Quantitative Studies on Structure-DPPH• Scavenging Activity Relationships of Food Phenolic Acids

Abstract
:1. Introduction
2. Results and Discussion
2.1. CoMFA and CoMSIA Model

| Statistics parameters | CoMFA model | CoMSIA model |
|---|---|---|
| q2 | 0.638 | 0.855 |
| r2 | 0.984 | 0.986 |
| s | 0.236 | 0.216 |
| F | 139.126 | 208.320 |
| PLS component | 5 | 5 |
| Field contribution | ||
| Steric | 0.505 | 0.058 |
| Electrostatic | 0.495 | 0.326 |
| Hydrophobic | 0.171 | |
| H-bond Donor | 0.140 | |
| H-bond Acceptor | 0.304 | |
| r2bs (10 runs) | 0.993 | 0.997 |
| SDbs | 0.007 | 0.003 |
| r2pred | 0.971 | 0.996 |
| r02 | 0.971 | 0.993 |
| k | 0.955 | 1.008 |
| (r2pred-r02)/r2pred | 0.000 | 0.003 |
{kind=link}
{kind=link}
{kind=link}
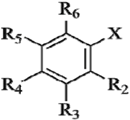
| Compds | R2 | R3 | R4 | R5 | R6 | X | Experimental pTEAC a | Predicted pTEAC a | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CoMFA | CoMSIA | |||||||||
| 1 b | H | OCH3 | OH | H | H | COOH | 2.127 | 2.252 | 2.497 | |
| 2 | H | OH | OH | H | H | COOH | 0.309 | 0.568 | 0.657 | |
| 3 | H | H | OH | H | H | COOH | 3.544 | 3.316 | 3.227 | |
| 4 | H | OCH3 | OCH3 | H | H | COOH | 3.700 | 3.928 | 3.764 | |
| 5 | OOCH3 | H | H | H | H | COOH | 3.558 | 3.530 | 3.579 | |
| 6 | OCH3 | H | H | H | H | COOH | 3.667 | 3.595 | 3.913 | |
| 7 | H | OCH3 | OH | OCH3 | H | COOH | 0.409 | 0.275 | 0.267 | |
| 8 | H | H | OCH3 | H | H | COOH | 3.608 | 3.464 | 3.475 | |
| 9 b | OH | H | H | OH | H | COOH | 0.168 | 0.135 | 0.149 | |
| 10 | OH | H | H | H | H | COOH | 3.535 | 3.624 | 3.572 | |
| 11 c | H | OH | OH | OH | H | COOH | 0.179 | 0.104 | 0.265 | |
| 12 | H | OCH3 | H | H | H | COOH | 3.633 | 3.675 | 3.424 | |
| 13 b | H | H | OH | H | H | CH2COOH | 3.482 | 3.297 | 3.602 | |
| 14 | OH | H | H | H | H | CH2COOH | 3.501 | 3.484 | 3.575 | |
| 15 | H | OCH3 | OH | H | H | CH2COOH | 0.876 | 0.891 | 0.7809 | |
| 16 b | OH | H | OH | H | H | CH=CHCOOH | 0.964 | 0.622 | 1.090 | |
| 17 | H | H | OH | H | H | CH=CHCOOH | 3.349 | 3.457 | 3.479 | |
| 18 b | H | OH | OH | H | H | CH=CHCOOH | 0.266 | 0.244 | 0.290 | |
| 19 | H | OH | OH | H | H | 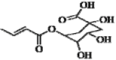 | 0.373 | 0.327 | 0.335 | |
| 20 | H | H | H | H | H | CH=CHCOOH | 3.594 | 3.468 | 3.483 | |
| 21 b | H | OH | OH | H | H | 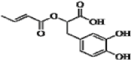 | 0.244 | 0.245 | 0.179 | |
| 22 b | OCH3 | H | H | H | H | CH=CHCOOH | 3.561 | 3.184 | 3.258 | |
| 23 | H | OCH3 | OH | H | H | CH=CHCOOH | 0.773 | 1.078 | 0.976 | |
| 24 | H | OCH3 | OH | OCH3 | H | CH=CHCOOH | 0.409 | 0.348 | 0.707 | |
2.2. External Validation of the CoMFA and CoMSIA Models
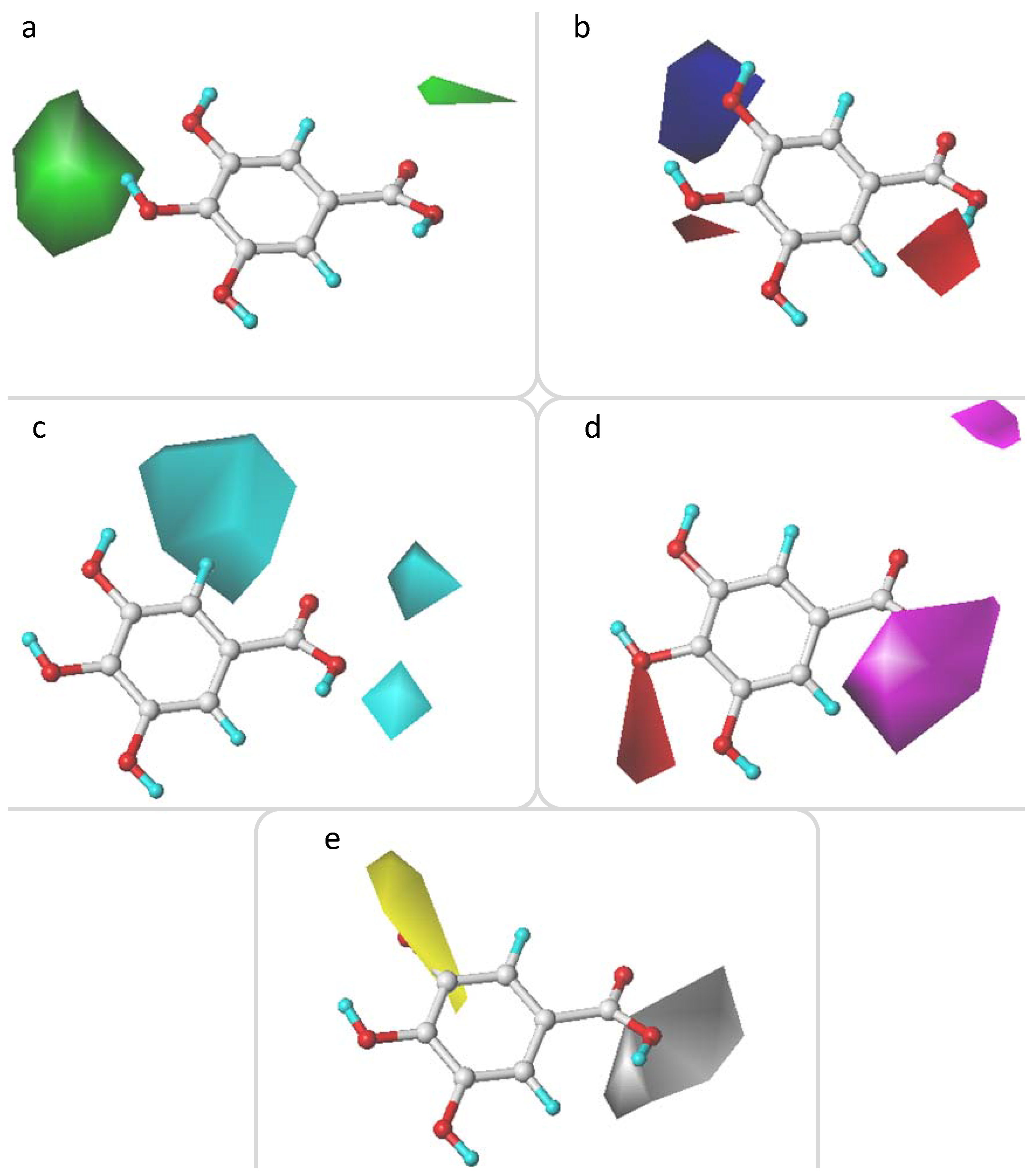
2.3. CoMFA and CoMSIA Contour Maps Analysis
2.3.1. CoMFA Contour Maps
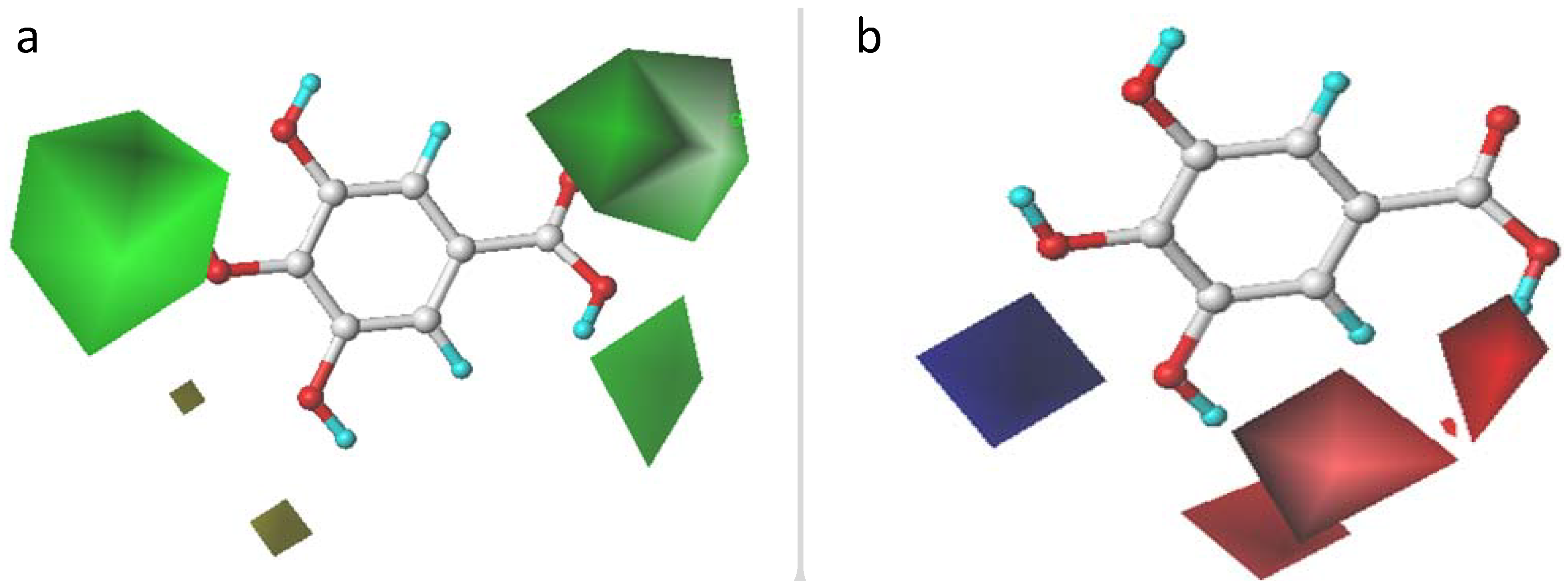
2.3.2. CoMSIA Contour Maps

2.4. Application of QSAR Results of Phenolic Acid Derivatives to Previous Studies

| No. | Compounds | para-OH a | meta-OH b | meta-OCH3c | ortho-OH d | X- | Bioactivity e | Ref. |
|---|---|---|---|---|---|---|---|---|
| EC50f (10−5 mol/L) | [19] | |||||||
| 1 | caffeic acid * | + | + | CH=CHCOOH | 2.6 ± 0.1 | |||
| 2 | sinapic acid | + | ++ | CH=CHCOOH | 4.5 ± 0.2 | |||
| 3 | ferulic acid | + | + | CH=CHCOOH | 4.9 ± 0.1 | |||
| 4 | umbellic acid * | + | + | CH=CHCOOH | 8.6 ± 0.1 | |||
| 5 | p-coumaric acid | + | CH=CHCOOH | 255 ± 64 | ||||
| Inhibition % | [18] | |||||||
| 1 | gallic acid | + | ++ | COOH | 75 ± 2 | |||
| 2 | 3,4-dihydroxyphenylacetic acid * | + | + | CH2COOH | 70.8 ± 0.3 | |||
| 3 | 2,3-dihydroxybenzoic acid * | + | + | COOH | 46 ± 3 | |||
| 4 | protocatechuic acid | + | + | COOH | 41.2 ± 0.6 | |||
| 5 | α-resorcylic acid * | ++ | COOH | 0.60 ± 0.08 | ||||
| 6 | o-hydroxybenzoic acid | + | COOH | 0.11 ± 0.07 | ||||
| 7 | β-resorcylic acid * | + | + | COOH | 0.11 ± 0.07 | |||
| 8 | m-hydroxybenzoic acid * | + | COOH | 0.07 ± 0.15 | ||||
| IC50f (μM) | [20] | |||||||
| 1 | dihydrosinapic acid * | + | ++ | CH2CH2COOH | 44.3 | |||
| 2 | dihydroferulic acid * | + | + | CH2CH2COOH | 77.0 | |||
| 3 | sinapic acid | + | ++ | CH=CHCOOH | 77.2 | |||
| 4 | ferulic acid | + | + | CH=CHCOOH | 113.9 | |||
| 5 | vanillic acid * | + | + | COOH | 250.0 | |||
| 6 | p-coumaric acid | + | CH=CHCOOH | 2130 | ||||
| TEAC g (mM) | ||||||||
| 1 | gallic acid | + | ++ | COOH | 3.92 ± 0.026 | |||
| 2 | syringic acid | + | ++ | COOH | 1.33 ± 0.012 | |||
| 3 | protocatechuic acid | + | + | COOH | 1.29 ± 0.007 | [4] | ||
| 4 | 2,4-dihydroxybenzoic acid * | + | + | COOH | 1.27 ± 0.011 | |||
| 5 | p-hydroxybenzoic acid | + | COOH | 0.059 ± 0.000 | ||||
| 6 | m-hydroxybenzoic acid * | + | COOH | 0.069 ± 0.000 | ||||
| 7 | o-hydroxybenzoic acid * | + | COOH | 0.052 ± 0.000 | ||||
| 8 | benzoic acid * | COOH | 0.006 ± 0.000 | |||||
| Inhibition % | [14] | |||||||
| 1 | syringic acid | + | ++ | COOH | 90 | |||
| 2 | ferulic acid | + | + | CH=CHCOOH | 60 | |||
| 3 | p-hydroxybenzoic acid | + | COOH | 2 | ||||
| EC50f (10−6 M) | [21] | |||||||
| 1 | gallic acid | + | ++ | COOH | 5.1 ± 0.1 | |||
| 2 | 2,5-dihydroxybenzoic acid * | + | + | COOH | 7.6 ± 0.2 | |||
| 3 | caffeic acid * | + | + | CH=CHCOOH | 12.1 ± 0.2 | |||
| 4 | syringic acid | + | ++ | COOH | 12.3 ± 0.0 | |||
| 5 | ferulic acid | + | + | CH=CHCOOH | 24.7 ± 0.4 | |||
| Inhibition % | [10] | |||||||
| 1 | dihydrocaffeic acid * | + | + | CH2CH2COOH | 93.9 | |||
| 2 | caffeic acid * | + | + | CH=CHCOOH | 76.6 | |||
| 3 | sinapic acid | + | ++ | CH=CHCOOH | 56.1 | |||
| 4 | ferulic acid | + | + | CH=CHCOOH | 30.9 | |||
| 5 | p-coumaric acid | + | CH=CHCOOH | 3.6 | ||||
| 6 | o-coumaric acid * | + | CH=CHCOOH | 3.5 | ||||
| 7 | m-coumaric acid * | + | CH=CHCOOH | 2.6 | ||||
| 8 | trans-cinnamic acid | CH=CHCOOH | 0.5 | |||||
| EC50f (μM) | [22] | |||||||
| 1 | gallic acid | + | ++ | COOH | 12.0 | |||
| 2 | protocatechuic acid | + | + | COOH | 15.0 |
3. Experimental
3.1. Experimental Design
3.2. DPPH Radical Scavenging Assay
3.3. Data Sets
3.4. Molecular Modeling and Alignment
3.5. Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Index Analysis (CoMSIA)
3.6. Partial Least Square (PLS) Analysis
4. Conclusions
Acknowledgments
References
- Herrmann, K. Occurrence and content of hydroxycinnamic and hydroxybenzoic acid compounds in foods. Crit. Rev. Food Sci. Nutr. 1989, 28, 315–347. [Google Scholar] [CrossRef]
- Shahidi, F.; Janitha, P.K.; Wanasundara, P.D. Phenolic antioxidants. Crit. Rev. Food Sci. Nutr. 1992, 32, 67–103. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Cai, Y.Z.; Mei, S.; Jie, X.; Luo, Q.; Corke, H. Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci. 2006, 78, 2872–2888. [Google Scholar] [CrossRef]
- Natella, F.; Nardini, M.; di Felice, M.; Scaccini, C. Benzoic and cinnamic acid derivatives as antioxidants: Structure-activity relation. J. Agric. Food Chem. 1999, 47, 1453–1459. [Google Scholar] [CrossRef]
- Ordoudi, S.A.; Tsimidou, M.Z.; Vafiadis, A.P.; Bakalbassis, E.G. Structure-DPPH• scavenging activity relationships: Parallel study of catechol and guaiacol acid derivatives. J. Agric. Food Chem. 2006, 54, 5763–5768. [Google Scholar] [CrossRef]
- Vajragupta, O.; Boonchoong, P.; Wongkrajang, Y. Comparative quantitative structure-activity study of radical scavengers. Bioorg. Med. Chem. 2000, 8, 2617–2628. [Google Scholar] [CrossRef]
- Yamagami, C.; Akamatsu, M.; Motohashi, N.; Hamada, S.; Tanahashi, T. Quantitative structure-activity relationship studies for antioxidant hydroxybenzalacetones by quantum chemical-and 3-D-QSAR(CoMFA) analyses. Bioorg. Med. Chem. Lett. 2005, 15, 2845–2850. [Google Scholar]
- Hyun, J.; Woo, Y.; Hwang, D.S.; Jo, G.; Eom, S.; Lee, Y.; Park, J.C.; Lim, Y. Relationships between structures of hydroxyflavones and their antioxidative effects. Bioorg. Med. Chem. Lett. 2010, 20, 5510–5513. [Google Scholar]
- Velkov, Z.A.; Kolev, M.K.; Tadjer, A.V. Modeling and statistical analysis of DPPH scavenging activity of phenolics. Collect. Czech. Chem. Commun. 2007, 72, 1461–1471. [Google Scholar] [CrossRef]
- Rastija, V.; Medic-Saric, M. QSAR study of antioxidant activity of wine polyphenols. Eur. J. Med. Chem. 2009, 44, 400–408. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef]
- Zhou, K.Q.; Yin, J.J.; Yu, L.L. ESR determination of the reactions between selected phenolic acids and free radicals or transition metals. Food Chem. 2006, 95, 446–457. [Google Scholar] [CrossRef]
- Kajiyama, T.; Ohkatsu, Y. Effect of meta-substituents of phenolic antioxidants - proposal of secondary substituent effect. Polym. Degrad. Stab. 2002, 75, 535–542. [Google Scholar] [CrossRef]
- Warren, J.; Tronic, T.; Mayer, J. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Foti, M.C.; Daquino, C.; Geraci, C. Electron-transfer reaction of cinnamic acids and their methyl esters with the DPPH center dot radical in alcoholic solutions. Eur. J. Org. Chem. 2004, 69, 2309–2314. [Google Scholar]
- Sroka, Z.; Cisowski, W. Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food Chem. Toxicol. 2003, 41, 753–758. [Google Scholar] [CrossRef]
- Abramovic, H.; Terpinc, P. A kinetic approach for evaluation of the antioxidant activity of selected phenolic acids. Food Chem. 2010, 121, 366–371. [Google Scholar] [CrossRef]
- Shimoji, Y.; Tamura, Y.; Nakamura, Y.; Nanda, K.; Nishidai, S.; Nishikawa, Y.; Ishihara, N.; Uenakai, K.; Ohigashi, H. Isolation and identification of DPPH radical scavenging compounds in Kurosu (Japanese unpolished rice vinegar). J. Agric. Food Chem. 2002, 50, 6501–6503. [Google Scholar] [CrossRef]
- Garcia-Parrilla, M.C.; Villano, D.; Fernandez-Pachon, M.S.; Moya, M.L.; Troncoso, A.M. Radical scavenging ability of polyphenolic compounds towards DPPH free radical. Talanta 2007, 71, 230–235. [Google Scholar] [CrossRef]
- Garrido, J.; Reis, B.; Martins, M.; Barreto, B.; Milhazes, N.; Garrido, E.M.; Silva, P.; Borges, F. Structure-Property-Activity Relationship of Phenolic Acids and Derivatives. Protocatechuic Acid Alkyl Esters. J. Agric. Food Chem. 2010, 58, 6986–6993. [Google Scholar]
- Cheng, Z.; Moore, J.; Yu, L. High-throughput relative DPPH radical scavenging capacity assay. J. Agric. Food Chem. 2006, 54, 7429–7436. [Google Scholar] [CrossRef]
- Clark, M.; Cramer, R.D.; Vanopdenbosch, N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analysis. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U. Comparative Molecular Similarity Index Analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aided Mol. Des. 1999, 13, 1–10. [Google Scholar] [CrossRef]
- Bush, B.L.; Nachbar, R.B., Jr. Sample-distance partial least squares: PLS optimized for many variables, with application to CoMFA. J. Comput. Aided Mol. Des. 1993, 7, 587–619. [Google Scholar] [CrossRef]
- Zhang, L.; Tsai, K.-C.; Du, L.; Fang, H.; Li, M.; Xu, W. How to Generate Reliable and Predictive CoMFA Models. Curr. Med. Chem. 2011, 18, 1–8. [Google Scholar] [CrossRef]
- Sample Availability: Samples of tested phenolic acids are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jing, P.; Zhao, S.-J.; Jian, W.-J.; Qian, B.-J.; Dong, Y.; Pang, J. Quantitative Studies on Structure-DPPH• Scavenging Activity Relationships of Food Phenolic Acids. Molecules 2012, 17, 12910-12924. https://doi.org/10.3390/molecules171112910
Jing P, Zhao S-J, Jian W-J, Qian B-J, Dong Y, Pang J. Quantitative Studies on Structure-DPPH• Scavenging Activity Relationships of Food Phenolic Acids. Molecules. 2012; 17(11):12910-12924. https://doi.org/10.3390/molecules171112910
Chicago/Turabian StyleJing, Pu, Shu-Juan Zhao, Wen-Jie Jian, Bing-Jun Qian, Ying Dong, and Jie Pang. 2012. "Quantitative Studies on Structure-DPPH• Scavenging Activity Relationships of Food Phenolic Acids" Molecules 17, no. 11: 12910-12924. https://doi.org/10.3390/molecules171112910