1D 13C-NMR Data as Molecular Descriptors in Spectra — Structure Relationship Analysis of Oligosaccharides

Abstract
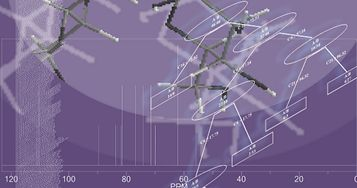
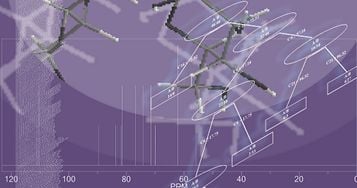
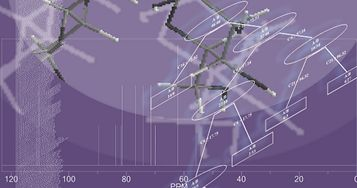
:
1. Introduction
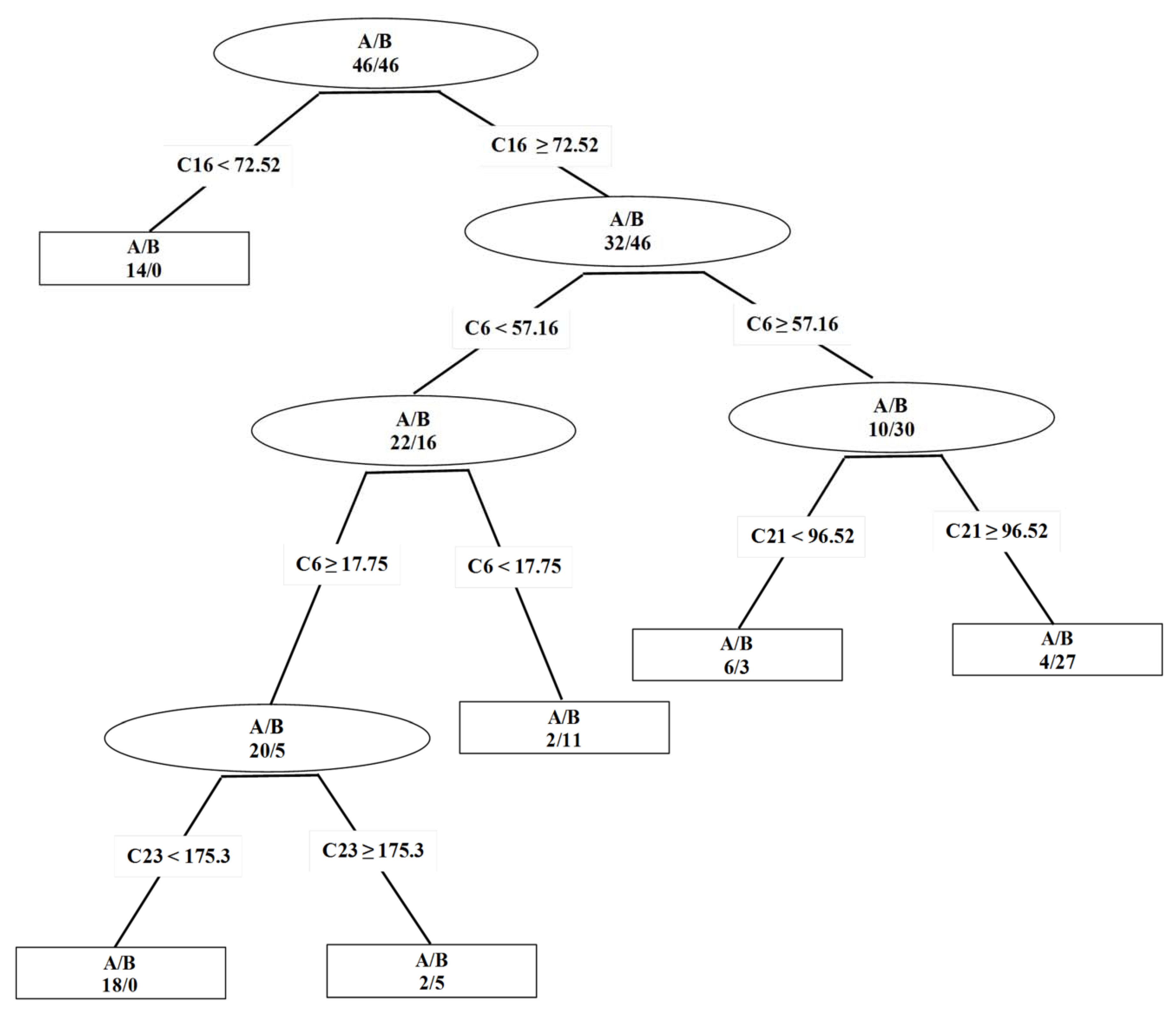
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| RF a | CT | CPGNN b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Training set / Test set | |||||||||||
| Model | Classes | Size | Correct pred. | Sensitivity c | Specificity d | Correct pred. | Sensitivity | Specificity | Correct pred. | Sensitivity | Specificity |
| Ano_F1 | A (α) | 46/12 | 33/6 | 0.71/0.50 | 0.75/0.67 | 43/7 | 0.93/0.58 | 0.78/0.58 | 27/5 | 0.59/0.42 | 0.59/0.56 |
| B (β) | 46/15 | 35/12 | 0.76/0.8 | 0.73/0.67 | 34/10 | 0.74/0.67 | 0.92/0.67 | 27/11 | 0.59/0.73 | 0.59/0.61 | |
| Ano_S2 | A (α) | 46/12 | 45/12 | 0.98/1 | 0.98/0.92 | 38/5 | 0.83/0.42 | 0.80/0.67 | 30/5 | 0.65/0.42 | 0.62/0.56 |
| B (β) | 46/15 | 45/14 | 0.98/0.93 | 0.98/1 | 37/10 | 0.81/0.50 | 0.82/0.59 | 28/11 | 0.61/0.73 | 0.64/0.61 | |
| Ano_R3 | A (α) | 46/16 | 34/12 | 0.74/0.75 | 0.77/0.92 | 38/11 | 0.83/0.69 | 0.93/1 | 23/8 | 0.5/ 0.5 | 0.74/0.89 |
| B (β) | 46/11 | 36/10 | 0.78/0.91 | 0.75/0.71 | 43/11 | 0.93/1 | 0.84/0.69 | 38/10 | 0.83/0.91 | 0.62/0.56 | |
| F_Link4 | A (1→2) | 33/13 | 30/10 | 0.91/0.77 | 0.83/1 | 28/10 | 0.85/0.77 | 0.78/0.62 | 8/4 | 0.24/0.31 | 0.53/0.8 |
| B (1→3) | 27/7 | 22/7 | 0.81/1 | 0.85/0.78 | 23/4 | 0.85/0.57 | 0.72/0.57 | 18/6 | 0.67/0.86 | 0.34/0.35 | |
| C (1→4) | 16/4 | 11/2 | 0.69/ 0.5 | 0.92/1 | 8/0 | 0.5/0 | 0.67/0 | 6/3 | 0.38/0.75 | 0.28/0.6 | |
| D (1→6) | 16/3 | 15/3 | 0.94/1 | 0.83/0.5 | 12/3 | 0.75/1 | 1/1 | 0/0 | 0/0 | 0/0 | |
| S_Link5 | A (1→2) | 8/1 | 1/0 | 0.12/0 | 1/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| B (1→3) | 17/12 | 9/7 | 0.53/0.58 | 1/1 | 12/8 | 0.70/0.67 | 0.86/1 | 7/6 | 0.41/0.5 | 0.32/0.86 | |
| C (1→4) | 51/13 | 50/13 | 0.98/1 | 0.74/0.72 | 47/11 | 0.92/0.85 | 0.75/0.73 | 39/12 | 0.76/0.92 | 0.57/0.6 | |
| D (1→6) | 16/1 | 13/1 | 0.81/1 | 0.93/0.5 | 13/1 | 0.81/1 | 0.87/0.25 | 0/0 | 0/0 | 0/0 | |
| Red_end6 | A (Glc) | 26/17 | 21/14 | 0.81/0.82 | 0.6/1 | 18/10 | 0.69/0.59 | 0.75/0.77 | 6/3 | 0.23/0.18 | 1/1 |
| B (Gal) | 18/5 | 8/5 | 0.44/1 | 0.5/0.71 | 8/2 | 0.44/0.4 | 0.89/1 | 6/2 | 0.33/0.4 | 0.17/0.14 | |
| C (Man) | 18/3 | 9/2 | 0.5/0.67 | 0.64/0.67 | 11/1 | 0.61/0.33 | 0.61/0.2 | 9/1 | 0.5/0.33 | 0.24/0.12 | |
| D (Rha) | 17/2 | 12/2 | 0.70/1 | 0.92/0.67 | 16/1 | 0.94/0.5 | 0.84/0.25 | 3/2 | 0.23/1 | 0.25/1 | |
| E (Fuc) | 13/0 | 10/0 | 0.77/--- | 0.71/--- | 13/0 | 1/--- | 0.59/--- | 1/0 | 0.08/--- | 1/--- | |
| M_residue7 | A (Glc) | 26/6 | 19/5 | 0.73/0.83 | 0.61/0.56 | 20/3 | 0.77/0.5 | 0.83/0.33 | 7/3 | 0.27/0.5 | 0.78/0.6 |
| B (Gal) | 19/5 | 9/4 | 0.47/0.8 | 0.64/0.8 | 8/1 | 0.42/0.2 | 0.53/0.17 | 11/2 | 0.58/0.4 | 0.34/0.2 | |
| C (Man) | 19/5 | 9/4 | 0.47/0.8 | 0.43/0.8 | 12/2 | 0.63/0.4 | 0.63/0.67 | 8/0 | 0.42/0 | 0.35/0 | |
| D (Rha) | 12/4 | 6/3 | 0.5/0.75 | 0.6/1 | 10/3 | 0.83/0.75 | 0.53/0.75 | 7/3 | 0.58/0.75 | 0.25/0.5 | |
| E (Fuc) | 16/7 | 9/4 | 0.56/0.57 | 0.56/0.8 | 11/4 | 0.69/0.57 | 0.73/0.8 | 0/1 | 0/0.14 | 0/1 | |
| F_residue8 | A (Glc) | 22/9 | 18/6 | 0.82/0.67 | 0.86/0.67 | 15/5 | 0.68/0.56 | 0.83/0.62 | 10/2 | 0.45/0.22 | 0.91/1 |
| B (Gal) | 18/3 | 14/2 | 0.78/0.67 | 0.82/0.28 | 11/1 | 0.61/0.33 | 0.69/0.17 | 8/2 | 0.44/0.67 | 0.38/0.17 | |
| C (Man) | 16/6 | 9/2 | 0.56/0.33 | 0.56/1 | 13/5 | 0.81/0.83 | 0.59/0.83 | 6/2 | 0.38/0.33 | 0.21/0.5 | |
| D (Rha) | 19/4 | 14/3 | 0.74/0.75 | 0.7/0.75 | 12/3 | 0.63/0.75 | 0.63/1 | 8/2 | 0.42/0.5 | 0.28/0.28 | |
| E (Fuc) | 17/5 | 15/3 | 0.88/0.6 | 0.83/0.6 | 14/3 | 0.82/0.6 | 0.82/0.75 | 1/1 | 0.06/0.2 | 0.33/0.5 | |
| Chain_Type | A (LT) | 39/8 | 29/7 | 0.74/0.88 | 0.83/0.88 | 37/8 | 0.95/1 | 0.80/0.67 | 26/6 | 0.67/0.75 | 0.81/1 |
| B (BT) | 53/19 | 47/18 | 0.89/0.95 | 0.82/0.95 | 44/15 | 0.83/0.79 | 0.96/1 | 47/19 | 0.89/1 | 0.78/0.90 | |
| Mean Predictability (%) a | |||||||
|---|---|---|---|---|---|---|---|
| Training set b | Test set | ||||||
| Model | RF | CT | CPGNN | RF | CT | CPGNN | |
| Anomeric Configurations | Ano_F1 | 73.91 | 83.70 | 58.70 | 65.00 | 62.5 | 57.50 |
| Ano_S2 | 97.83 | 81.52 | 63.04 | 96.67 | 54.17 | 57.50 | |
| Ano_R3 | 76.09 | 88.04 | 66.30 | 82.95 | 84.38 | 70.45 | |
| Linkage Types | F_Link4 | 83.72 | 73.76 | 32.10 | 81.73 | 58.52 | 47.87 |
| S_Link5 | 61.18 | 61.00 | 29.41 | 64.58 | 62.82 | 35.58 | |
| Residues | Red_end6 | 64.54 | 73.78 | 26.35 | 87.25 | 45.54 | 47.74 |
| M_residue7 | 54.81 | 66.85 | 37.05 | 75.10 | 48.43 | 41.25 | |
| F_residue8 | 75.55 | 71.21 | 35.08 | 60.33 | 61.44 | 43.06 | |
| Chain_Type | 81.52 | 88.94 | 77.67 | 91.12 | 89.47 | 87.50 | |

| Training set / Test set | ||||||
|---|---|---|---|---|---|---|
| Model | Classes | Size | Correct pred. | Sensitivity a | Specificity b | Mean Predictability c (%) |
| Ano_F1 | A (α) | 105/30 | 89/25 | 0.85/0.83 | 0.88/0.78 | 86.32/82.69 |
| B (β) | 99/39 | 87/32 | 0.88/0.82 | 0.84/0.86 | ||
| Ano_S2 | A (α) | 46/12 | 38/10 | 0.83/0.83 | 0.74/0.83 | 84.78/90 |
| B (β) | 46/15 | 33/13 | 0.72/0.87 | 0.80/0.87 | ||
| X (NA) | 112/42 | 112/42 | 1/1 | 1/1 | ||
| Ano_R3 | A (α) | 102/39 | 84/31 | 0.82/0.79 | 0.88/0.94 | 85.78/88.08 |
| B (β) | 102/30 | 91/28 | 0.89/0.93 | 0.83/0.78 | ||
| F_Link4 | A (1→2) | 33/13 | 30/10 | 0.91/0.77 | 0.79/1 | 84.99/85.38 |
| B (1→3) | 27/7 | 21/7 | 0.78/1 | 0.88/0.78 | ||
| C (1→4) | 16/4 | 11/2 | 0.69/0.5 | 0.85/0.67 | ||
| D (1→6) | 16/3 | 14/3 | 0.88/1 | 0.82/0.6 | ||
| X (NA) | 112/42 | 112/42 | 1/1 | 1/1 | ||
| S_Link5 | A (1→2) | 36/13 | 22/10 | 0.61/0.77 | 0.88/0.83 | 82.32/85.16 |
| B (1→3) | 48/21 | 38/15 | 0.79/0.71 | 0.84/0.94 | ||
| C (1→4) | 71/26 | 69/24 | 0.97/0.92 | 0.748/0.77 | ||
| D (1→6) | 49/9 | 45/9 | 0.92/1 | 0.98/0.9 | ||
| Red_end6 | A (Glc) | 72/38 | 60/33 | 0.83/0.87 | 0.71/0.75 | 75.70/74.96 |
| B (Gal) | 58/13 | 39/9 | 0.67/0.69 | 0.72/0.75 | ||
| C (Man) | 44/16 | 37/7 | 0.73/0.44 | 0.86/0.7 | ||
| D (Rha) | 17/2 | 12/2 | 0.70/1 | 0.92/0.67 | ||
| E (Fuc) | 13/0 | 11/0 | 0.87/--- | 0.69/--- | ||
| M_residue7 | A (Glc) | 26/6 | 20/5 | 0.77/0.83 | 0.69/0.56 | 62.27/79.25 |
| B (Gal) | 19/5 | 9/4 | 0.47/0.8 | 0.5/0.8 | ||
| C (Man) | 19/5 | 7/4 | 0.37/0.8 | 0.37/0.8 | ||
| D (Rha) | 12/4 | 6/3 | 0.5/0.75 | 0.67/1 | ||
| E (Fuc) | 16/7 | 10/4 | 0.62/0.57 | 0.59/0.8 | ||
| X (NA) | 112/42 | 112/42 | 1/1 | 1/1 | ||
| F_residue8 | A (Glc) | 74/33 | 65/31 | 0.88/0.94 | 0.86/0.76 | 79.64/67.50 |
| B (Gal) | 44/7 | 31/2 | 0.70/0.28 | 0.74/0.33 | ||
| C (Man) | 50/20 | 39/12 | 0.78/0.6 | 0.78/1 | ||
| D (Rha) | 19/4 | 14/3 | 0.74/0.75 | 0.67/0.75 | ||
| E (Fuc) | 17/5 | 15/4 | 0.88/0.8 | 0.88/0.67 | ||
| Chain_Type | A (LT) | 39/8 | 28/7 | 0.72/0.88 | 0.82/0.88 | 86.82/94.08 |
| B (BT) | 53/19 | 47/18 | 0.89/0.95 | 0.81/0.95 | ||
| X (NA) | 112/42 | 112/42 | 1/1 | 1/1 | ||
| Model | RF | CT | ||
|---|---|---|---|---|
| Trisaccharides | Di- and trisaccharides | |||
| Ano_F1 | C12; C11; C9; C22; C14; C13; C23; C15; C16; C17 | C23; C19; C20; C12; C15; C18; C21; C13; C11; C22 | C12; C9 (2×); C21 | |
| Ano_S2 | C15; C14; C13; C22; C16; C6; C10; C23; C17; C7 | C12; C6; C10; C9; C8; C11; C7; C21; C13; C14 | C14; C6; C22 | |
| Ano_R3 | C16; C6; C21; C10; C14; C18; C11; C17; C5; C8 | C22; C18; C16; C20; C19; C17; C14; C23; C21; C6 | C16; C6 (2×); C21; C2 | |
| F_Link4 | C8; C7; C20; C6; C19; C23; C10; C14; C16; C11 | C8; C7; C6; C10; C21; C12; C11; C9; C20; C13 | C8; C20; C7; C11; C6; C17 | |
| S_Link5 | C8; C7; C19; C6; C22; C20; C5; C18; C9; C23 | C21; C13; C14; C15; C8; C22; C20; C12; C23; C19 | C8 (2×); C22; C17 | |
| Red_end6 | C6; C5; C22; C19; C20; C7; C9; C10; C14; C18 | C22; C15; C14; C16; C20; C13; C19; C21; C18; C17 | C6; C12; C5; C20; C23 | |
| M_residue7 | C6; C7; C15; C5; C16; C11; C23; C9; C10; C18 | C10; C7; C6; C9; C8; C12; C11; C21; C15; C5 | C16; C6 (2×); C7; C18; C23 | |
| F_residue8 | C7; C5; C6; C10; C15; C16; C8; C9; C11; C21 | C14; C12; C16; C15; C23; C17; C20; C5; C21; C13 | C16; C9 (2×); C7; C8; C5 | |
| Chain_Type | C7; C23; C20; C14; C5; C8; C6; C21; C18; C12 | C8; C21; C7; C6; C9; C10; C11; C12; C14; C23 | C7; C5; C23 (2×); C8 | |
3. Experimental
3.1. Data Set and Descriptors
3.2. Selection of Training and Test Sets for the Trisaccharides Model
3.3. Random Forest (RF) [33,34,35]
3.4. Classification Tree (CT) [35,38]
3.5. Counterpropagation Neural Network (CPGNN) [39]
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Herget, S.; Toukach, P.V.; Ranzinger, R.; Hull, W.E.; Knirel, Y.A.; von der Lieth, C-W. Statistical analysis of the bacterial carbohydrate structure data base (BCSDB): Characteristics and diversity of bacterial carbohydrates in comparison with mammalian glycans. BMC Struct. Biol. 2008, 8, 35. [Google Scholar] [CrossRef]
- Bubb, W.A. NMR spectroscopy in the study of carbohydrates: Characterizing the structural complexity. Concepts Magn. Reson. 2003, 19A, 1–19. [Google Scholar]
- Duus, J.Ø.; Gotfredsen, C.H.; Bock, K. Carbohydrate structural determination by NMR spectroscopy: Modern methods and limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef]
- Vliegenthart, J.F.G.; Woods, R.J. NMR Spectroscopy and Computer Modeling of Carbohydrates—Recent Advances; American Chemical Society: Washington, DC, USA, 2006; pp. 1–19. [Google Scholar]
- Toukash, F.V.; Shashkov, A.S. Computer-assisted structural analysis of regular glycopolymers on the basis of 13C-NMR data. Carbohydr.Res. 2001, 335, 101–114. [Google Scholar]
- Maes, E.; Bonachera, F.; Strecker, G.; Guerardel, Y. SOACS index: An easy NMR-based query for glycan retrieval. Carbohydr.Res. 2009, 344, 322–330. [Google Scholar]
- Pereira, F. Prediction of the anomeric configuration, type of linkage, and residues in disaccharides from 1D 13C-NMR data. Carbohydr.Res. 2011, 346, 960–972. [Google Scholar] [CrossRef]
- Jansson, P.E.; Kenne, L.; Wildmalm, G. CASPER—A Computer program used for structural analysis of carbohydrates. J. Chem. Inf. Comput. Sci. 1991, 31, 508–516. [Google Scholar]
- Stenutz, R.; Erbing, B.; Widmalm, G.; Jansson, P.E.; Nimmich, W. The structure of the capsular polysaccharide from klebsiella type 52, using the computerised approach CASPER and NMR spectroscopy. Carbohydr.Res. 1997, 302, 79–84. [Google Scholar]
- Stenutz, R.; Jansson, P.E.; Widmalm, G. Computer-assisted structural analysis of oligo- and polysaccharides: An extension of CASPER to multibranched structures. Carbohydr.Res. 1998, 306, 11–17. [Google Scholar]
- Jansson, P.E.; Stenutz, R.; Widmalm, G. Sequence determination of oligosaccharides and regular polysaccharides using NMR spectroscopy and a novel web-based version of the computer program CASPER. Carbohydr.Res. 2006, 341, 1003–1010. [Google Scholar]
- CASPER website. Available online: http://www.casperold.organ.su.se/casper/ (accessed on 23 February 2012).
- Hiltunen, Y.; Heiniemi, E.; Ala-Korpela, M. Lipoprotein-lipid quantification by neural-network analysis of 1H-NMR data from human blood plasma. J. Magn. Reson. B 1995, 106, 191–194. [Google Scholar]
- Bienfait, B. Applications of high-resolution self-organizing maps to retrosynthetic and QSAR analysis. J. Chem. Inf. Comput. Sci. 1994, 34, 890–898. [Google Scholar]
- Novic, M.; Zupan, J. Investigation of infrared spectra-structure correlation using kohonen and counterpropagation neural network. J. Chem. Inf. Comput. Sci. 1995, 35, 454–466. [Google Scholar]
- Munk, M.E.; Madison, M.S.; Robb, E.W. The neural network as a tool for multispectral interpretation. J. Chem. Inf. Comput. Sci. 1996, 36, 231–238. [Google Scholar]
- Rufino, A.R.; Brant, A.J.C.; Santos, J.B.O.; Ferreira, M.J.P.; Emerenciano, V.P. Simple method for identification of skeletons of aporphine alkaloids from 13C-NMR data using artificial neural networks. J. Chem. Inf. Comput. Sci. 2005, 45, 645–651. [Google Scholar]
- Emerenciano, V.P.; Alvarenga, S.A.V.; Scotti, M.T.; Ferreira, M.J.P.; Stefani, R.; Nuzillard, J.M. Automatic identification of terpenoid skeletons by feed-forward neural networks. Anal.Chim. Acta 2006, 579, 217–226. [Google Scholar]
- Dominik, M. NeuroCarb: Artificial neural networks for NMR structure elucidation of oligosaccharides.
- Shashkov, A.S.; Nifanťev, N.E.; Amochaeva, V.Y.; Kochetkov, N.K. 1H and 13C-NMR data for 2-O-, 3-O-and 2,3-di-O-glycosylated methyl α- and β-D-galactopyranosides. Magn.Reson. Chem. 1993, 31, 599–605. [Google Scholar]
- Usui, T.; Mizuno, T.; Kato, K.; Tomoda, M. 13C-NMR spectra of gluco-mamnooligosaccharides and structurally related glucomannan. Agric. Biol. Chem. 1979, 43, 863–865. [Google Scholar]
- Baumann, H.; Erbing, B.; Jansson, P.E.; Kenne, L. NMR and conformational studies of some 3-O, 4-O-, and 3,4-di-O-glycopyranosyl-substituted methyl α-D-galactopyranosides. J. Chem. Soc. Perkin Trans. 1989, 1, 2153–2165. [Google Scholar]
- Usui, T.; Yamaoka, N.; Matsuda, K.; Tuzimura, K.; Sugiyama, H.; Seto, S. 13C-Nuclear magnetic resonance spectra of glucobioses, glucotrioses, and glucans. J. Chem. Soc. Perkin Trans. 1973, 1, 2425–2432. [Google Scholar]
- Jansson, P.E.; Kjellberg, A.; Rundlölf, T.; Widmalm, G. Synthesis, NMR spectroscopy and conformational studies of two vicinally disubstituted trisaccharides. J. Chem. Soc. Perkin Trans. 1996, 2, 33–37. [Google Scholar]
- Baumann, H.; Erbing, B.; Jansson, P.E.; Kenne, L. Synthesis, NMR, and conformational studies of some 3,4-di-O-glycopyranosyl- substituted methyl α-D-galactopyranosides. J. Chem. Soc. Perkin Trans. 1989, 1, 2167–2168. [Google Scholar]
- Roslund, M.U.; Säwén, E.; Landström, J.; Rönnols, J.; Jonsson, K.H.M.; Lundborg, M.; Svensson, M.V.; Widmalm, G. Complete 1H and 13C-NMR chemical shift assignments of mono-, di-, and trisaccharides as basis for NMR chemical shift predictions of polysaccharides using the computer program CASPER. Carbohydr. Res. 2011, 346, 1311–1319. [Google Scholar] [CrossRef]
- Urashima, T.; Bubb, W.A.; Messer, M.; Tsuji, Y.; Taneda, Y. Studies of the neutral trisaccharides of goat (Capra hircus) colostrum and of the one- and two-dimensional 1H and 13C-NMR spectra of 6'-N-acetylglucosaminyllactose. Carbohydr.Res. 1994, 262, 173–184. [Google Scholar] [CrossRef]
- Bock, K.; Duus, J.Ø.; Norman, B.; Pedersen, S. Assignment of structures to oligosaccharides produced by enzymic degradation of a β-D-glucan from barley by 1H- and 13C-NMR spectroscopy. Carbohydr. Res. 1991, 211, 219–233. [Google Scholar]
- Flugge, L.A.; Blank, J.T.; Petillo, P.A. Isolation, modification, and NMR assignments of a series of cellulose oligomers. J. Am. Chem. Soc. 1999, 121, 7228–7238. [Google Scholar]
- Jansson, P.E.; Stenutz, R.; Widmalm, G. Sequence determination of oligosaccharides and regular polysaccharides using NMR spectroscopy and a novel web-based version of the computer program CASPER. Carbohydr. Res. 2006, 341, 1003–1010. [Google Scholar]
- Loß, A.; Stenutz, R.; Schwarzer, E.; von der Lieth, C.W. GlyNest and CASPER: Two independent approaches to estimate 1H and 13C NMR shifts of glycans available through a common web-Interface. Nucleic Acids Res. 2006, 34, W733–W737. [Google Scholar]
- Kohonen, T. Self-Organization and Associative Memory; Springer: Berlin, Germany, 1988. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar]
- Zhang, Q.Y.; Aires-de-Sousa, J. Random forest prediction of mutagenicity from empirical physicochemical descriptors. J. Chem. Inf. Model. 2007, 47, 1–8. [Google Scholar]
- R Development Core Team. R. A language and environment for statistical computing. ISBN 3-900051-07.-0; R Foundation for Statistical Computing: Vienna, Austria, 2004. Available online: http://www.r-project.org/ (accessed on 23 February 2012).
- Liaw, A.; Weiner, M. randomForest (R software for random forest). Fortran original (Breiman,L.; Cutler,A.),R port (Liaw,A.; Wiener,M.). Version 4.6-6. Available online: http://stat-www.berkeley.edu/users/breiman/RandomForests/ (accessed on 23 February 2012).
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees; Chapman & Hall/CRC: Boca Raton, FL, USA, 2000. [Google Scholar]
- Zupan, J.; Gasteiger, J. Neural Networks in Chemistry and Drug Design; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Aires-de-Sousa, J. JATOON: Java tools for neural networks. Chemometr.Intell. Lab. Syst. 2002, 61, 167–173. [Google Scholar]
- JATOON applets. Available online: http://joao.airesdesousa.com/jatoon/ (accessed on 23 February 2012).
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pereira, F. 1D 13C-NMR Data as Molecular Descriptors in Spectra — Structure Relationship Analysis of Oligosaccharides. Molecules 2012, 17, 3818-3833. https://doi.org/10.3390/molecules17043818
Pereira F. 1D 13C-NMR Data as Molecular Descriptors in Spectra — Structure Relationship Analysis of Oligosaccharides. Molecules. 2012; 17(4):3818-3833. https://doi.org/10.3390/molecules17043818
Chicago/Turabian StylePereira, Florbela. 2012. "1D 13C-NMR Data as Molecular Descriptors in Spectra — Structure Relationship Analysis of Oligosaccharides" Molecules 17, no. 4: 3818-3833. https://doi.org/10.3390/molecules17043818