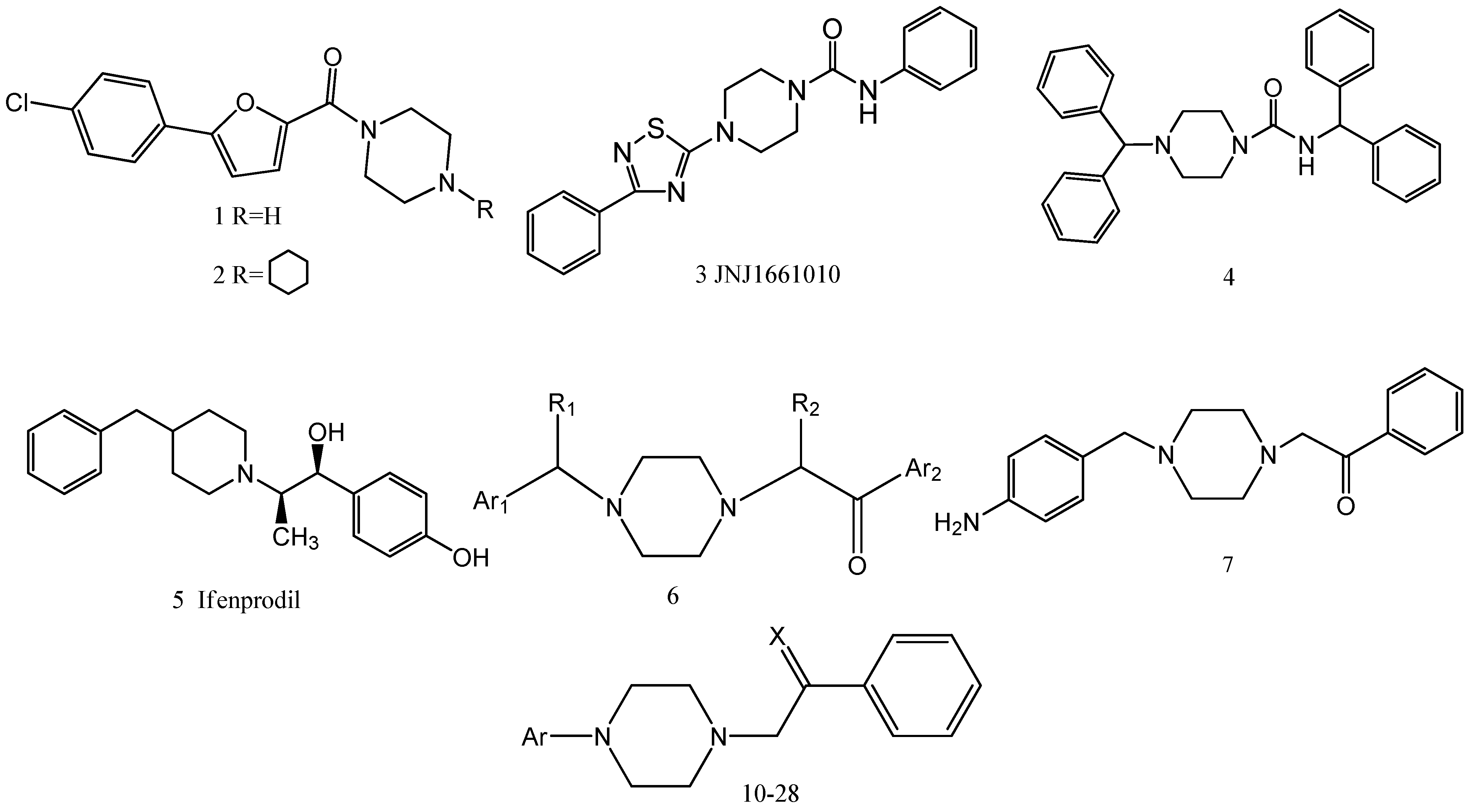
Design, Synthesis and Biological Activity Evaluation of Arylpiperazine Derivatives for the Treatment of Neuropathic Pain

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemistry

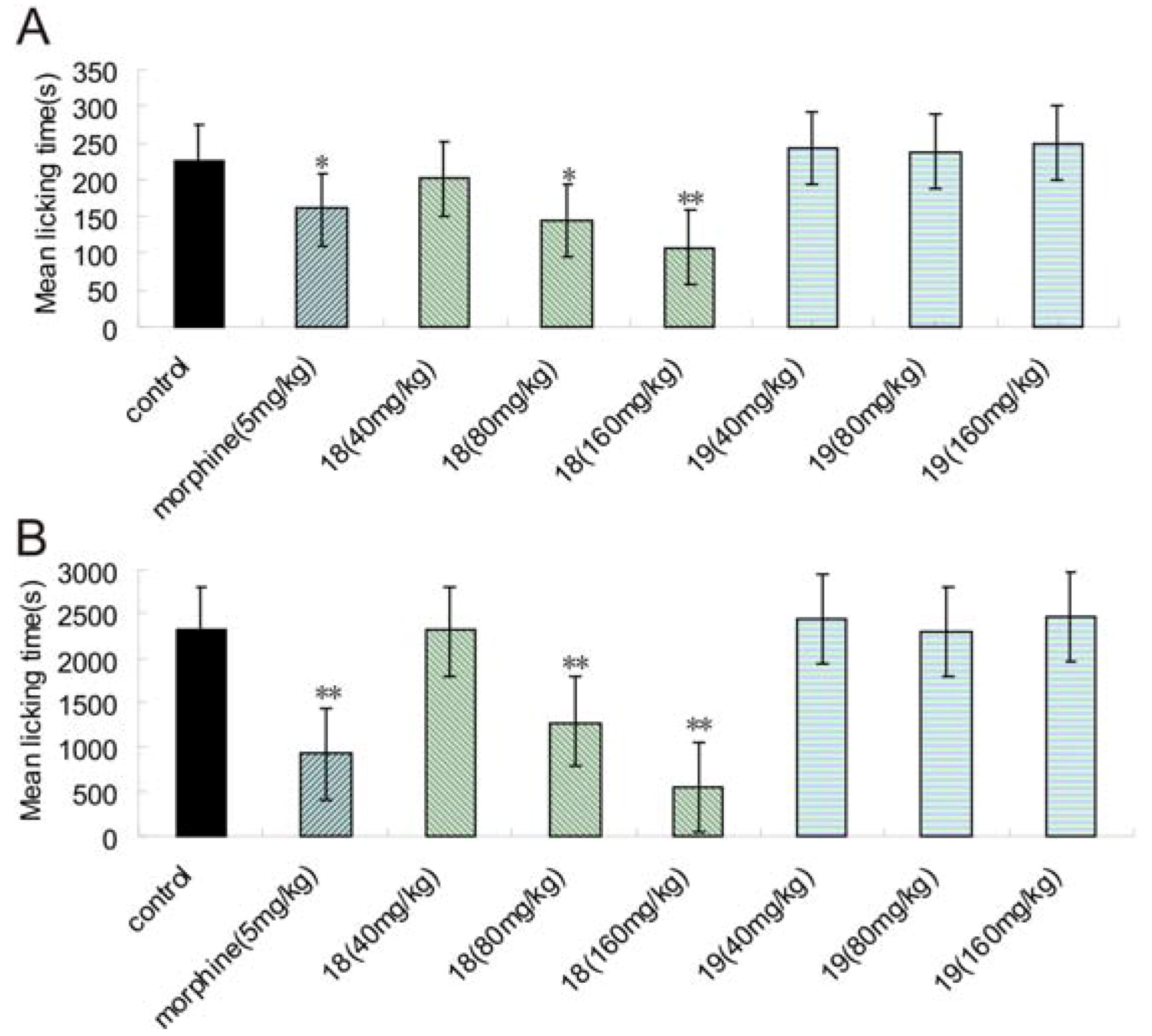
2.2. Hot Plate Test and Writhing Test

{kind=link}
{kind=link}
{kind=link}
| Compound a | Ar | R | Dose (mg/kg) | Licking latency (s) b | Writhing | |||
|---|---|---|---|---|---|---|---|---|
| Before treatment | After treatment (60 min) | Increased rate of latency (%) | Writhes (per 15 min) | Inhibition (%) | ||||
| 10 | 2-OCH3-Ph | - | 10 | 14.1 ± 1.3 | 29.0 ± 0.9 * | 105.7 | 12.1 ± 1.1 | 43.3 |
| 20 | 13.0 ± 0.9 | 37.9 ± 1.3 * | 191.5 | 13.2 ± 3.9 | 38.0 | |||
| 40 | 13.7 ± 1.5 | 29.4 ± 1.5 * | 114.6 | 6.4 ± 2.2 ** | 70.0 | |||
| 11 | 3-OCH3-Ph | - | 10 | 15.5 ± 0.6 | 16.7 ± 2.1 | 7.7 | 7.2 ± 3.6 * | 66.4 |
| 20 | 17.3 ± 0.9 | 19.0 ± 1.2 | 9.8 | 24.4 ± 9.9 | -14.5 | |||
| 40 | 18.4 ± 1.3 | 27.4 ± 4.3 | 48.9 | 4.9 ± 1.3 * | 77.1 | |||
| 12 | 4-OCH3-Ph | - | 10 | 18.3 ± 1.2 | 14.0 ± 0.8 | -23.5 | 17.9 ± 4.2 | 15.8 |
| 20 | 14.5 ± 1.5 | 17.0 ± 1.2 | 17.2 | 19.7 ± 2.8 | 7.3 | |||
| 40 | 12.4 ± 1.4 | 22.0 ± 0.9 | 77.4 | 20.2 ± 2.9 | 5.0 | |||
| 13 | 2-Cl-Ph | - | 10 | 15.6 ± 1.2 | 18.4 ± 1.6 | 17.9 | 11.9 ± 2.7 | 44.3 |
| 20 | 19.9 ± 1.3 | 20.2 ± 2.1 | 1.5 | 14.8 ± 3.6 | 30.5 | |||
| 40 | 15.2 ± 2.1 | 17.6 ± 2.3 | 15.8 | 13.0 ± 4.4 | 38.9 | |||
| 14 | 3-Cl-Ph | - | 10 | 15.9 ± 1.6 | 13.9 ± 1.2 | -12.6 | 9.6 ± 3.8 | 55.0 |
| 20 | 20.2 ± 2.1 | 23.1 ± 1.3 | 14.3 | 7.2 ± 3.4 * | 66.4 | |||
| 40 | 18.1 ± 0.9 | 25.3 ± 2.3 | 39.8 | 6.3 ± 2.2 * | 70.2 | |||
| 15 | 4-Cl-Ph | - | 10 | 16.0 ± 1.7 | 15.8 ± 2.3 | -1.3 | 5.5 ± 1.3 * | 74.1 |
| 20 | 15.5 ± 1.6 | 22.1 ± 1.3 | 42.6 | 8.9 ± 1.0 | 58.0 | |||
| 40 | 18.9 ± 1.4 | 27.1 ± 3.4 | 43.4 | 3.7 ± 1.5 * | 82.4 | |||
| 16 | 2,3-di-Cl-Ph | - | 10 | 17.4 ± 1.3 | 17.9 ± 1.8 | 2.9 | 9.8 ± 3.6 | 54.1 |
| 20 | 19.5 ± 1.5 | 23.3 ± 2.1 | 19.5 | 12.2 ± 2.6 | 42.6 | |||
| 40 | 18.3 ± 1.6 | 26.6 ± 3.9 | 45.4 | 28.6 ± 6.6 | -34.4 | |||
| 17 | 4-F-Ph | - | 10 | 15.5 ± 1.8 | 17.3 ± 0.9 | 11.6 | 32.3 ± 3.2 | -51.8 |
| 20 | 17.3 ± 1.9 | 23.7 ± 1.5 | 37.0 | 25.2 ± 5.7 | -18.3 | |||
| 40 | 18.4 ± 2.1 | 19.8 ± 2.6 | 7.6 | 27.5 ± 2.9 | -29.2 | |||
| 18 | 3-CF3-Ph | - | 10 | 17.6 ± 1.3 | 27.5 ± 2.6 | 56.3 | 5.8 ± 2.0 * | 73.0 |
| 20 | 19.2 ± 2.4 | 39.8 ± 2.4 * | 107.3 | 5.3 ± 1.5 * | 75.2 | |||
| 40 | 16.2 ± 2.3 | 35.0 ± 3.1 * | 116.0 | 4.5 ± 1.4 * | 78.7 | |||
| 19 | 2,3-di-CH3-Ph | - | 10 | 19.3 ± 1.3 | 26.6 ± 3.6 | 37.8 | 5.3 ± 2.0 * | 75.2 |
| 20 | 15.7 ± 1.4 | 33.2 ± 1.8 * | 111.5 | 7.7 ± 2.2 * | 63.7 | |||
| 40 | 16.3 ± 2.3 | 38.2 ± 2.1 * | 134.4 | 10.3 ± 3.2 | 51.8 | |||
| 20 | 6-methoxy-benzo[d]-thiazole | - | 10 | 19.6 ± 2.8 | 21.0 ± 2.3 | 7.1 | 6.9 ± 2.0 * | 67.6 |
| 20 | 16.9 ± 1.6 | 18.1 ± 2.4 | 7.1 | 11.2 ± 3.3 | 47.2 | |||
| 40 | 18.0 ± 1.9 | 26.4 ± 3.1 | 46.7 | 7.3 ± 1.8 * | 65.7 | |||
| 21 | 6-methyl-benzo[d]-thiazole | - | 10 | 18.7 ± 1.6 | 19.1 ± 3.1 | 2.1 | 11.4 ± 2.4 | 46.3 |
| 20 | 17.6 ± 2.8 | 16.3 ± 1.5 | -7.4 | 15.9 ± 4.8 | 25.9 | |||
| 40 | 16.2 ± 2.3 | 24.1 ± 1.6 | 48.8 | 9.3 ± 5.7 | 56.5 | |||
| 22 | 4-methyl-benzo[d]-thiazole | - | 10 | 15.0 ± 1.3 | 21.9 ± 2.1 | 46.0 | 32.5 ± 6.1 | -52.8 |
| 20 | 15.9 ± 3.1 | 17.1 ± 2.6 | 7.5 | 12.6 ± 3.7 | 40.7 | |||
| 40 | 18.5 ± 1.8 | 22.5 ± 2.4 | 21.6 | 9.5 ± 4.2 | 55.6 | |||
| 23 | 6-chloro-benzo[d]-thiazole | - | 10 | 16.3 ± 1.3 | 24.9 ± 1.9 | 52.8 | 15.4 ± 6.3 | 27.8 |
| 20 | 18.5 ± 1.8 | 26.2 ± 3.8 | 41.6 | 33.7 ± 5.7 | -58.3 | |||
| 40 | 20.6 ± 1.9 | 25.0 ± 2.6 | 5.9 | 13.0 ± 4.3 | 38.9 | |||
| 24 | 4-chloro-benzo[d]-thiazole | - | 10 | 18.6 ± 2.1 | 28.5 ± 1.0 * | 53.2 | 5.8 ± 2.1 * | 75.2 |
| 20 | 17.5 ± 1.3 | 28.6 ± 0.6 * | 63.4 | 21.5 ± 5.9 | -0.9 | |||
| 40 | 16.1 ± 1.7 | 22.4 ± 2.5 | 39.1 | 37.3 ± 9.1 | -75.0 | |||
| 25 | 2-pyrimidine | - | 10 | 17.3 ± 1.8 | 24.5 ± 3.7 | 41.6 | 11.2 ± 3.4 | 47.3 |
| 20 | 15.3 ± 2.0 | 15.1 ± 1.9 | -1.3 | 8.0 ± 2.1 | 43.9 | |||
| 40 | 20.3 ± 1.6 | 24.7 ± 1.8 | 21.7 | 7.6 ± 5.2 * | 64.2 | |||
| 26 | 4-OCH3-Ph | H | 10 | 13.0 ± 1.9 | 16.6 ± 2.1 | 27.7 | 38.7 ± 6.6 | -81.6 |
| 20 | 11.4 ± 2.4 | 19.1 ± 1.6 | 67.5 | 43.7 ± 5.1 | -105.3 | |||
| 40 | 17.4 ± 1.3 | 23.8 ± 1.7 | 36.8 | 25.8 ± 7.7 | -21.1 | |||
| 27 | 2-OCH3-Ph | H | 10 | 18.9 ± 2.4 | 25.4 ± 2.1 | 34.4 | 29.2 ± 9.9 | -37.1 |
| 20 | 15.0 ± 2.3 | 20.6 ± 1.6 | 37.3 | 23.3 ± 6.4 | -9.4 | |||
| 40 | 13.8 ± 1.8 | 21.9 ± 1.4 | 58.7 | 11.7 ± 3.1 | 45.1 | |||
| 28 | 2-OCH3-Ph | C2H5 | 10 | 17.0 ± 1.5 | 24.1 ± 2.3 | 45.3 | 16.4 ± 3.3 | 22.9 |
| 20 | 19.0 ± 1.5 | 27.3 ± 4.8 | 43.7 | 11.1 ± 2.4 | 47.8 | |||
| 40 | 17.6 ± 1.4 | 26.4 ± 1.3 | 50.0 | 34.5 ± 10.2 | -62.2 | |||
| saline | - | - | - | 16.4 ± 2.3 | 19.2 ± 1.8 | 17.1 | 21.3 ± 3.7 | - |
| acetylsalicylic acid | - | - | 100 | - | - | - | 5.2 ± 0.3 ** | 75.6 |
| morphine | - | - | 5 | 15.4 ± 1.6 | 36.3 ± 2.1 * | 135.7 | - | |
2.3. Acute Toxicity
| Compound a | LD50 (mg/kg) |
|---|---|
| 10 | 477.0 (357.2-637.1) |
| 18 | > 2,000 |
| 19 | > 2,000 |
2.4. Exploratory Locomotor Activity
| Compound | Dose (mg/kg, po) | Average speed (cm/s) | ||||
|---|---|---|---|---|---|---|
| Before treatment | After treatment | |||||
| 30 min | 60 min | 90 min | 120 min | |||
| Control | - | 8.48 ± 3.02 | 7.95 ± 2.85 | 7.64 ± 2.94 | 7.09 ± 2.89 | 7.98 ± 2.89 |
| clonazepam | 15 | 7.42 ± 2.62 | 3.28 ± 2.03 ** | 3.16 ± 2.32 ** | 3.41 ± 3.75 ** | 2.99 ± 1.89 ** |
| 18 | 40 | 8.56 ± 2.89 | 7.94 ± 2.50 | 7.75 ± 1.72 | 7.51 ± 1.74 | 7.88 ± 3.01 |
| 80 | 7.63 ± 1.9 | 7.54 ± 3.05 | 7.03 ± 3.55 | 7.75 ± 2.95 | 7.46 ± 2.47 | |
| 160 | 7.49 ± 2.51 | 7.94 ± 2.72 | 7.56 ± 3.65 | 7.47 ± 2.17 | 6.98 ± 2.27 | |
| 19 | 40 | 7.16 ± 2.07 | 6.75 ± 3.95 | 6.71 ± 2.02 | 5.62 ± 2.40 | 5.64 ± 3.13 |
| 80 | 8.36 ± 3.23 | 7.83 ± 3.40 | 6.82 ± 3.26 | 7.44 ± 2.61 | 7.68 ± 2.43 | |
| 160 | 8.12 ± 2.61 | 6.36 ± 3.00 | 6.81 ± 2.84 | 6.88 ± 3.05 | 6.55 ± 2.99 | |
2.5. Formalin Test
2.6. Spared Nerve Injury (SNI) and Chronic Constriction Injury (CCI) Test

| Compound | Dose (mg/kg, po) | Number of rats | Mechanical allodynia (g) | |||
|---|---|---|---|---|---|---|
| Baseline | Before administration (decreased rate of pain threshold %) a | Single administration c (increased rate of pain threshold %) b | Repeated administration c (increased rate of pain threshold %) b | |||
| Sham | - | 10 | 55.1 ± 4.4 | 49.7 ± 12.4 ** | 48.1 ± 11.1 ** | 55.8 ± 8.6 ** |
| 9.7 | -3.3 | 12.3 | ||||
| Control | - | 10 | 61.86 ± 5.48 | 29.7 ± 4.3 ## | 30.9 ± 4.8 ## | 28.7 ± 3.8 ## |
| 52.0 | 3.9 | -3.2 | ||||
| Gabapentin | 40 | 10 | 60.9 ± 5.6 | 28.5 ± 5.3 ## | 38.1 ± 5.4 * | 40.6 ± 7.9 ** |
| 53.3 | 33.7 | 42.7 | ||||
| 18 | 40 | 10 | 60.3 ± 6.0 | 28.7 ± 4.1 ## | 35.2 ± 8.6 | 30.5 ± 8.5 |
| 52.3 | 22.3 | 5.9 | ||||
| 80 | 10 | 60.8 ± 3.2 | 28.4 ± 3.6 ## | 37.8 ± 5.5 * | 36.8 ± 4.8 * | |
| 53.3 | 33.2 | 29.6 | ||||
| 160 | 10 | 63.4 ± 4.2 | 28.7 ± 5.1 ## | 40.3 ± 5.4 ** | 38.1 ± 8.5 * | |
| 54.7 | 40.4 | 32.8 | ||||
| 19 | 40 | 10 | 61.8 ± 5.8 | 28.4 ± 7.2 ## | 32.9 ± 9.1 | 34.5 ± 2.3 |
| 54.1 | 15.8 | 21.6 | ||||
| 80 | 10 | 55.3 ± 13.3 | 29.0 ± 5.4 ## | 33.9 ± 8.2 | 30.2 ± 6.7 | |
| 47.5 | 16.7 | 4.0 | ||||
| 160 | 10 | 58.8 ± 5.8 | 28.3 ± 5.6 ## | 30.0 ± 5.4 | 33.1 ± 3.3 | |
| 51.8 | 5.8 | 16.8 | ||||
| Compound | Dose (mg/kg, po) | Number of rats | Mechanical allodynia (g) | |||
|---|---|---|---|---|---|---|
| Baseline | Before administration (decreased rate of pain threshold %) a | Single administration c (increased rate of pain threshold %) b | repeated administration c (increased rate of pain threshold %) b | |||
| sham | - | 10 | 56.0 ± 5.9 | 51.2 ± 9.0 ** | 53.4 ± 9.8 ** | 49.0 ± 7.3 ** |
| 8.7 | 4.4 | -4.2 | ||||
| control | - | 10 | 58.3 ± 6.7 | 31.25 ± 5.1 ## | 31.8 ± 5.5 ## | 31.7 ± 6.4 ## |
| 46.5 | 2.1 | 1.73 | ||||
| Gabapentin | 40 | 10 | 59.44 ± 6.20 | 31.4 ± 4.4 ## | 39.1 ± 4.9 ** | 37.3 ± 7.5 * |
| 47.2 | 24.6 | 18.7 | ||||
| 18 | 40 | 10 | 56.7 ± 4.7 | 34.1 ± 2.4 ## | 36.2 ± 4.8 | 33. 6 ± 6.7 |
| 39.9 | 6.3 | -1.5 | ||||
| 80 | 10 | 58.7 ± 8.2 | 31.9 ± 3.5 ## | 37.5 ± 8.3 * | 38.0 ± 6.2 * | |
| 45.68 | 17.6 | 19.2 | ||||
| 160 | 10 | 59.0 ± 4.4 | 32.1 ± 6.0 ## | 35.9 ± 6.5 | 32.9 ± 4.0 | |
| 45.6 | 11.9 | 2.6 | ||||
| 19 | 40 | 10 | 61.6 ± 4.9 | 31.3 ± 3.4 ## | 32.9 ± 2.7 | 33.4 ± 3.4 |
| 49.2 | 5.1 | 6.9 | ||||
| 80 | 10 | 56.4 ± 5.6 | 31.1 ± 4.8 ## | 37.1 ± 3.5 * | 32.0 ± 4.7 | |
| 44.9 | 19.3 | 2.8 | ||||
| 160 | 10 | 59.6 ± 5.2 | 31.1 ± 4.6 ## | 32.1 ± 3.2 | 29.8 ± 7.2 | |
| 47.8 | 3.2 | -4.3 | ||||
| Compound | Dose (mg/kg, po) | Number of rats | Latency (s) | |||
|---|---|---|---|---|---|---|
| Baseline | Before administration (decreased rate of latency %) a | Single administration c (increased rate of latency %) b | Repeated administration c (increased rate of latency %) b | |||
| Sham | - | 10 | 14.7 ± 2.9 | 13.2 ± 2.7 * | 13.41 ± 3.4 * | 14.2 ± 4.1 * |
| 9.9 | 1.4 | 7.2 | ||||
| Control | - | 10 | 15.1 ± 2.9 | 10.2 ± 3.1 ## | 10.4 ± 3.4 ## | 11.2 ± 3.3 ## |
| 32.5 | 2.5 | 9.5 | ||||
| Gabapentin | 40 | 10 | 15.7 ± 3.6 | 11.9 ± 3.5 # | 17.9 ± 7.6 ** | 17.0 ± 13.9 |
| 24.6 | 50.5 | 42.9 | ||||
| 18 | 40 | 10 | 13.0 ± 1.7 | 8.7 ± 2.3 ## | 13.4 ± 4.3 * | 14.5 ± 3.9 * |
| 33.1 | 53.5 | 66.4 | ||||
| 80 | 10 | 15.5 ± 3.4 | 10.5 ± 3.7 ## | 13.4 ± 4.1 | 11.2 ± 3.0 | |
| 32.34 | 27.7 | 6.7 | ||||
| 160 | 10 | 15.5 ± 5.5 | 9.3 ± 4.3 ## | 18.6 ± 9.1 ** | 13.8 ± 7.3 | |
| 39.9 | 99.3 | 48.0 | ||||
| 19 | 40 | 10 | 15.7 ± 2.4 | 9.6 ± 2.1 ## | 11.5 ± 3.9 | 13.5 ± 8.1 |
| 39.0 | 20.4 | 40.9 | ||||
| 80 | 10 | 12.9 ± 1.9 | 8.7 ± 2.7 ## | 10.5 ± 2.9 | 10.5 ± 1.7 | |
| 33.1 | 21.2 | 21.4 | ||||
| 160 | 10 | 13.0 ± 1.9 | 9.2 ± 2.3 ## | 11.5 ± 2.6 | 13.1 ± 4.2 | |
| 29.3 | 25.6 | 42.0 | ||||
2.7. Mechanism of Action
2.8. Structure-Activity Relationships (SAR)
| Compound | Concentration (mol/L) | Emax10 μM a (%) | |||||
|---|---|---|---|---|---|---|---|
| μ | δ | κ | 5-HT2A | 5-HT1A | 5-HT uptake | ||
| Naloxone | 10-5 | 100 | 100 | 100 | - | - | - |
| Aripiprazole | 10-5 | - | - | - | 100 | - | - |
| 5-HT | 10-5 | - | - | - | - | 100 | - |
| Duloxetine | 10-5 | - | - | - | - | - | 90.6 |
| w-conotoxinGVIA | 10-5 | - | - | - | - | - | - |
| 18 | 10-5 | 18.1 | 0 | 2.9 | 57.7 | 96.9 | 59.3 |
3. Experimental
3.1. General
3.1.1. General Procedure for the Synthesis of Compounds 10-25 [20]
3.1.2. General Procedure for the Synthesis of Compounds 26-28
3.2. Pharmacology
3.2.1. Animals
3.2.1.1. Acetic Acid-Induced Abdominal Constrictions Assay [21]

3.2.1.2. Hot Plate Test [22]

3.2.1.3. Acute Toxicity Study (LD50) [23]
3.2.1.4. Exploratory Locomotor Activity [24]
3.2.1.5. Formalin Test [28]
3.3. Spared Nerve Injury (SNI) Neuropathy Assay
3.3.1. Group and Design
3.3.2. Surgery [31,41]
3.3.3. Mechanical Withdrawal Threshold [41]
3.4. Chronic Constriction Injury (CCI) Neuropathy Assay
3.4.1. Group and Design
3.4.2. Surgey [32]
3.4.3. Behavioral Testing
3.4.3.1. Mechanical Withdrawal Threshold (MWT) [42]
3.4.3.2. Thermal Withdrawal Latency (TWL) [43]
3.5. 5-HT1A Binding Assay [44]

3.6. 5-HT2A Binding Assay [45]

3.7. 5-HT Uptake Binding Assay [46]
3.8. μ-Opioid Receptor Binding Assay [47]
3.9. k-Opioid Receptor Binding Assay [47]
3.10. δ-Opioid Receptor Binding Assay [47]
3.11. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Woolf, C.J. The pathophysiology of peripheral neuropathic pain-abnormal peripheral input and abnormal central processing. Acta Neurochir. Suppl. (Wien) 1993, 58, 125–130. [Google Scholar]
- Galer, B.S. Neuropathic pain of peripheral origin: Advances in pharmacologic treatment. Neurology 1995, 45, S17–S25. [Google Scholar] [CrossRef]
- Attal, N.; Martinez, V. Recent developments in the pharmacological management of neuropathic pain. Eur. Neurol. J. 2010, 2, 25–30. [Google Scholar]
- Naef, M.; Curatolo, M.; Petersen-Felix, S.; Arendt-Nielsen, L.; Zbinden, A.; Brenneisen, R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain 2003, 105, 79–88. [Google Scholar] [CrossRef]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl. J. Med. 2000, 23, 1520–1528. [Google Scholar]
- Mattia, C.; Coluzzi, F. Antidepressants in chronic neuropathic pain. Mini Rev. Med. Chem. 2003, 3, 773–784. [Google Scholar] [CrossRef]
- Gutierrez-Alvarez, A.M.; Beltrán-Rodríguez, J.; Moreno, C.B. Antiepileptic drugs in treatment of pain caused by diabetic neuropathy. J. Pain Symptom Manag. 2007, 34, 201–208. [Google Scholar] [CrossRef]
- White, P.F. The changing role of non-opioid analgesic techniques in the management of postoperative pain. Anesth. Analg. 2005, 101, 5–22. [Google Scholar] [CrossRef]
- Gilron, I.; Coderre, T.J. Emerging drugs in neuropathic pain. Expert Opin. Emerg. Drugs 2007, 12, 113–126. [Google Scholar] [CrossRef]
- Chong, M.S.; Brandner, B. Neuropathic agents and pain. New strategies. Biomed. Pharmacother. 2006, 60, 318–322. [Google Scholar] [CrossRef]
- Gan, L.L.; Cai, J.L.; Zhou, C.H. Advances in piperazine-containing compounds as receptor ligands. Chin. Pharm. J. 2009, 44, 1361–1368. [Google Scholar]
- Gan, L.L.; Lu, Y.H.; Zhou, C.H. Advances in the research of piperazine compounds as enzyme inhibitors. Chin. J. Biochem. Pharm. 2009, 30, 127–131. [Google Scholar]
- Willems, L.I.; Ilzerman, A.P. Small molecule antagonists for chemokine CCR3 receptors. Med. Chem. Res. 2010, 30, 778–817. [Google Scholar]
- Karbarz, M.J.; Luo, L.; Chang, L.; Tham, C.S.; Palmer, J.A.; Wilson, S.J.; Wennerholm, M.L.; Brown, S.M.; Scott, B.P.; Apodaca, R.L.; et al. Biochemical and biological properties of 4-(3-phenyl-[1,2,4]thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a mechanism-based inhibitor of fatty acid amide hydrolase. Anesth. Analg. 2009, 108, 316–329. [Google Scholar] [CrossRef]
- Drizin, I.; Gregg, R.J.; Scanio, M.J.; Shi, L.; Gross, M.F.; Atkinson, R.N.; Thomas, J.B.; Johnson, M.S.; Carroll, W.A.; Marron, B.E.; et al. Discovery of potent furan piperazine sodium channel blockers for treatment of neuropathic pain. Bioorg. Med. Chem. 2008, 16, 6379–6386. [Google Scholar]
- Pajouhesh, H.; Feng, Z.P.; Ding, Y.B.; Zhang, L.Y.; Pajouhesh, H.; Morrison, J.L.; Belardetti, F.; Tringham, E.; Simonson, E.; Vanderah, T.W.; et al. Structure-activity relationships of diphenylpiperazine N-type calcium channel inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1378–1383. [Google Scholar]
- Borza, I.; Domany, G. NR2B selective NMDA antagonists: The evolution of the ifenprodil-type pharmacophore. Curr. Top. Med. Chem. 2006, 6, 687–695. [Google Scholar] [CrossRef]
- Li, J.Q.; Huang, L.Y.; Chen, J.X.; Weng, Z.J.; Zhang, C.N. Design and synthesis of aralkyl-ketone piperazine derivatives and their antalgic activities. Yao Xue Xue Bao 2007, 42, 1166–1175, in Chinese.. [Google Scholar]
- Li, J.Q.; Huang, L.Y.; Chen, J.X.; Weng, Z.J.; Zhang, C.N. Synthesis and central none-opioid analgesic activity of SIPI5047. Yao Xue Xue Bao 2008, 43, 611–618. (in Chinese).. [Google Scholar]
- Zhang, W.; Curran, D.P.; Chen, C.H.-T. Use of fluorous silica gel to separate fluorous thiol quenching derivatives in solution-phase parallel synthesis. Tetrahedron 2002, 58, 3871–3875. [Google Scholar] [CrossRef]
- Coolier, H.O.J.; Dinneen, L.C.; Johnson, C.A.; Schneider, C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br. J. Pharmacol. Chemother. 1968, 32, 295–310. [Google Scholar]
- Kuraishi, Y.; Harada, Y.; Aratani, S.; Satoh, M.; Takagi., H. Separate involvement of the spinal noradrenergic and serotonergic systems in morphine analgesia: The differences in mechanical and thermal algesic tests. Brain Res. 1983, 273, 245–252. [Google Scholar] [CrossRef]
- Litchfield, J.T.; Wilcoxon, F. A simplified method of evaluating dose-effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
- Butini, S.; Gemma, S.; Campiani, G.; Franceschini, S.; Trotta, F.; Borriello, M.; Ceres, N.; Ros, S.; Coccone, S.S.; Bernetti, M.; et al. Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: Design, synthesis, and effects on behavior. J. Med. Chem. 2009, 52, 151–169. [Google Scholar]
- Hurley, R.W.; Chatterjea, D.; Rose Feng, M.H.; Taylor, C.P.; Hammond, D.L. Gabapentin and pregabalin can interact synergistically with naproxen to produce antihyperalgesia. Anesthesiology 2002, 97, 1263–1273. [Google Scholar]
- Pedersen, L.H.; Blackburn-Munro, G. Pharmacological characterisation of place escape/avoidance behaviour in the rat chronic constriction injury model of neuropathic pain. Psychopharmacology 2006, 185, 208–217. [Google Scholar] [CrossRef]
- Hayashida, K.; Parker, R.; Eisenach, J.C. Oral gabapentin activates spinal cholinergic circuits to reduce hypersensitivity after peripheral nerve injury and interacts synergistically with oral donepezil. Anesthesiology 2007, 106, 1213–1219. [Google Scholar] [CrossRef]
- Bars, D.L.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Dworkin, R.H. An overview of neuropathic pain: Syndromes, symptoms, signs, and several mechanisms. Clin. J. Pain. 2002, 18, 343–349. [Google Scholar] [CrossRef]
- Butera., J.A. Current and emerging targets to treat neuropathic pain. J. Med. Chem. 2007, 50, 2543–2546. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Mico, J.A.; Berrocoso, E.; Ortega-Alvaro, A.; Gibert-Rahola, J.; Rojas-Corrales, M.O. The role of 5-HT1A receptors in research strategy for extensive pain treatment. Curr. Top. Med. Chem. 2006, 6, 1997–2003. [Google Scholar] [CrossRef]
- Fasmer, O.B.; Berge, O.G.; Post, C.; Hole, K. Effects of the putative 5-HT1A receptor agonist 8-OH-2-(di-n-propylamino)tetralin on nociceptive sensitivity in mice. Pharmacol. Biochem. Behav. 1986, 25, 883–888. [Google Scholar] [CrossRef]
- Bardin, L.; Tarayre, J.P.; Malfetes, N.; Koek, W.; Colpaert, F.C. Profound, non-opioid analgesia produced by the high-efficacy 5-HT1A agonist F 13640 in the formalin model of tonic nociceptive pain. Pharmacology 2003, 67, 182–194. [Google Scholar] [CrossRef]
- Deseure, K.R.; Adriaensen, H.F.; Colpaert, F.C. Effects of the combined continuous administration of morphine and the high-efficacy 5-HT1A agonist, F 13640 in a rat model of trigeminal neuropathic pain. Eur. J. Pain 2004, 8, 547–554. [Google Scholar] [CrossRef]
- Kiss, I.; Degryse, A.D.; Bardin, L.; Gomez de, S.I.A.; Colpaert, F.C. The novel analgesic, F 13640, produces intra- and postoperative analgesia in a rat model of surgical pain. Eur. J. Pharmacol. 2005, 523, 29–39. [Google Scholar] [CrossRef]
- Paluchowska, M.; Mokrosz, M.J.; Charakchieva-Minol, S. Duszyńska, B.; Kozioł, A.; Wesołowska, A.; Stachowicz, K.; Chojnacka-Wójcik, E. Novel 4-alkyl-1-arylpiperazines and 1,2,3,4-tetrahydroisoquinolines containing diphenylmethylamino or diphenylmethoxy fragment with differentiated 5-HT1A/5-HT2A/D2 receptor activity. Pol. J. Pharmacol. 2003, 55, 543–552. [Google Scholar]
- Obniska, J.; Pawlowski, M.; Kolaczkowski, M.; Czopek, A.; Duszynska, B.; Klodzinska, A.; Tatarczynska, E.; Chojnacka-Wojcik, E. Synthesis and 5-HT1A/5-HT2A receptor affinity of new N-[(4-arylpiperazin-1yl)-propyl] derivatives of 3-spirocyclohexanepyrrolidine-2,5-dione and 3-spiro-β-tetralonepyrrolidine-2,5-dione. Pol. J. Pharmacol. 2003, 55, 553–557. [Google Scholar]
- Obniska, J.; Kolaczkowski, M.; Charakchieva-Minol, S.; Nedza, K.; Dybala, M.; Bojarski, A.J.; Synthesis, anticonvulsant properties and 5-HT1A/5-HT2A receptor affinity of new N-[(4-arylpiperazin-1yl)-propyl]-2-aza-spiro[4.4]-nonane and [4.5]decane-1,3-dione derivatives. Pharmacol. Rep. 2005, 57, 336–344. [Google Scholar]
- Erichsen, H.K.; Blackburn-Munro, G. Pharmacological characterisation of the spared nerve injury model of neuropathic pain. Pain 2002, 98, 151–161. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.J. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Frecentese, F.; Fiorino, F.; Perissutti, E.; Severino, B.; Magli, E.; Esposito, A.; Angelis, F.D.; Massarelli, P.; Nencini, C.; Viti, B.; et al. Efficient microwave combinatorial synthesis of novel indolic arylpiperazine derivatives as serotoninergic ligands. Eur. J. Med. Chem. 2010, 45, 752–759. [Google Scholar]
- Brea, J.; Rodrigo, J.; Carrieri, A.; Sanz, F.; Cadavid, M.I.; Enguix, M.J.; Villazon, M.; Mengod, G.; Caro, Y.; Masaguer, C.F.; et al. New serotonin 5-HT(2A), 5-HT(2B), and 5-HT(2C) receptor antagonists: Synthesis, pharmacology, 3D-QSAR, and molecular modeling of (aminoalkyl)benzo and heterocycloalkanoes. J. Med. Chem. 2002, 45, 54–71. [Google Scholar] [CrossRef]
- Hatzenbuhler, N.T.; Baudy, R.; Evrard, D.A.; Failli, A.; Harrison, B.L.; Lenicek, S.; Mewshaw, R.E.; Saab, A.; Shah, U.; Sze, J.; et al. Advances toward new antidepressants with dual serotonin transporter and 5-HT1A receptor affinity within a class of 3-aminochroman derivatives. Part 2. J. Med. Chem. 2008, 51, 6980–7004. [Google Scholar]
- Shinkai, H.; Ito, T.; Iida, T.; Kitao, Y.; Yamada, H.; Uchida, I. 4-Aminoquinolines: Novel nociceptin antagonists with analgesic activity. J. Med. Chem. 2000, 43, 4667–4677. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, Y.; Wang, G.; Xu, X.; Liu, B.-F.; Li, J.; Zhang, G. Design, Synthesis and Biological Activity Evaluation of Arylpiperazine Derivatives for the Treatment of Neuropathic Pain. Molecules 2011, 16, 5785-5806. https://doi.org/10.3390/molecules16075785
Chen Y, Wang G, Xu X, Liu B-F, Li J, Zhang G. Design, Synthesis and Biological Activity Evaluation of Arylpiperazine Derivatives for the Treatment of Neuropathic Pain. Molecules. 2011; 16(7):5785-5806. https://doi.org/10.3390/molecules16075785
Chicago/Turabian StyleChen, Yin, Guan Wang, Xiangqing Xu, Bi-Feng Liu, Jianqi Li, and Guisen Zhang. 2011. "Design, Synthesis and Biological Activity Evaluation of Arylpiperazine Derivatives for the Treatment of Neuropathic Pain" Molecules 16, no. 7: 5785-5806. https://doi.org/10.3390/molecules16075785