Solvent-Free Synthesis, DNA-Topoisomerase II Activity and Molecular Docking Study of New Asymmetrically N,N'-Substituted Ureas

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterisation
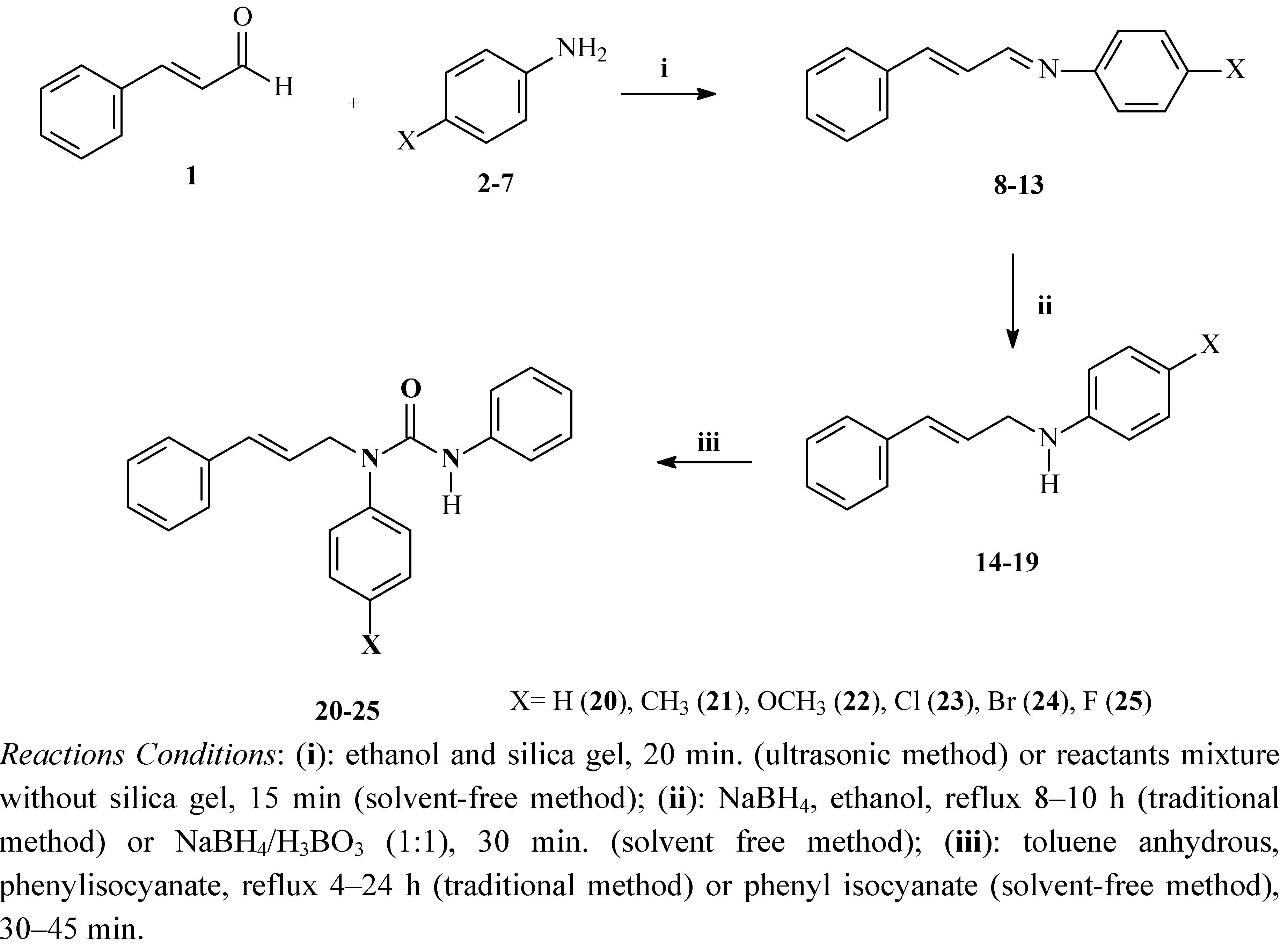

{kind=link}
{kind=link}
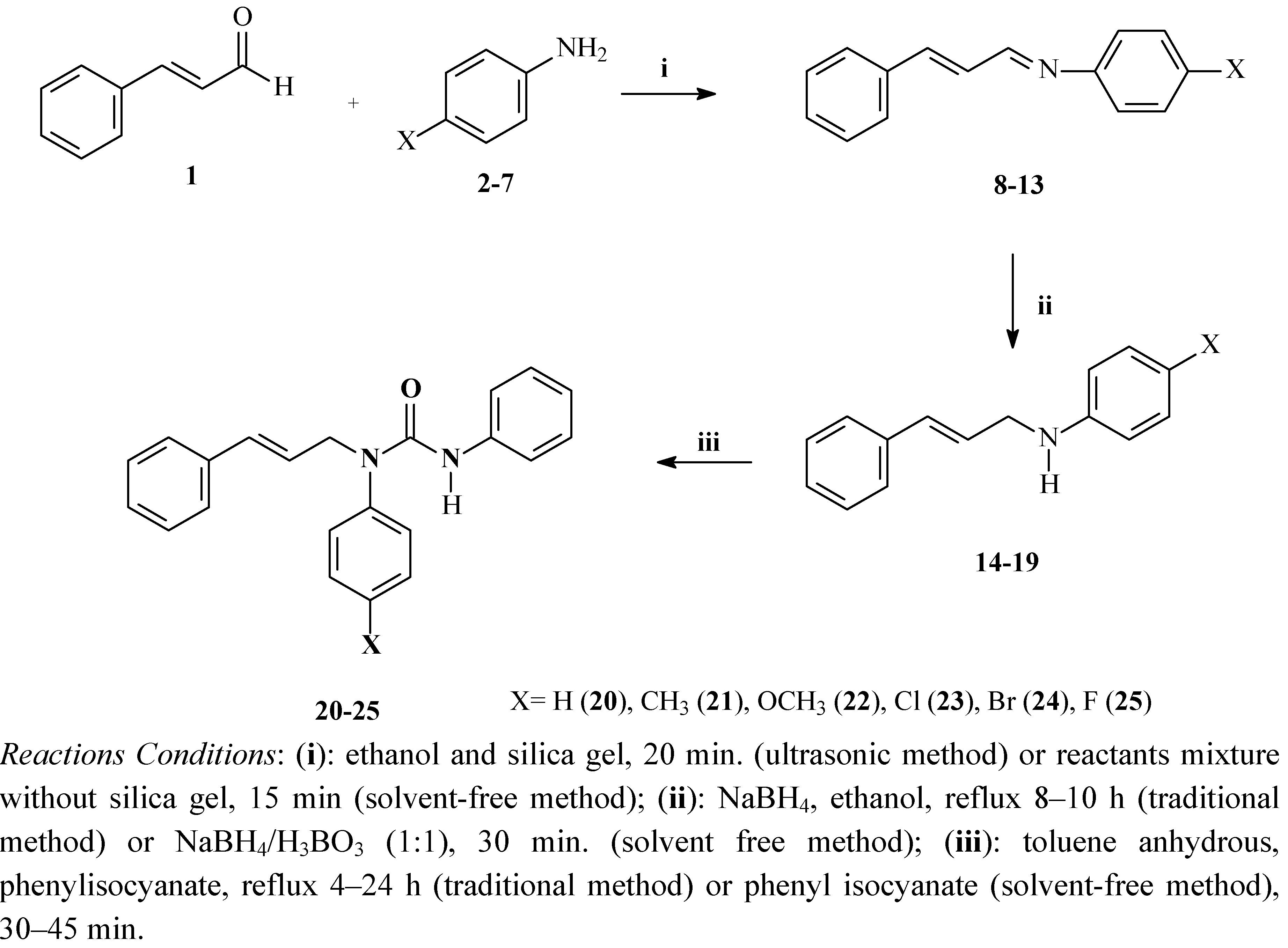{kind=link}
| Schiff base | X | MP (°C) | Ultrasonic irradiation Yield (%) 20 min | Solvent free condition Yield (%) 15 min |
|---|---|---|---|---|
| 8 | H | 107 a | 70 | 100 |
| 9 | 4-CH3 | 77 b | 70 | 98 |
| 10 | 4-OCH3 | 120–121 c | 85 | 100 |
| 11 | 4-Cl | 104–106 d | 90 | 97 |
| 12 | 4-Br | 120–121 d | 90 | 100 |
| 13 | 4-F | 69–71 d | 75 | 98 |
| Allyl amine | X | MP (°C) | Reflux procedure Yield (%) 8–10 h | Solvent free conditions Yield (%) 30 min |
|---|---|---|---|---|
| 14 | H | Oil a | 60 | 96 |
| 15 | 4-CH3 | Oil b | 45 | 99 |
| 16 | 4-OCH3 | 67 c | 55 | 99 |
| 17 | 4-Cl | 83 c | 72 | 98 |
| 18 | 4-Br | 86–87 c | 75 | 98 |
| 19 | 4-F | 60 c | 70 | 99 |
| Urea | X | MP (°C) | Reflux procedure Yield (%) | Time (h) | Solvent free conditions Yield (%) 30 min |
|---|---|---|---|---|---|
| 20 | H | oil | 26 | 4 | >99 |
| 21 | 4-CH3 | 65–68 | 30 | 4 | >99 |
| 22 | 4-OCH3 | oil | 40 | 24 | 98 |
| 23 | 4-Cl | 98–100 | 64 | 4 | >99 |
| 24 | 4-Br | oil | 30 | 4 | >99 |
| 25 | 4-F | 75–78 | 25 | 4 | >99 |
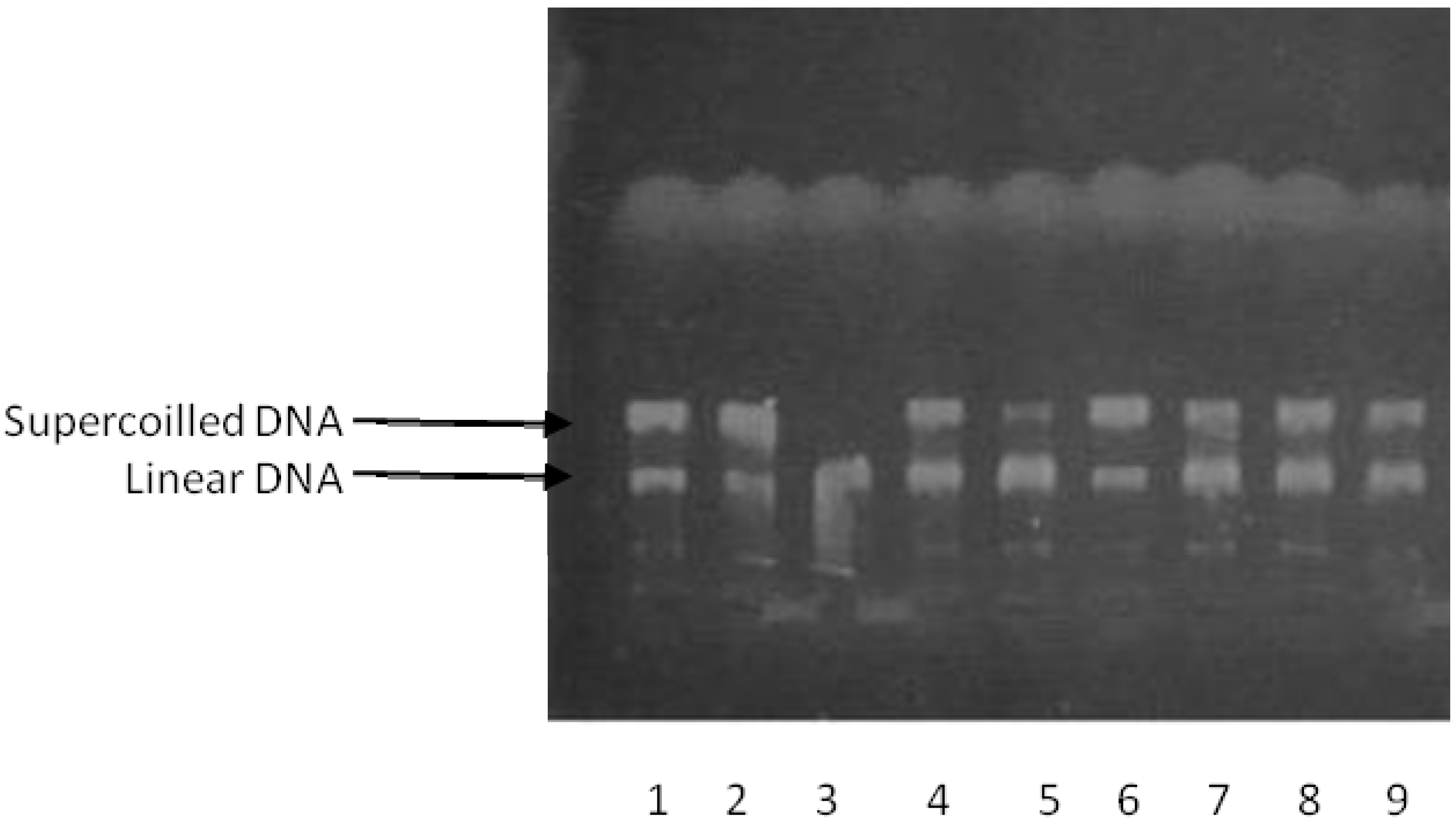
2.2. DNA-Topoisomerase Assays
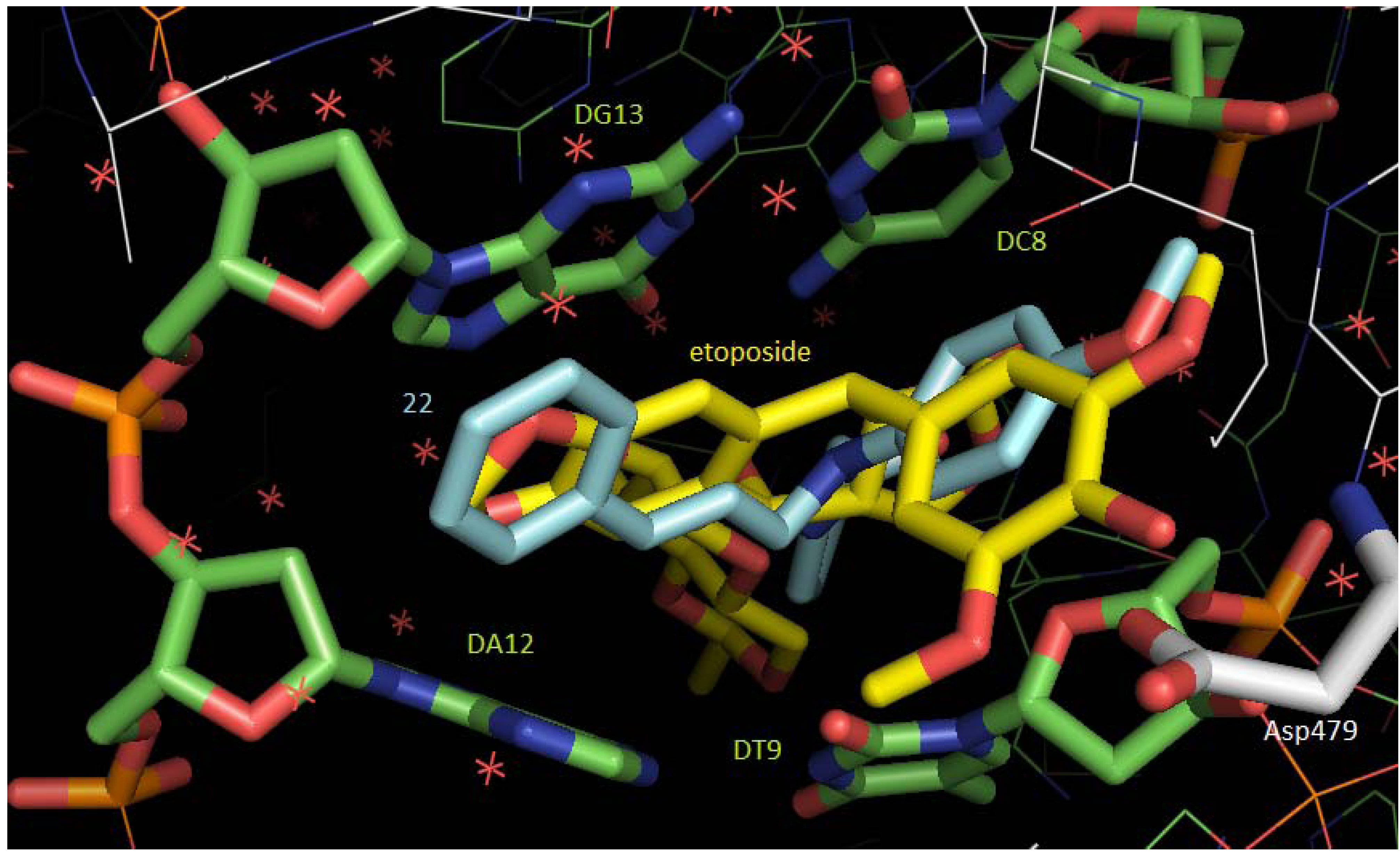
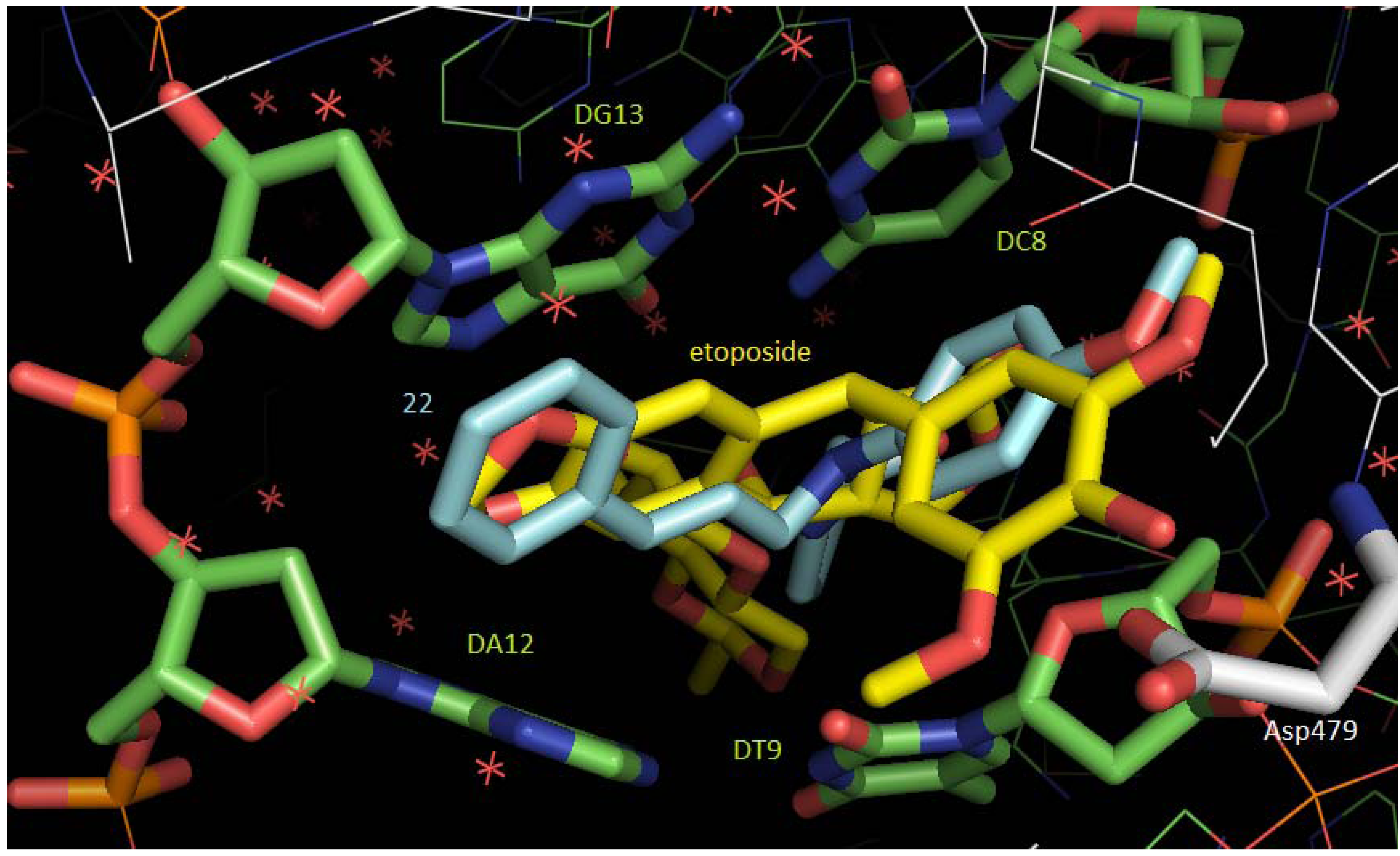
2.3. Molecular Docking Studies
| Compound | X | Fitness score into DNA binding site | Fitness score into ATP binding site |
|---|---|---|---|
| 20 | H | 79.54 | 64.65 |
| 21 | 4-CH3 | 83.94 | 71.29 |
| 22 | 4-OCH3 | 84.29 | 69.08 |
| 23 | 4-Cl | 82.17 | 67.01 |
| 24 | 4-Br | 80.10 | 66.90 |
| 25 | 4-F | 81.39 | 69.81 |
| ETP | - | 111.26 | - |
| ATP analogous | - | - | 143.37 |
3. Experimental
3.1. General
3.2. Procedure for the Preparation of N-(p-X-Phenyl)-N-[(E, 2E)-3-phenyl-2-propenylidene]imines 8–13
3.2.1. Ultrasonic-Assisted Method Using Ethanol as the Solvent
3.2.2. Solvent Free Method
3.3. Procedure for the Preparation of 4-(p-X-Phenyl)-N-[(2E)-3-phenyl-2-propenyl]benzenamines 14–19
3.3.1. Using Traditional Reflux
3.3.2. Solvent Free Reactions
3.4. Procedure for the Preparation of Ureas 20–25
3.4.1. Using Traditional Reflux
3.4.2. Solvent Free Reactions
3.5. Materials for DNA-Relaxation Assays
3.6. Procedure for the DNA-Topoisomerase Assay
3.7. Docking Studies
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Tanaka, K. Solvent-free Organic Synthesis (Green Chemistry), 1st ed; Wiley-VCH: Weinhein, Germany, 2003. [Google Scholar]
- Mallouk, S.; Bougrin, K.; Laghzizil, A.; Benhida, R. Microwave-assisted and efficient solvent-free Knoevenagel condensation: A sustainable protocol using porous calcium hydroxyapatite as catalyst. Molecules 2010, 15, 818–823. [Google Scholar]
- Monguchi, Y.; Fujita, Y.; Hashimoto, S.; Ina, M.; Takahashi, T.; Ito, R.; Nozaki, K.; Maegawa, T.; Sajiki, H. Palladium on carbon-catalyzed solvent-free and solid-phase hydrogenation and Suzuki-Miyaura reaction. Tetrahedron 2011, 67, 8628–8634. [Google Scholar]
- Veeranarayana Reddy, M.V.; Sekhar Reddy, G.C.; Jeong, Y.T. Microwave-assisted, Montmorillonite K-10 catalyzed three-component synthesis of 2H-indazolo[2,1-b]phthalazine-triones under solvent-free conditions. Tetrahedron 2012. [Google Scholar] [CrossRef]
- Kaltenbach, R.F.; Patel, M.; Waltermire, R.E.; Harris, G.D.; Stone, B.R.P.; Klabe, R.M.; Garber, S.; Bacheler, L.T.; Cordova, B.C.; Logue, K.; et al. Synthesis, antiviral activity and pharmacokinetics of P1/P1'substituted 3-aminoindazole cyclic urea HIV protease inhibitors. Bioorg. Med. Chem. 2003, 13, 605–608. [Google Scholar] [CrossRef]
- Džimbeg, G.; Zorc, B.; Kralj, M.; Ester, K.; Pavelić, K.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; Mintas, M. The novel primaquine derivatives of N-alkyl, Cycloalkyl or aryl urea: Synthesis, cytostatic and antiviral activity evaluations. Eur. J. Med. Chem. 2008, 43, 1180–1887. [Google Scholar] [CrossRef]
- Li, H.-Q.; Zhu, T.-T.; Yan, T.; Luo, Y.; Zhu, H.-L. Design, Synthesis and structure-activity relationships of antiproliferative 1,3-disubstituted urea derivatives. Eur. J. Med. Chem. 2009, 44, 453–459. [Google Scholar] [CrossRef]
- Esteves-Souza, A.; Pissinate, K.; Nascimento, M.G.; Grynberg, N.F.; Echevarria, A. Synthesis, Cytotoxicity, and DNA-topoisomerase inhibitory activity of new asymmetric ureas and thioureas. Bioorg. Med. Chem. 2006, 14, 492–499. [Google Scholar] [CrossRef]
- Brown, J.R.; North, E.J.; Hurdle, J.G.; Morisseau, C.; Scarborough, J.S.; Sun, D.; Korduláková, J.; Scherman, M.S.; Jones, V.; Grzegorzewicz, A.; et al. The structure–activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg. Med. Chem. 2011, 19, 5585–5595. [Google Scholar] [CrossRef]
- Scherman, M.S.; North, E.J.; Jones, V.; Hess, T.N.; Grzegorzewicz, A.; Kasagami, T.; Kim, I.H.; Lenaerts, A.J.; Lee, R.E.; Jackson, M.; et al. Screening a library of 1600 adamantyl ureas for anti-Micobacterium tuberculosis activity in vitro and for better physical-chemical properties for bioavailability. Bioorg. Med. Chem. 2012, 20, 3255–3262. [Google Scholar] [CrossRef]
- Pommier, Y. Diversity of DNA topoisomerases I and inhibitors. Biochimie 1998, 80, 255–270. [Google Scholar] [CrossRef]
- Kellner, U.; Rudolph, P.; Parwaresch, R. Human DNA-topoisomerases–diagnostic and therapeutic implications for cancer. Onkologie 2000, 23, 424–430. [Google Scholar] [CrossRef]
- Reis, C.M.; Pereira, D.S.; Paiva, R.O.; Kneipp, L.; Echevarria, A. Microwave-assisted synthesis of new N1,N4-substituted thiosemicarbazones. Molecules 2011, 16, 10668–10684. [Google Scholar] [CrossRef]
- Rodrigues-Santos, C.E.; Echevarria, A. An efficient and fast synthesis of 4-aryl-3,4-dihydrocoumarins by (CF3SO3)3Y catalysis under microwave irradiation. Tetrahedron Lett. 2007, 48, 4505–4508. [Google Scholar] [CrossRef]
- Rodrigues-Santos, C.E.; Echevarria, A. Convenient syntheses of pyrazolo[3,4-b]pyridin-6-ones using either microwave or ultrasound irradiation. Tetrahedron Lett. 2011, 52, 336–340. [Google Scholar] [CrossRef]
- Esteves-Souza, A.; Echevarria, A.; Vencato, I.; Jimeno, M.L.; Elguero, J. Unexpected formation of bis-pyrazolyl derivatives by solid support coupled with microwave irradiation. Tetrahedron 2001, 57, 6147–6153. [Google Scholar] [CrossRef]
- Oberg, K.M. Enantioselective rhodium-catalyzed [4+2] cycloaddition of α,β-unsaturated imines and isocyanates. J. Am. Chem. Soc. 2011, 133, 4785–4787. [Google Scholar] [CrossRef]
- Yasser, M.S.A.A.; Madkour, H.M.F.; Ali, D.; Yasinzai, M. Antileishmanial, antimicrobial and antifungal activities of some new aryl azomethines. Molecules 2010, 15, 660–671. [Google Scholar] [CrossRef]
- Bennett, J.S.; Charles, K.L.; Miner, M.R.; Heuberger, C.F.; Spina, E.J.; Bartels, M.F.; Foreman, T. Ethyl lactate as a tunable solvent for the synthesis of aryl aldimines. Green Chem. 2009, 11, 166–168. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, S.; Maurya, R.A. Single nucleotide-catalyzed biomimetic reductive amination. Adv. Synth. Catal. 2010, 352, 2227–2232. [Google Scholar] [CrossRef]
- Nagaiah, K.; Naveen Kumar, V.; Srinivasa Rao, R.; Reddy, B.V.S.; Narsaiah, A.V.; Yadav, J.S. Efficient protocol for reductive amination of aldehydes and ketones with sodium borohydride in an ionic liquid/H2O system. Synth. Commun. 2006, 36, 3345–3352. [Google Scholar] [CrossRef]
- Ohshima, T.; Miyamoto, Y.; Ipposhi, J.; Nakahara, Y.; Utsunomiya, M.; Mashima, K. Platinum-catalyzed direct amination of allylic alcohols under mild conditions: Ligand and microwave effects, Substrate scope, And mechanistic study. J. Am. Chem. Soc. 2009, 131, 14317–14328. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring function for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein-ligand docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Guzen, K.P.; Guarezemini, A.S.; Órfão, A.T.G.; Cella, R.; Pereira, C.M.P.; Stefani, H.A. Eco-friendly synthesis of imines by ultrasound irradiation. Tetrahedron Lett. 2007, 48, 1845–1848. [Google Scholar]
- Cho, B.T.; Kang, S.K. Direct and indirect reductive amination of aldehydes and ketones with solid acid-activated sodium borohydride under solvent-free conditions. Tetrahedron 2005, 61, 5725–5734. [Google Scholar] [CrossRef]
- TopoGEN. Available online: http://www.topogen.com (accessed on 17 August 2011).
- Stewart, J.J.P. Optimization of parameters for semi-empirical methods. I Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar]
- Verdonk, M.L.; Cole, J.C.; Hartshom, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aid. Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein–ligand interactions. Proteins 2005, 61, 272–287. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Esteves-Souza, A.; Rodrigues-Santos, C.E.; Del Cistia, C.D.N.; Silva, D.R.d.; Sant'Anna, C.M.R.; Echevarria, A. Solvent-Free Synthesis, DNA-Topoisomerase II Activity and Molecular Docking Study of New Asymmetrically N,N'-Substituted Ureas. Molecules 2012, 17, 12882-12894. https://doi.org/10.3390/molecules171112882
Esteves-Souza A, Rodrigues-Santos CE, Del Cistia CDN, Silva DRd, Sant'Anna CMR, Echevarria A. Solvent-Free Synthesis, DNA-Topoisomerase II Activity and Molecular Docking Study of New Asymmetrically N,N'-Substituted Ureas. Molecules. 2012; 17(11):12882-12894. https://doi.org/10.3390/molecules171112882
Chicago/Turabian StyleEsteves-Souza, Andressa, Claudio E. Rodrigues-Santos, Catarina De Nigris Del Cistia, Daniel Rosa da Silva, Carlos Maurício R. Sant'Anna, and Aurea Echevarria. 2012. "Solvent-Free Synthesis, DNA-Topoisomerase II Activity and Molecular Docking Study of New Asymmetrically N,N'-Substituted Ureas" Molecules 17, no. 11: 12882-12894. https://doi.org/10.3390/molecules171112882