The Final Link: Tapping the Power of Chemical Genetics to Connect the Molecular and Biologic Functions of Mitotic Protein Kinases

Abstract
:1. Introduction
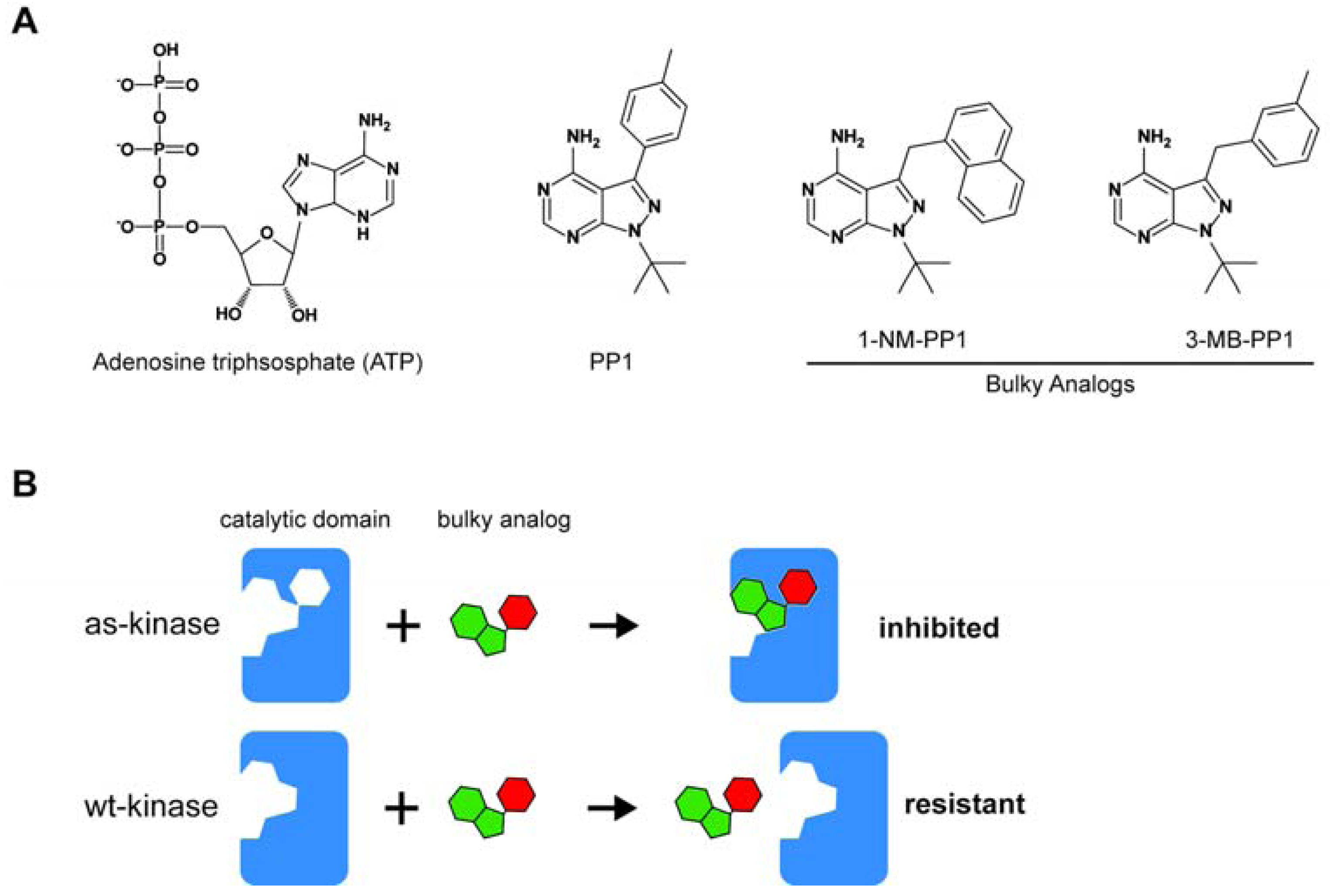
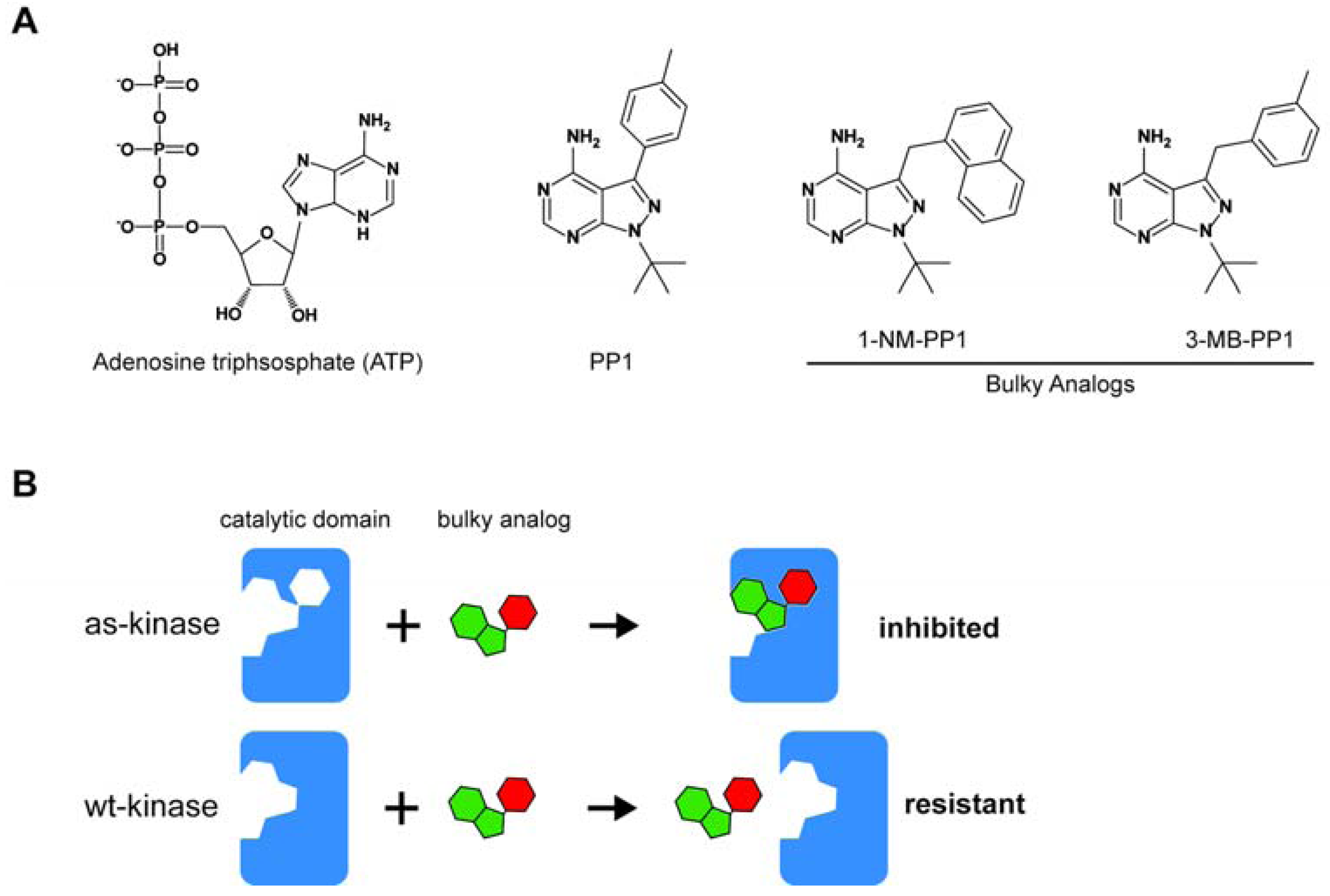
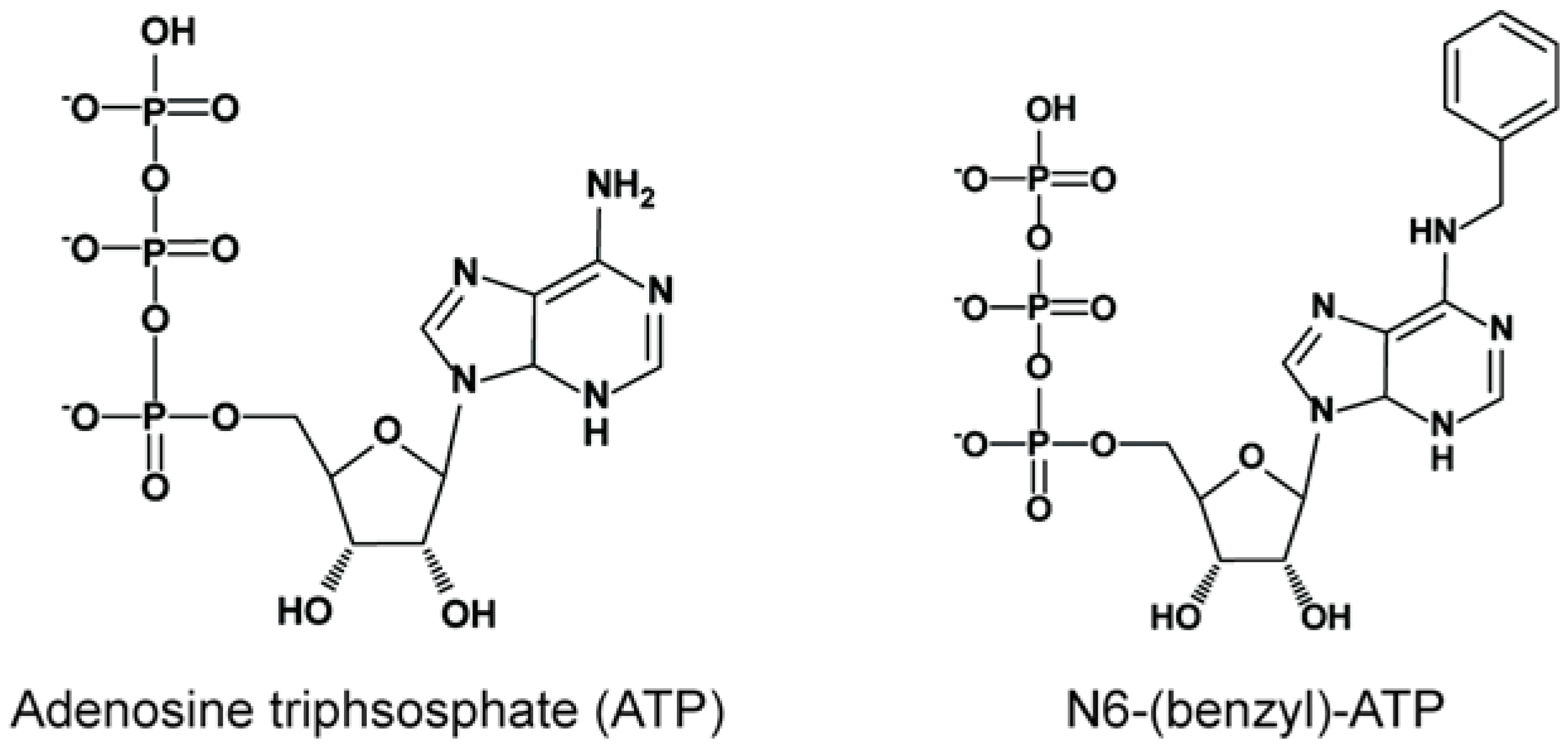
2. Strategies to Identify the Substrates of Analog-Sensitive Mitotic Kinases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Authors | as-kinase | Organism | Proteins Identified |
|---|---|---|---|---|
| Chemical inhibition in intact cells | Oppermann et al. [28] | Plk1 | human | 382 |
| Koch et al. [21] | Ark1/Aurora | fission yeast | 42 | |
| Holt et al. [29] | Cdk1 | budding yeast | 308 | |
| Substrate labeling in cell extracts | Hengeveld et al. [20] | Aurora B | human | 58 |
| Blethrow et al. [30] | Cdk1 | human | >70 | |
| Larochelle et al. [31] | Cdk7 | human | 7 |
2.1. Substrate Identification using Chemical Inhibitors
Considerations
2.2. Substrate Identification Using Signature Labels
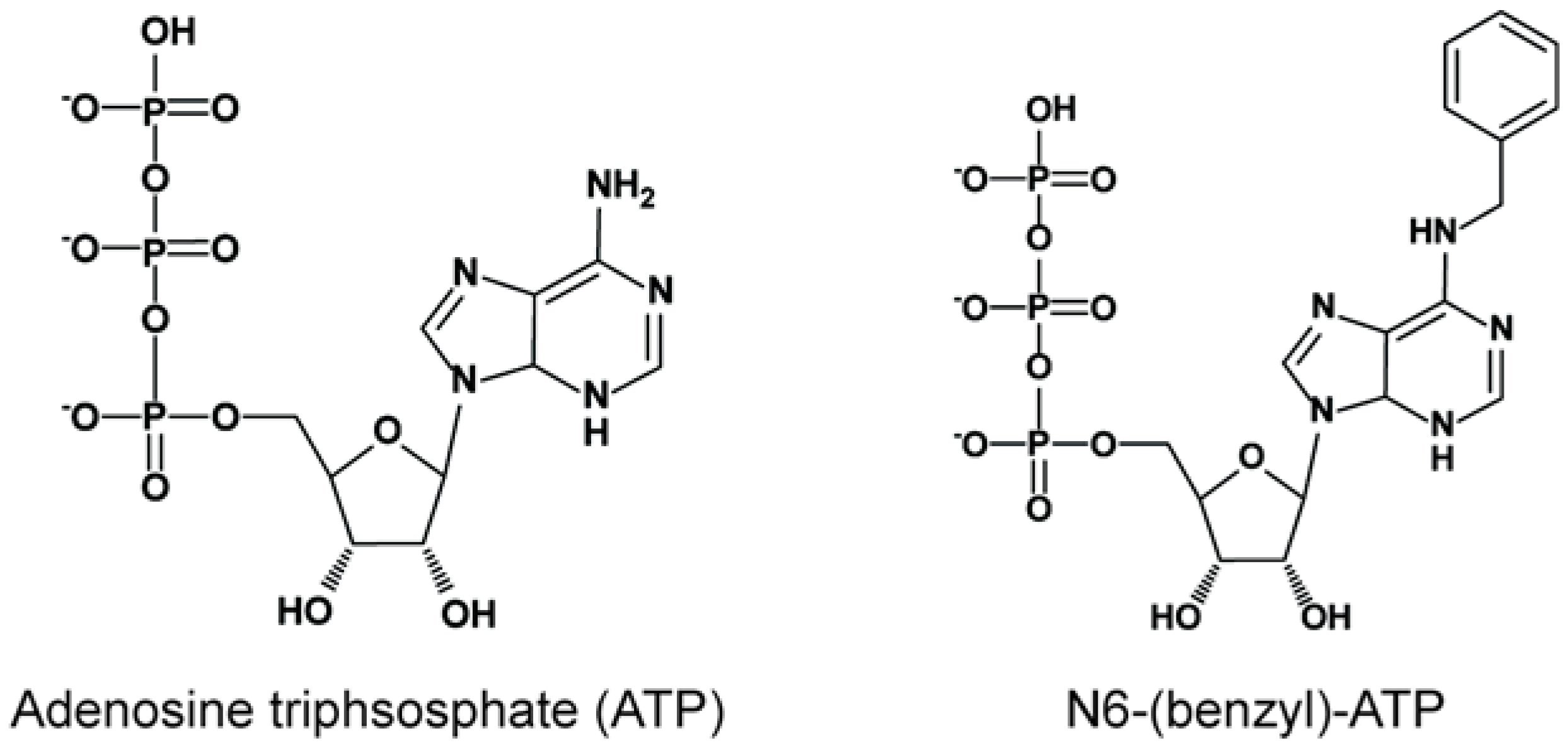
Considerations
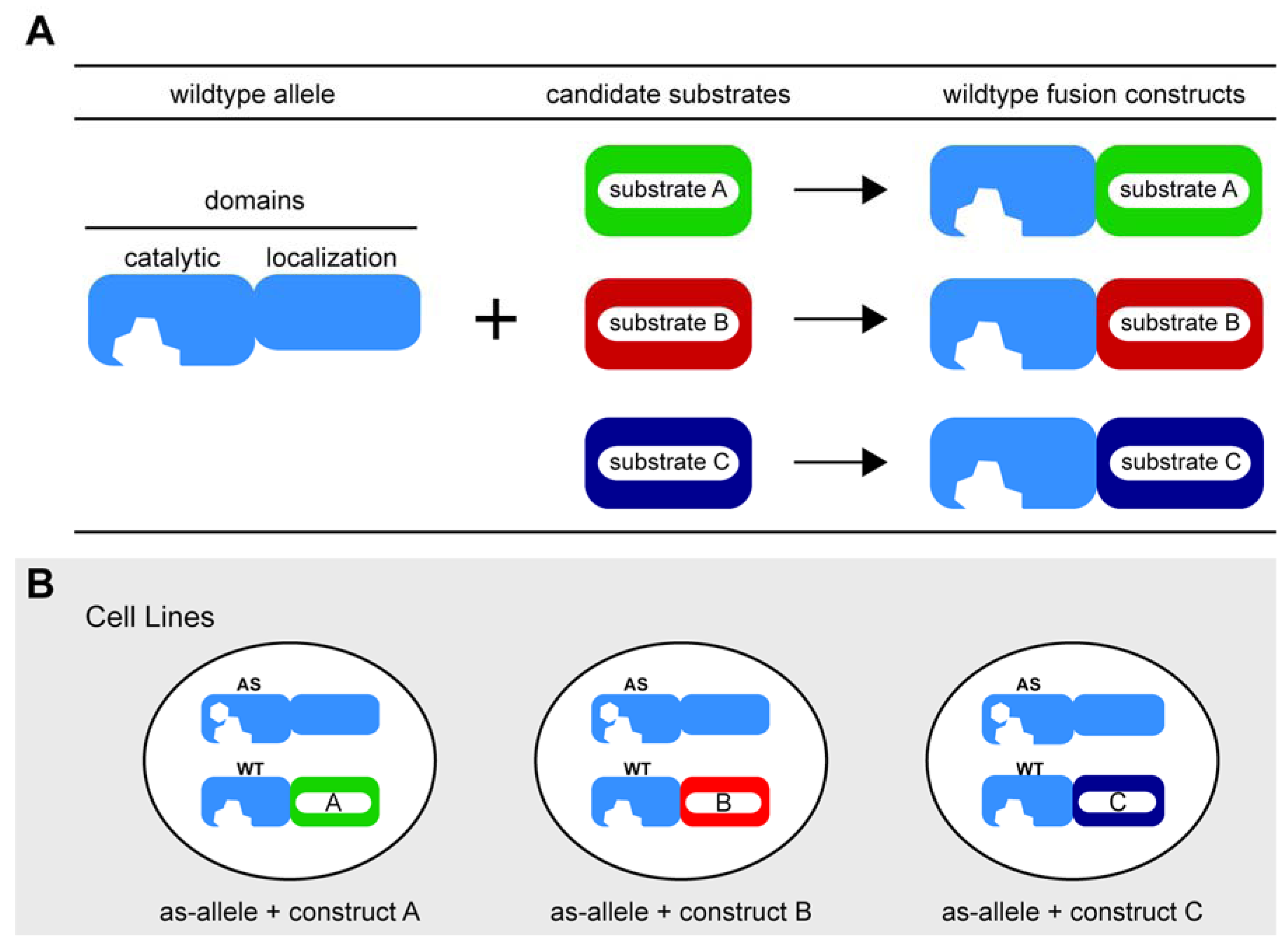
3. Current Strategies to Link Substrates to Kinase Function
3.1. Classic Genetic Complementation: Replacement with Non-Phosphorylatable Mutants
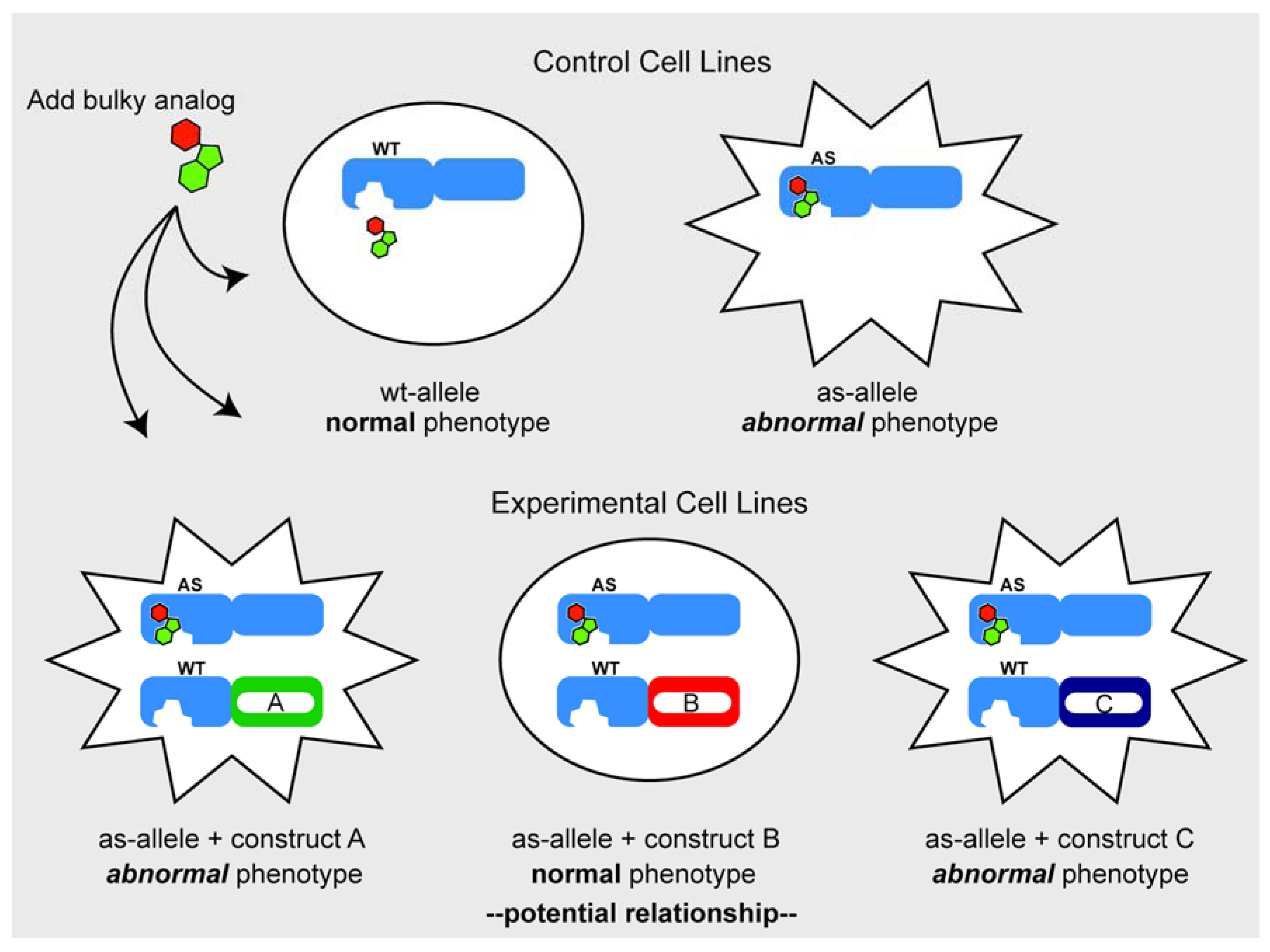
3.2. Chemical Genetic Complementation: A New Twist
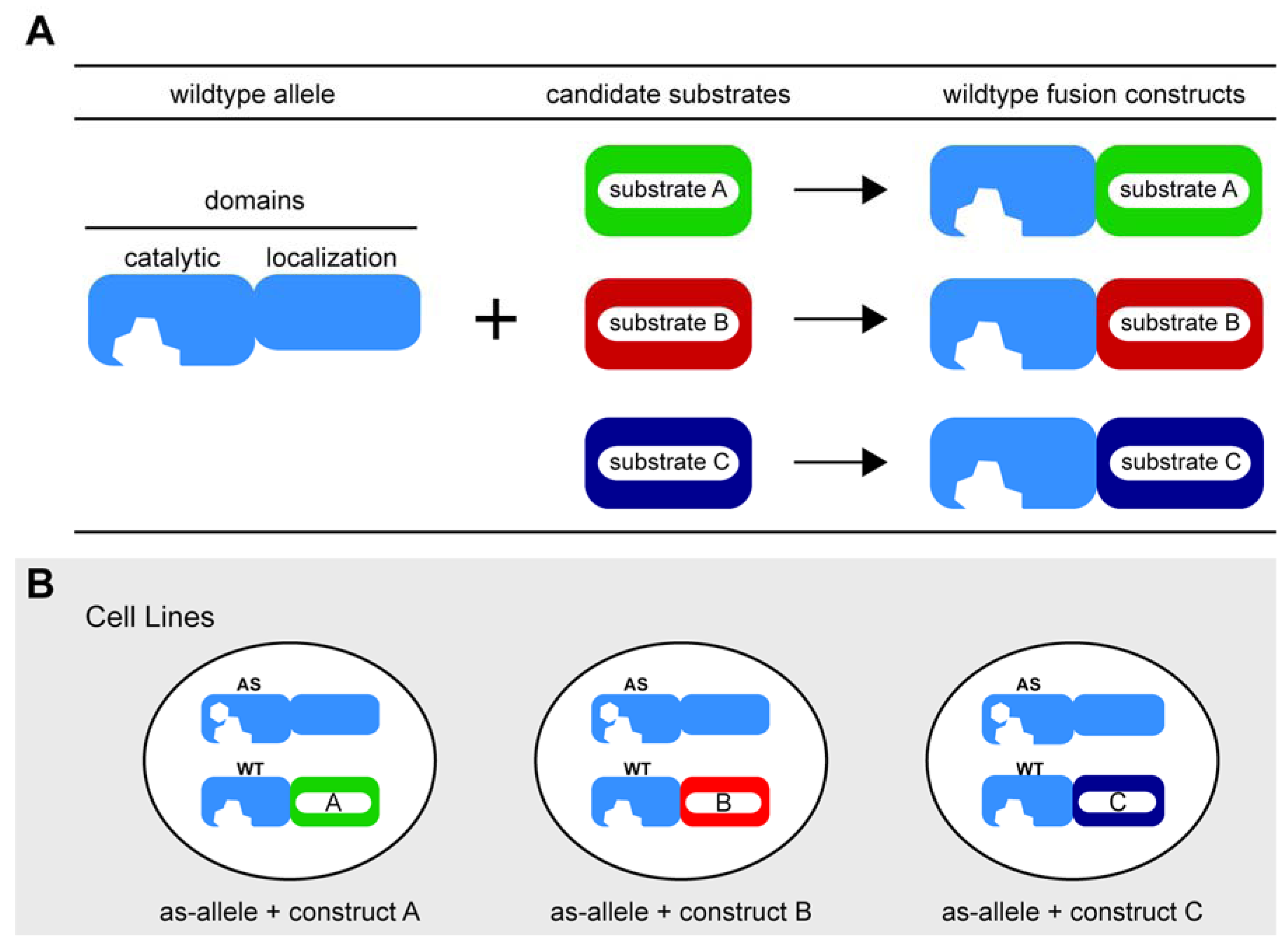
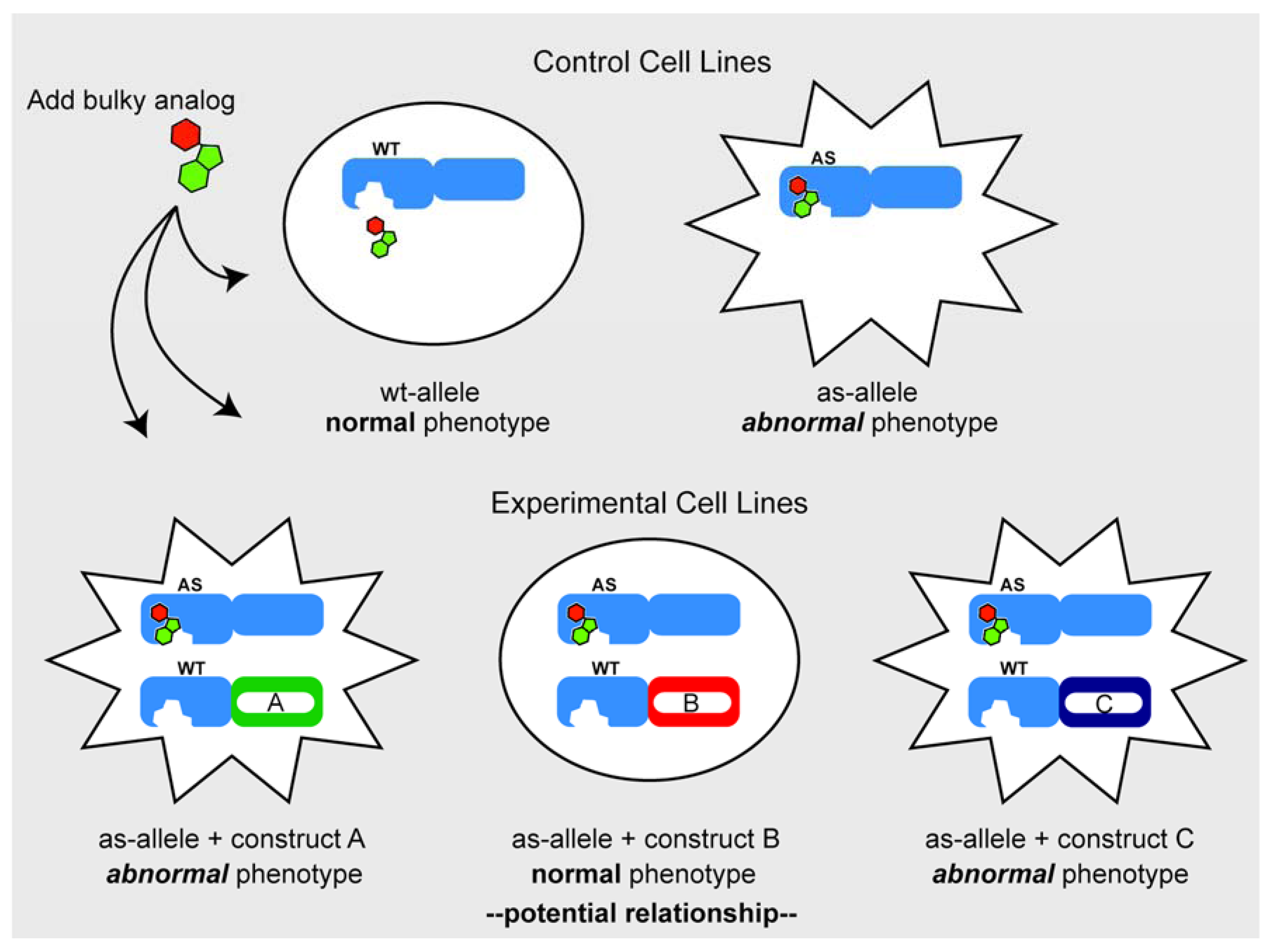
Considerations
4. Fractionating the Problem: Separating Multiple Kinase Functions
5. Perspectives
Acknowledgments
References
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef]
- Siegel, J.J.; Amon, A. New Insights into the Troubles of Aneuploidy. Annu. Rev. Cell Dev. Biol. 2012, in press. [Google Scholar]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef]
- Parsons, G.G.; Spencer, C.A. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol. Cell. Biol. 1997, 17, 5791–5802. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Dulla, K.; Daub, H.; Hornberger, R.; Nigg, E.A.; Körner, R. Quantitative site-specific phosphorylation dynamics of human protein kinases during mitotic progression. Mol. Cell. Proteomics 2010, 9, 1167–1181. [Google Scholar] [CrossRef]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Körner, R.; Greff, Z.; Kéri, G.; Stemmann, O.; Mann, M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Horiuchi, D.; Huskey, N.E.; Kusdra, L.; Wohlbold, L.; Merrick, K.A.; Zhang, C.; Creasman, K.J.; Shokat, K.M.; Fisher, R.P.; Goga, A. Chemical-genetic analysis of cyclin dependent kinase 2 function reveals an important role in cellular transformation by multiple oncogenic pathways. Proc. Natl. Acad. Sci. USA 2012, 109, E1019–E1027. [Google Scholar]
- Merrick, K.A.; Wohlbold, L.; Zhang, C.; Allen, J.J.; Horiuchi, D.; Huskey, N.E.; Goga, A.; Shokat, K.M.; Fisher, R.P. Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol. Cell 2011, 42, 624–636. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Echalier, A.; Cot, E.; Camasses, A.; Hodimont, E.; Hoh, F.; Jay, P.; Sheinerman, F.; Krasinska, L.; Fisher, D. An integrated chemical biology approach provides insight into cdk2 functional redundancy and inhibitor sensitivity. Chem. Biol. 2012, 19, 1028–1040. [Google Scholar] [CrossRef]
- Bishop, A.C.; Shah, K.; Liu, Y.; Witucki, L.; Kung, C.; Shokat, K.M. Design of allele-specific inhibitors to probe protein kinase signaling. Curr. Biol. 1998, 8, 257–266. [Google Scholar] [CrossRef]
- Bishop, A.C.; Ubersax, J.A.; Petsch, D.T.; Matheos, D.P.; Gray, N.S.; Blethrow, J.; Shimizu, E.; Tsien, J.Z.; Schultz, P.G.; Rose, M.D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. [Google Scholar] [CrossRef]
- Frank, C.J.; Hyde, M.; Greider, C.W. Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Mol. Cell 2006, 24, 423–432. [Google Scholar] [CrossRef]
- Hochegger, H.; Dejsuphong, D.; Sonoda, E.; Saberi, A.; Rajendra, E.; Kirk, J.; Hunt, T.; Takeda, S. An essential role for Cdk1 in S phase control is revealed via chemical genetics in vertebrate cells. J. Cell Biol. 2007, 178, 257–268. [Google Scholar] [CrossRef]
- Cipak, L.; Zhang, C.; Kovacikova, I.; Rumpf, C.; Miadokova, E.; Shokat, K.M.; Gregan, J. Generation of a set of conditional analog-sensitive alleles of essential protein kinases in the fission yeast Schizosaccharomyces pombe. Cell Cycle 2011, 10, 3527–3532. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.-E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef]
- Hengeveld, R.C.C.; Hertz, N.T.; Vromans, M.J.M.; Zhang, C.; Burlingame, A.L.; Shokat, K.M.; Lens, S.M.A. Development of a chemical genetic approach for human aurora B kinase identifies novel substrates of the chromosomal passenger complex. Mol. Cell. Proteomics 2012, 11, 47–59. [Google Scholar] [CrossRef]
- Koch, A.; Krug, K.; Pengelley, S.; Macek, B.; Hauf, S. Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci. Signal. 2011, 4, rs6. [Google Scholar] [CrossRef]
- Jones, M.H.; Huneycutt, B.J.; Pearson, C.G.; Zhang, C.; Morgan, G.; Shokat, K.; Bloom, K.; Winey, M. Chemical genetics reveals a role for Mps1 kinase in kinetochore attachment during mitosis. Curr. Biol. 2005, 15, 160–165. [Google Scholar] [CrossRef]
- Maciejowski, J.; George, K.A.; Terret, M.-E.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V. Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J. Cell Biol. 2010, 190, 89–100. [Google Scholar] [CrossRef]
- Sliedrecht, T.; Zhang, C.; Shokat, K.M.; Kops, G.J.P.L. Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS One 2010, 5, e10251. [Google Scholar]
- Burkard, M.E.; Randall, C.L.; Larochelle, S.; Zhang, C.; Shokat, K.M.; Fisher, R.P.; Jallepalli, P.V. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4383–4388. [Google Scholar]
- Snead, J.L.; Sullivan, M.; Lowery, D.M.; Cohen, M.S.; Zhang, C.; Randle, D.H.; Taunton, J.; Yaffe, M.B.; Morgan, D.O.; Shokat, K.M. A coupled chemical-genetic and bioinformatic approach to Polo-like kinase pathway exploration. Chem. Biol. 2007, 14, 1261–1272. [Google Scholar] [CrossRef]
- Koch, A.; Hauf, S. Strategies for the identification of kinase substrates using analog-sensitive kinases. Eur. J. Cell Biol. 2010, 89, 184–193. [Google Scholar] [CrossRef]
- Oppermann, F.S.; Grundner-Culemann, K.; Kumar, C.; Gruss, O.J.; Jallepalli, P.V.; Daub, H. Combination of chemical genetics and phosphoproteomics for kinase signaling analysis enables confident identification of cellular downstream targets. Mol. Cell. Proteomics 2012, 11, O111.012351. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef]
- Blethrow, J.D.; Glavy, J.S.; Morgan, D.O.; Shokat, K.M. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. USA 2008, 105, 1442–1447. [Google Scholar]
- Larochelle, S.; Batliner, J.; Gamble, M.J.; Barboza, N.M.; Kraybill, B.C.; Blethrow, J.D.; Shokat, K.M.; Fisher, R.P. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat. Struct. Mol. Biol. 2006, 13, 55–62. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, As a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Alexander, J.; Lim, D.; Joughin, B.A.; Hegemann, B.; Hutchins, J.R.A.; Ehrenberger, T.; Ivins, F.; Sessa, F.; Hudecz, O.; Nigg, E.A.; et al. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signal. 2011, 4, ra42. [Google Scholar] [CrossRef]
- Santamaria, A.; Wang, B.; Elowe, S.; Malik, R.; Zhang, F.; Bauer, M.; Schmidt, A.; Sillje, H.H.W.; Koerner, R.; Nigg, E.A. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteomics 2010, 10, M110.004457. [Google Scholar]
- Grosstessner-Hain, K.; Hegemann, B.; Novatchkova, M.; Rameseder, J.; Joughin, B.A.; Hudecz, O.; Roitinger, E.; Pichler, P.; Kraut, N.; Yaffe, M.B.; et al. Quantitative phospho-proteomics to investigate the Polo-like kinase 1-dependent phospho-proteome. Mol. Cell. Proteomics 2011, 10, M111.008540. [Google Scholar] [CrossRef]
- Lienhard, G.E. Non-functional phosphorylations? Trends Biochem. Sci. 2008, 33, 351–352. [Google Scholar] [CrossRef]
- Kraybill, B.C.; Elkin, L.L.; Blethrow, J.D.; Morgan, D.O.; Shokat, K.M. Inhibitor scaffolds as new allele specific kinase substrates. J. Am. Chem. Soc. 2002, 124, 12118–12128. [Google Scholar] [CrossRef]
- Allen, J.J.; Li, M.; Brinkworth, C.S.; Paulson, J.L.; Wang, D.; Hübner, A.; Chou, W.H.; Davis, R.J.; Burlingame, A.L.; Messing, R.O.; et al. A semisynthetic epitope for kinase substrates. Nat. Methods 2007, 4, 511–516. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Woodbury, E.L.; Quang, P.N.; Paraz, M.; Blethrow, J.D.; Shah, K.; Shokat, K.M.; Morgan, D.O. Targets of the cyclin-dependent kinase Cdk1. Nature 2003, 425, 859–864. [Google Scholar] [CrossRef]
- Niu, S.; Wang, Z.; Ge, D.; Zhang, G.; Li, Y. Prediction of functional phosphorylation sites by incorporating evolutionary information. Protein Cell 2012, 3, 675–690. [Google Scholar] [CrossRef]
- Heinrich, S.; Windecker, H.; Hustedt, N.; Hauf, S. Mph1 kinetochore localization is crucial and upstream in the hierarchy of spindle assembly checkpoint protein recruitment to kinetochores. J. Cell Sci. 2012, in press. [Google Scholar]
- Ito, D.; Saito, Y.; Matsumoto, T. Centromere-tethered Mps1 pombe homolog (Mph1) kinase is a sufficient marker for recruitment of the spindle checkpoint protein Bub1, but not Mad1. Proc. Natl. Acad. Sci. USA 2012, 109, 209–214. [Google Scholar] [CrossRef]
- Jelluma, N.; Dansen, T.B.; Sliedrecht, T.; Kwiatkowski, N.P.; Kops, G.J.P.L. Release of Mps1 from kinetochores is crucial for timely anaphase onset. J. Cell Biol. 2010, 191, 281–290. [Google Scholar] [CrossRef]
- Liu, D.; Vader, G.; Vromans, M.J.M.; Lampson, M.A.; Lens, S.M.A. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 2009, 323, 1350–1353. [Google Scholar] [CrossRef]
- Lee, K.; Rhee, K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J. Cell Biol. 2011, 195, 1093–1101. [Google Scholar] [CrossRef]
- Kishi, K.; van Vugt, M.A.T.M.; Okamoto, K.; Hayashi, Y.; Yaffe, M.B. Functional dynamics of Polo-like kinase 1 at the centrosome. Mol. Cell. Biol. 2009, 29, 3134–3150. [Google Scholar] [CrossRef]
- Liu, D.; Davydenko, O.; Lampson, M.A. Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J. Cell Biol. 2012, 198, 491–499. [Google Scholar] [CrossRef]
- Burkard, M.E.; Maciejowski, J.; Rodriguez-Bravo, V.; Repka, M.; Lowery, D.M.; Clauser, K.R.; Zhang, C.; Shokat, K.M.; Carr, S.A.; Yaffe, M.B.; et al. Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol. 2009, 7, e1000111. [Google Scholar] [CrossRef]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 2006, 5, 749–757. [Google Scholar]
- Burkard, M.E.; Santamaria, A.; Jallepalli, P.V. Enabling and Disabling Polo-like Kinase 1 Inhibition through Chemical Genetics. ACS Chem. Biol. 2012, 7, 978–981. [Google Scholar] [CrossRef]
- Paschal, C.R.; Maciejowski, J.; Jallepalli, P.V. A stringent requirement for Plk1 T210 phosphorylation during K-fiber assembly and chromosome congression. Chromosoma 2012, in press. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lera, R.F.; Burkard, M.E. The Final Link: Tapping the Power of Chemical Genetics to Connect the Molecular and Biologic Functions of Mitotic Protein Kinases. Molecules 2012, 17, 12172-12186. https://doi.org/10.3390/molecules171012172
Lera RF, Burkard ME. The Final Link: Tapping the Power of Chemical Genetics to Connect the Molecular and Biologic Functions of Mitotic Protein Kinases. Molecules. 2012; 17(10):12172-12186. https://doi.org/10.3390/molecules171012172
Chicago/Turabian StyleLera, Robert F., and Mark E. Burkard. 2012. "The Final Link: Tapping the Power of Chemical Genetics to Connect the Molecular and Biologic Functions of Mitotic Protein Kinases" Molecules 17, no. 10: 12172-12186. https://doi.org/10.3390/molecules171012172