Lipases as Tools in the Synthesis of Prodrugs from Racemic 9-(2,3-Dihydroxypropyl)adenine

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of Lipases
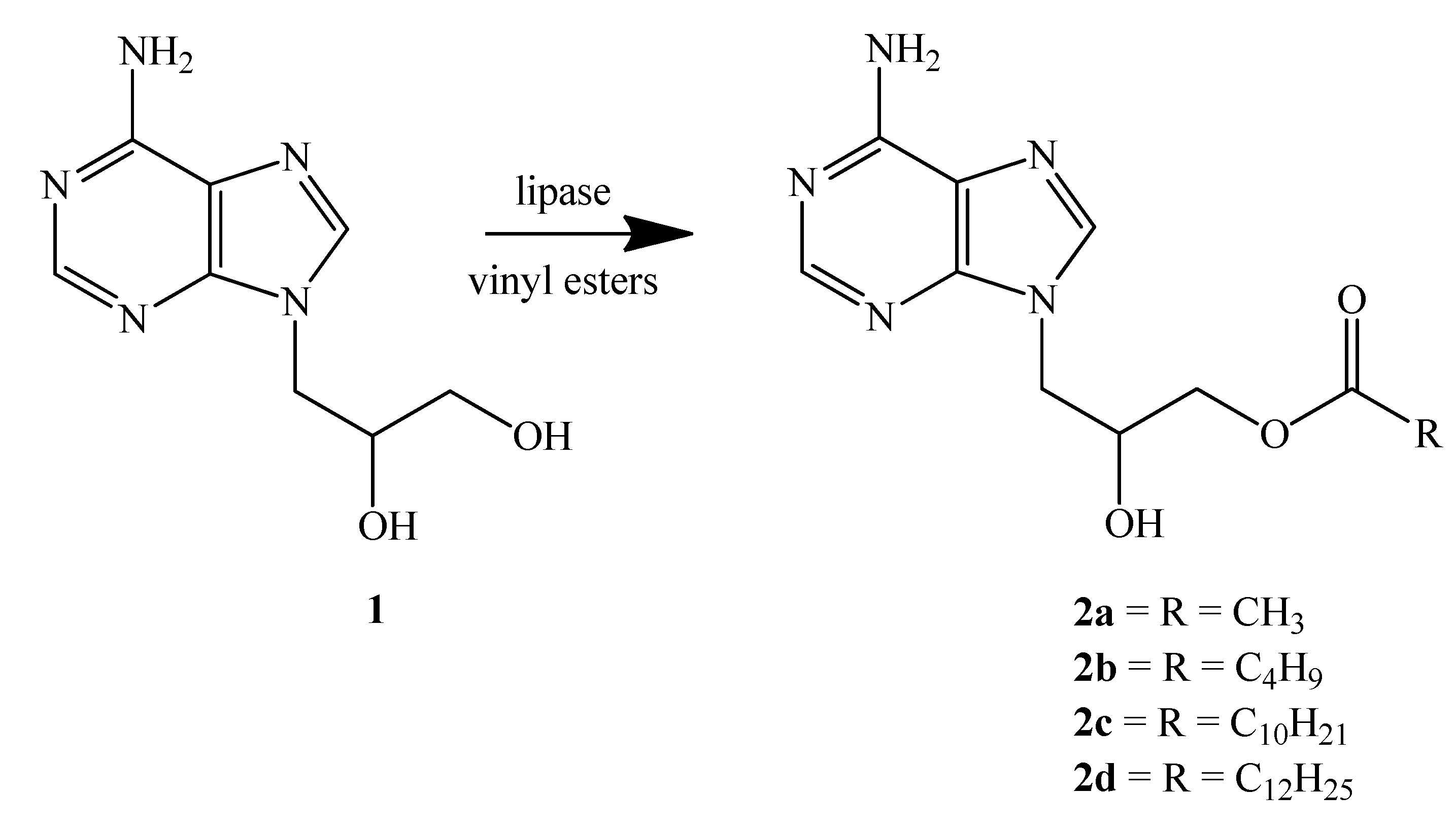
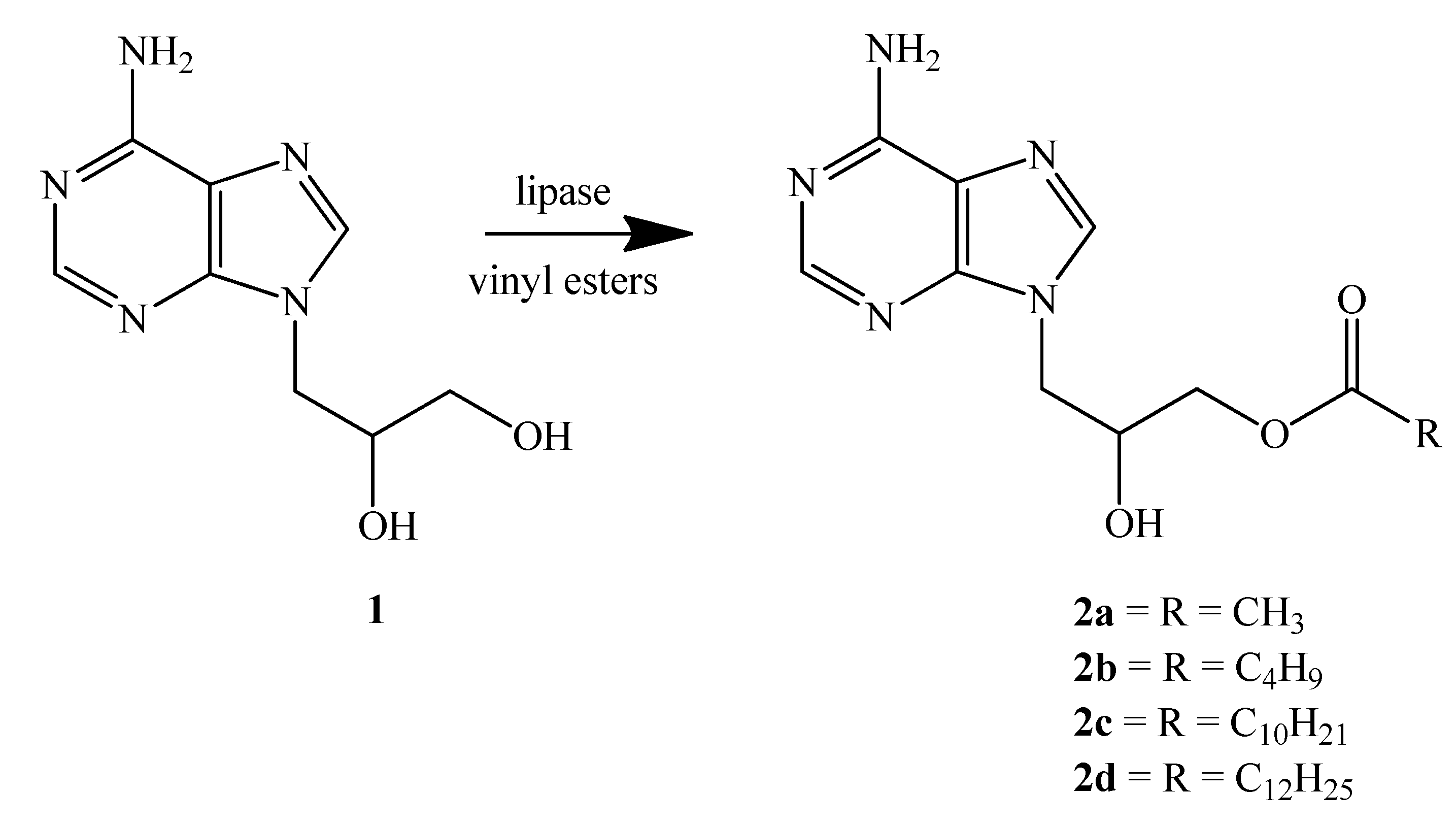
2.2. Lipases from G. Candidum and Their Immobilization

{kind=link}
| Enzyme | Protein content of free lipase solution before immobilization (mg/mL) | Protein content of free lipase solution after immobilization (mg/mL) | Protein loading yield (%) | Specificactivity of lipase solution before immobilization (U/mg protein) | Specific activity of immobilized lipase (U/g chitosan beads) |
|---|---|---|---|---|---|
| Extracellular lipase | 2.82 | 1.86 | 33 | 0.145 | 0.052 |
| Released lipase | 9.16 | 4.70 | 48.6 | 0.291 | 0.125 |
2.3. Enzymatic Transesterification
| Source of lipase | Vinyl ester * | Chemical yield (%) |
|---|---|---|
| Geotrichum candidum–acetone powder | VA | 49 |
| VB | 37 | |
| VD | 30 | |
| VL | 28 | |
| Geotrichum candidum–chitosan beads (extracellular lipase) | VA | 25 |
| VB | 20 | |
| VD | 17 | |
| VL | 8 | |
| Geotrichum candidum–chitosan beads (cell-bound lipase) | VA | 20 |
| VB | 13 | |
| VD | 13 | |
| VL | 10 | |
| Candida antarctica | VA | 66 |
| VB | 46 | |
| VD | 40 | |
| VL | 25 | |
| Aspergillus niger | VA | 42 |
| VB | 40 | |
| VD | 38 | |
| VL | 30 | |
| Pseudomonas fluorescens | VA | 51 |
| VB | 72 | |
| VD | 49 | |
| VL | 42 | |
| Hog pancreas | VA | 58 |
| VB | 49 | |
| VD | 36 | |
| VL | 28 |
2.4. Reusability of Immobilized Lipases from G. candidum
3. Experimental
3.1. Microorganism and Chemicals
3.2. Preparation of Lipases from Geotrichum candidum 4013 [19]
3.3. Extraction of Enzyme from the Cell Wall—Preparation of Released Lipase [8,21]
3.4. Immobilization of Lipases On Chitosan Beads [19,35]
3.5. Enzyme Activity Assay
| Lipase from | Activity declared by Sigma a | Activity determination spectrophotometrically b |
|---|---|---|
| Aspergillus niger | 4 U/g | 2.1 U/g |
| Candida antarctica | 3.0 U/mg | 2.62 U/g |
| Pseudomonas fluorescens | 42.5 U/mg | 1.8 U/g |
| Hog pancreas | 2.4 U/mg | 0.569 U/g |
| Geotrichum candidum-acetone powder | - | 0.083 U/g |
3.6. Transesterification
3.7. Analytical Methods
Preparative TLC
4. Conclusions
Acknowledgments
References
- Krishna, S.H.; Persson, M.; Bornscheuer, U.T. Enantioselective transesterification of a tertiary alcohol by lipase A from Candida antarctica. Tetrahedron: Asymmetry 2002, 13, 2693–2696. [Google Scholar] [CrossRef]
- Chenevert, R.; Pelchat, N.; Jacques, F. Stereoselective enzymatic acylations (transesterifications). Curr. Org. Chem. 2006, 10, 1067–1094. [Google Scholar] [CrossRef]
- Paravidino, M.; Hanefeld, U. Enzymatic acylation: Assessing the greenness of different acyl donors. Green Chem. 2011, 13, 2651–2657. [Google Scholar] [CrossRef]
- Bizerra, A.M.C.; Montenegro, T.G.C.; Lemos, T.L.G.; Oliveira, M.C.F.; Mattos, M.C.; Lavandera, I.; Gotor-Fernández, V.; Gonzalo, G.; Gotor, V. Enzymatic regioselective production of chloramphenicol esters. Tetrahedron 2011, 67, 2858–2862. [Google Scholar]
- Stella, V.J.; Charman, W.N.A.; Naringrekar, V.H. Prodrugs: Do they have advantages in clinical practice? Drugs 1985, 29, 455–473. [Google Scholar] [CrossRef]
- Warren, M.S.; Rautio, J. Prodrugs Designed to target transporters for oral drug delivery. In Prodrugs and Targeted Delivery, 1st; Rautio, J., Ed.; Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2011; Volume 47, pp. 133–153. [Google Scholar]
- Stránský, K.; Zarevúcka, M.; Kejík, Z.; Wimmer, Z.; Macková, M.; Demnerová, K. Substrate specifity, regioselectivity and hydrolytic activity of lipases activated from Geotrichum sp. Biochem. Eng. J. 2007, 34, 209–216. [Google Scholar] [CrossRef]
- Hlavsová, K.; Zarevúcka, M.; Wimmer, Z.; Macková, M.; Sovová, H. Geotrichum candidum 4013: Extracellular lipase versus cell-bound lipase from the single strain. J. Mol. Catal. B-Enzym. 2009, 61, 188–193. [Google Scholar]
- Krečmerová, M. Nucleoside and nucleotide Analogues for the Treatment of Herpesvirus Infections: Current Stage and New Prospects in the Field of Acyclic Nucleoside Phosphonates. In Herpesviridae—A Look into This Unique Family of Viruses; George, D., Magel, S.T., Eds.; Intech: Rijeka, Croatia, 2012; pp. 245–270. [Google Scholar]
- De Clercq, E.; Descamps, J.; De Somer, P.; Holý, A. (S)-9-(2,3-Dihydroxypropyl)adenine: An Aliphatic Nucleoside Analog with Broad-Spectrum Antiviral Activity. Science 1978, 200, 563–565. [Google Scholar]
- De Clercq, E.; Holý, A. Alkyl Esters of 3-Adenin-9-yl-2-hydroxypropanoic Acid: A New Class of Broad-spectrum Antiviral Agents. J. Med. Chem. 1985, 28, 282–287. [Google Scholar] [CrossRef]
- Holý, A.; Votruba, I.; De Clercq, E. Studies on S-adenosyl-L-homocysteine hydrolase. 5. Synthesis and antiviral activity of stereoisomeric eritadenines. Collect. Czech. Chem. Commun. 1982, 47, 1392–1407. [Google Scholar] [CrossRef]
- Toida, J.; Arikawa, Y.; Kondou, K.; Fukuzawa, M.; Sekiguchi, J. Purification and characterization of triacylglycerol lipase from Aspergillus oryzae. Biosci. Biotechnol. Biochem. 1998, 62, 759–763. [Google Scholar] [CrossRef]
- Berger, M.; Schneider, M. Lipase catalyzed preparation of isomerically pure monoglycerides and diglycerides. Biol. Chem. Hoppe-Seyler 1991, 372, 526–526. [Google Scholar]
- Berger, M.; Schneider, M. Regioselectivity of lipases in organic-solvents. Biotechnol. Lett. 1991, 13, 333–338. [Google Scholar] [CrossRef]
- Trodler, P.; Nieveler, J.; Rusnak, M.; Schmid, R.D.; Pleiss, J. Rational design of a new one-step purification strategy for Candida antarctica lipase B by ion-exchange chromatography. J. Chromatogr. A 2008, 1179, 161–167. [Google Scholar] [CrossRef]
- Rangheard, M.S.; Langrand, G.; Triantaphylides, C.; Baratti, J. Multi-competitive enzymatic-reactions in organic media — A simple test for the determination of lipase fatty-acid specificity. Biochim. Biophys. Acta 1989, 1004, 20–28. [Google Scholar] [CrossRef]
- Brabcová, J.; Zarevúcka, M.; Macková, M. Difference in hydrolytic activities of two crude lipases from Geotrichum candidum 4013. Yeast 2010, 27, 1029–1038. [Google Scholar] [CrossRef]
- Zarevúcka, M.; Kejík, Z.; Šaman, D.; Wimmer, Z.; Demnerová, K. Enantioselective properties of induced lipases from Geotrichum. Enzyme Microb. Technol. 2005, 37, 481–486. [Google Scholar] [CrossRef]
- Hlavsová, K.; Wimmer, Z.; Xanthakis, E.; Bernášek, P.; Sovová, H.; Zarevúcka, M. Lipase activity enhancement by SC-CO2 treatment. Z. Naturforsch. B: Chem. Sci. 2008, 63, 779–784. [Google Scholar]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilization. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef]
- Theil, F.; Weidner, J.; Ballschuh, S.; Kunath, A.; Schick, H. Kinetic resolution of acyclic 1,2-diols using a sequential lipase-catalyzed transesterification in organic solvents. J. Org. Chem. 1994, 59, 388–393. [Google Scholar] [CrossRef]
- Henegar, K.E.; Ashford, S.W.; Baughman, T.A.; Sih, J.C.; Gu, R.L. Practical asymmetric synthesis of (S)-4-ethyl-7,8-dihydro-4- hydroxy-1H-pyrano[3,4-f]indolizine- 3,6,10(4H)-trione, a key intermediate for the synthesis of irinotecan and other camptothecin analogs. J. Org. Chem. 1997, 62, 6588–6597. [Google Scholar]
- Chênevert, R.; Simard, M.; Bergeron, M.; Dasser, M. Chemoenzymatic formal synthesis of (S)-(−)-phosphonotrixin. Tetrahedron: Asymmetry 2004, 15, 1889–1892. [Google Scholar] [CrossRef]
- Serra, S. Lipase-mediated resolution of substituted 2-aryl-propanols: Application to the enantioselective synthesis of phenolic sesquiterpenes. Tetrahedron: Asymmetry 2011, 22, 619–628. [Google Scholar] [CrossRef]
- Djadchenko, M.A.; Pivnitsky, K.K.; Theil, F.; Schick, H. Enzymes in organic synthesis. Part 3. Synthesis of enantiomerically pure prostaglandin intermediates by enzyme-catalyzed transesterification of (1SR,2RS,5SR,6RS)-bicyclo[3.3.0]octane-2,6-diol with trichloroethyl acetate in an organic solvent. J. Chem. Soc. Perkin Trans. 1989, 1, 2001–2002. [Google Scholar]
- Guo, Z.W.; Wu, S.H.; Chen, C.S.; Girdaukas, G.; Sih, C.J. Sequential biocatalytic kinetic resolutions. J. Am. Chem. Soc. 1990, 112, 4942–4945. [Google Scholar] [CrossRef]
- Theil, F.; Schick, H.; Lapitskaya, M.A.; Pivnitsky, K.K. Enzymes in organic-synthesis. 4. Investigation of the pancreatin-catalyzed acylation of cis-cyclopent-2-ene-1,4-diol with various trichloroethyl and vinyl alkanoates. Liebigs Ann. Chem. 1991, 195–200. [Google Scholar]
- Theil, F.; Schick, H.; Winter, G.; Reck, G. Lipase-catalyzed transesterification of meso-cyclopentane diols. Tetrahedron 1991, 47, 7569–7582. [Google Scholar] [CrossRef]
- Santaniello, E.; Ferraboschi, P.; Grisenti, P. An efficient chemo-enzymatic approach to the enantioselective synthesis of 2-methyl-1,3-propamedical derivatives. Tetrahedron Lett. 1990, 31, 5657–5660. [Google Scholar]
- Burgess, K.; Henderson, I. Biocatalytic desymmetrizations of pentitol derivatives. Tetrahedron Lett. 1991, 32, 5701–5704. [Google Scholar] [CrossRef]
- Hung, T.C.; Giridhar, R.; Chiou, S.H.; Wu, W.T. Binary immobilization of Candida rugosalipase on chitosan. J. Mol. Catal. B-Enzym. 2003, 26, 69–78. [Google Scholar] [CrossRef]
- Yi, S.S.; Noh, J.M.; Lee, Y.S. Amino acid modified chitosan beads: Improved polymer supports for immobilization of lipase from Candida rugosa. J. Mol. Catal. B-Enzym. 2009, 57, 123–129. [Google Scholar] [CrossRef]
- Shafei, M.S.; Allam, R.F. Production and immobilization of partially purified lipase from Penicillium chrysogenum. Mal. J. Microbiol. 2010, 6, 196–202. [Google Scholar]
- Nasratun, M.; Said, H.A.; Noraziah, A.; Alla, A.N.A. Immobilization of lipase from Candida rugosa on chitosan beads for transesterification reaction. Am. J. Appl. Sci. 2009, 6, 1653–1657. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brabcová, J.; Blažek, J.; Janská, L.; Krečmerová, M.; Zarevúcka, M. Lipases as Tools in the Synthesis of Prodrugs from Racemic 9-(2,3-Dihydroxypropyl)adenine. Molecules 2012, 17, 13813-13824. https://doi.org/10.3390/molecules171213813
Brabcová J, Blažek J, Janská L, Krečmerová M, Zarevúcka M. Lipases as Tools in the Synthesis of Prodrugs from Racemic 9-(2,3-Dihydroxypropyl)adenine. Molecules. 2012; 17(12):13813-13824. https://doi.org/10.3390/molecules171213813
Chicago/Turabian StyleBrabcová, Jana, Jiří Blažek, Lucie Janská, Marcela Krečmerová, and Marie Zarevúcka. 2012. "Lipases as Tools in the Synthesis of Prodrugs from Racemic 9-(2,3-Dihydroxypropyl)adenine" Molecules 17, no. 12: 13813-13824. https://doi.org/10.3390/molecules171213813
APA StyleBrabcová, J., Blažek, J., Janská, L., Krečmerová, M., & Zarevúcka, M. (2012). Lipases as Tools in the Synthesis of Prodrugs from Racemic 9-(2,3-Dihydroxypropyl)adenine. Molecules, 17(12), 13813-13824. https://doi.org/10.3390/molecules171213813