bromopyridines: Two alternative strategies for functionalization. Synlett 2004, 2319–2322. [Google Scholar]
- Cheng, J.; Xu, L.; Stevens, E.D.; Trudell, M.L.; Izenwasser, S.; Wade, D. Stereoselective synthesis of conformationally constrained tropane analogues: 6-Chloro-2,5-diazatetracyclo [8.5.0.02,13.04,9]pentadeca-4,6,8-triene-11-one and 6-Chloro-2,7-diazatetracyclo[8.5.0.02,13.04,9] pentadeca-4,6,8-triene-11-one. J. Heterocycl. Chem. 2004, 41, 569–574. [Google Scholar] [CrossRef]
- Chao, H.J.; Tuerdi, H.; Herpin, T.; Roberge, J.Y.; Liu, Y.; Lawrence, R.M.; Rehfuss, R.P.; Clark, C.G.; Qiao, J.X.; Gungor, T.; et al. Urea antagonists of p2y1 receptor useful in the treatment of thrombotic conditions. WO 2005/113511 A1, 2005. [Google Scholar]
- Collins, I.; Caldwell, J.J.; Oliver, A.W.; Raynham, T.M.; London, C.; Welsh, E.J.; Matijssen, C.A.J. WO 2009/053694 A1, 2009.
- Fischer, G.M.; Isomäki-Krondahl, M.; Göttker-Schnetmann, I.; Daltrozzo, E.; Zumbusch, A. Pyrrolopyrrole cyanine dyes: A new class of near-infrared dyes and fluorophores. Chem. Eur. J. 2009, 15, 4857–4864. [Google Scholar]
- Rangisetty, J.B.; Gupta, V.H.B.; Prasad, A.L.; Srinivas, P.; Sridhar, N.; Parimoo, P.; Veeranjaneyulu, A. Synthesis of new arylaminoquinoxalines and their antimalarial activity in mice. J. Pharm. Pharmcol. 2001, 53, 1409–1413. [Google Scholar]
- Galal, S.A.; Abdelsamie, A.S.; Tokuda, H.; Suzuki, N.; Lida, A.; ElHefnawi, M.M.; Ramadan, R.A.; Atta, M.H.E.; El Diwani, H.I. Part I: Synthesis, cancer chemopreventive activity and molecular docking study of novel quinoxaline derivatives. Eur. J. Med. Chem. 2011, 46, 327–340. [Google Scholar]
- Daub, W. Process for preparing 2,6-dicholoroquinoxaline. DE 3925969 A1, 1989. [Google Scholar]
- Milbank, J.B.J.; Pryde, D.C.; Tran, T.D. Hepatitis c virus inhibitors. WO Patent 2011/004276 A1, 2011. [Google Scholar]
- Huchel, U.; Tiwari, P.; Schmidt, R.R. N-Aryl-O-glycosyl haloacetimidates as glycosyl donors. J. Carbohydr. Chem. 2010, 29, 61–75. [Google Scholar]
- Ohta, A.; Akita, Y.; Hara, M. Syntheses and reactions of some 2,5-disubstituted pyrazine monoxides. Chem. Pharm. Bull. 1979, 27, 2027–2041. [Google Scholar]
- Ohta, A.; Kojima, A.; Aoyagi, Y. Emeheterone: Synthesis and structural revision. Heterocycles 1990, 31, 1655–1662. [Google Scholar] [CrossRef]
- Sample Availability: Electronic Supplementary Information (ESI) available: NMR spectra (1H, 13C, and 19F when applicable) of all compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, H.; Wen, K.; Wang, L.; Xiang, Y.; Xu, X.; Shen, Y.; Sun, Z. Large-Scale Solvent-Free Chlorination of Hydroxy-Pyrimidines, -Pyridines, -Pyrazines and -Amides Using Equimolar POCl3. Molecules 2012, 17, 4533-4544. https://doi.org/10.3390/molecules17044533
Wang H, Wen K, Wang L, Xiang Y, Xu X, Shen Y, Sun Z. Large-Scale Solvent-Free Chlorination of Hydroxy-Pyrimidines, -Pyridines, -Pyrazines and -Amides Using Equimolar POCl3. Molecules. 2012; 17(4):4533-4544. https://doi.org/10.3390/molecules17044533
Chicago/Turabian StyleWang, Han, Kun Wen, Le Wang, Ye Xiang, Xiaocheng Xu, Yongjia Shen, and Zhihua Sun. 2012. "Large-Scale Solvent-Free Chlorination of Hydroxy-Pyrimidines, -Pyridines, -Pyrazines and -Amides Using Equimolar POCl3" Molecules 17, no. 4: 4533-4544. https://doi.org/10.3390/molecules17044533
APA StyleWang, H., Wen, K., Wang, L., Xiang, Y., Xu, X., Shen, Y., & Sun, Z. (2012). Large-Scale Solvent-Free Chlorination of Hydroxy-Pyrimidines, -Pyridines, -Pyrazines and -Amides Using Equimolar POCl3. Molecules, 17(4), 4533-4544. https://doi.org/10.3390/molecules17044533