Quantitative Mass Spectrometry for Bacterial Protein Toxins — A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis

Abstract
:1. Introduction


2. Mass Spectrometric Detection of Toxin Activity
2.1. MALDI-TOF MS for peptides

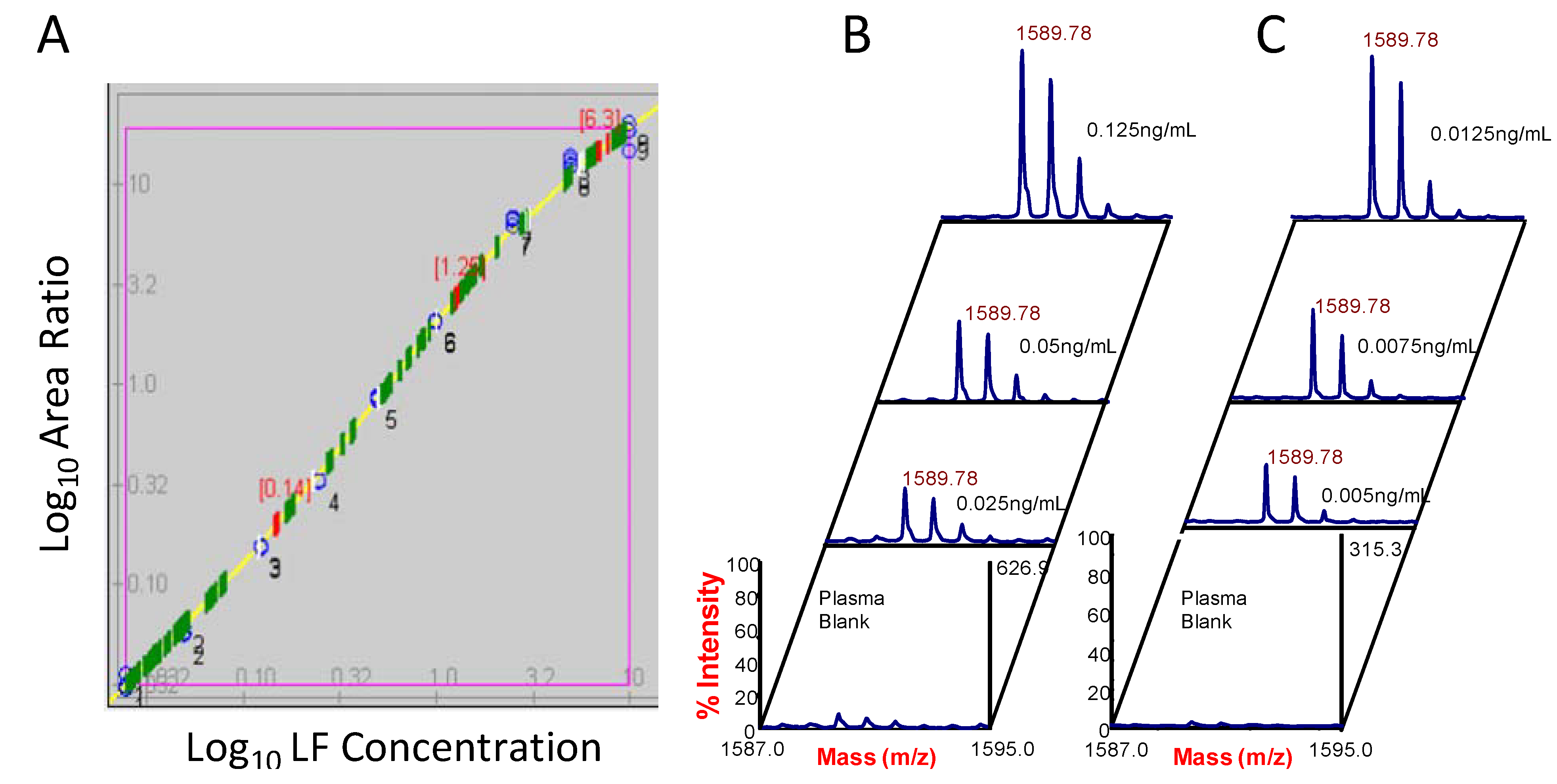
2.2. Isotope dilution quantification for MALDI-TOF MS

{kind=link}
{kind=link}
{kind=link}
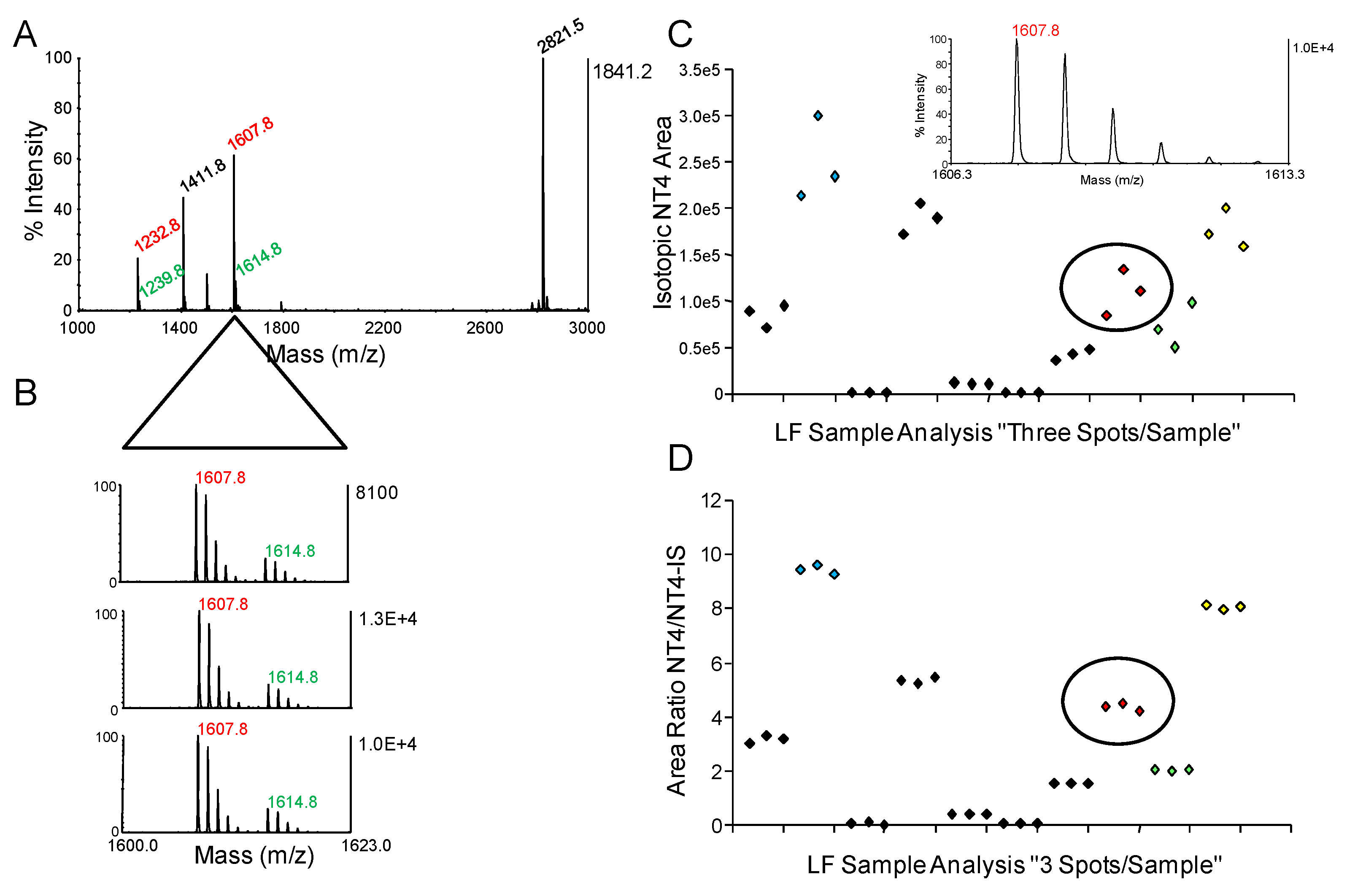{kind=link}
{kind=link}
{kind=link}
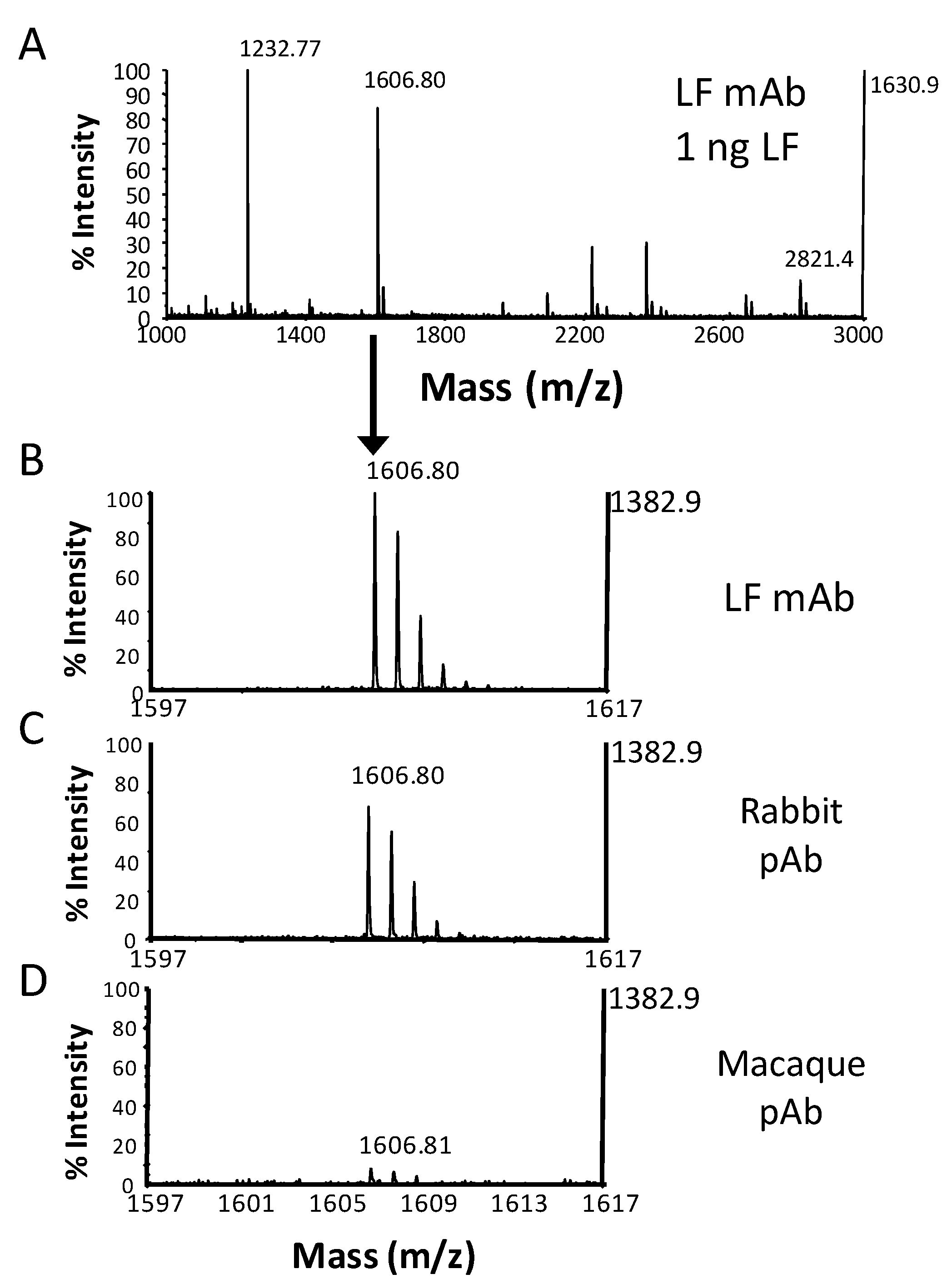{kind=link}
{kind=link}
| Name | Sequence | [M + H]+ | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 ↓ P1’ | ||||||||||||||||||||||||||||||||||||||||||||||
| MAPKK-11-25 | (M1) | M | P | K | K | K | P | T | P | I | Q | L | N | P | A | P | D | G | S | A | V | N | G | T | S | S | ||||||||||||||||||||
| MAPKK-21-27 | (M2) | M | L | A | R | R | K | P | V | L | P | A | L | T | I | N | P | T | I | A | E | G | P | S | P | T | S | E | ||||||||||||||||||
| MAPKK-3b17-43 | (M3) | G | K | S | K | R | K | K | D | L | R | I | S | C | MS | K | P | P | A | P | N | P | T | P | P | R | N | |||||||||||||||||||
| MAPKK-436-62 | (M4a) | S | M | Q | G | K | R | K | A | L | K | L | N | F | A | N | P | P | F | K | S | T | A | R | F | T | L | N | ||||||||||||||||||
| MAPKK-449-75 | (M4b) | A | N | P | P | F | K | S | T | A | R | F | T | L | N | P | N | P | T | G | V | Q | N | P | H | I | E | R | ||||||||||||||||||
| MAPKK-65-31 | (M6) | K | G | K | K | R | N | P | G | L | K | I | P | K | E | A | F | E | Q | P | Q | T | S | S | T | P | P | R | ||||||||||||||||||
| MAPKK-735-61 | (M7a) | S | P | Q | R | P | R | P | T | L | Q | L P | L | A | N | D | G | G | S | R | S | P | S | S | E | S | S | |||||||||||||||||||
| MAPKK-767-93 | (M7b) | P | P | A | R | P | R | H | M | L | G | L P | S | T | L | F | T | P | R | S M E S | I | E | I | D | ||||||||||||||||||||||
| Consensus | S | P | A | R | R | K | K | T | L | P/K | L/I P | L | N/AN/PP/F | P/T | P | A | S | T | P | S | P | T | S | |||||||||||||||||||||||
| LF-1 (Core ) | (R | R | K | K | V | Y | P | Y | P M | E) P | T | I | A | 1751.9 | ||||||||||||||||||||||||||||||||
| LF-2 | A | R | R | R | K | K | V | Y | P | Y | P M | E | P | T | I | A | K | 1880.1 | ||||||||||||||||||||||||||||
| LF-3 | S | P | A | R | R | K | K | V | Y | P | Y | P M | E | N | P | T | P | R | S | T | P | S | P | T | 2857.5 | |||||||||||||||||||||
| LF-4 | S | K | A | R | R | K | K | V | Y | P | Y | P M | E | N | F | P | P | S | T | A | R | P | T | 2821.5 | ||||||||||||||||||||||
| LF-5 | S | K | A | R | R | K | K | V | Y | P | Y | P X | E | N | F | P | P | S | T | A | R | P | T | 2804.2 | ||||||||||||||||||||||
| NT4/5 | S | K | R | K | K | V | Y | P | 1232.8 | |||||||||||||||||||||||||||||||||||||
| CT4 | S | Y | P M | E | N | F | P | P | S | T | A | R | P | T | 1607.8 | |||||||||||||||||||||||||||||||
| NT4/5-IS | K | (A+7) | R | R | K | K | V | Y | P | 1239.8 | ||||||||||||||||||||||||||||||||||||
| CT4-IS | Y | P M | E | N | F | P | P | S | T | (A+7) | R | P | T | 1614.8 | ||||||||||||||||||||||||||||||||
| CT5 | Y | P X | E | N | F | P | P | S | T | A | R | P | T | 1589.8 | ||||||||||||||||||||||||||||||||
| CT5-IS | Y | P X | E | N | F | P | P | S | T | (A+7) | R | P | T | 1596.8 | ||||||||||||||||||||||||||||||||

2.3. LC-ESI-MS/MS for peptides

2.4. LC-ESI-MS/MS for small molecules
3. Toxin-Specific Substrate Design
3.1. Peptide substrate design

3.2. Substrate design for other toxin activities
4. Enzyme Reaction Optimization
4.1. Reaction buffer optimization of endoprotease/peptide substrate reactions
4.2. Reaction buffer optimization of adenylate cyclase activities

5. Toxin Purification and Enrichment
6. Toxin Quantification

7. Discussion
8. Conclusions
References
- Rossetto, O.; de Bernard, M.; Pellizzari, R.; Vitale, G.; Caccin, P.; Schiavo, G.; Montecucco, C. Bacterial toxins with intracellular protease activity. Clin.Chim. Acta 2000, 291, 189–199. [Google Scholar] [CrossRef]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007, 9, 555–565. [Google Scholar] [CrossRef]
- Boyer, A.E.; Moura, H.; Woolfitt, A.R.; Kalb, S.R.; McWilliams, L.G.; Pavlopoulos, A.; Schmidt, J.G.; Ashley, D.L.; Barr, J.R. From the mouse to the mass spectrometer: detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal.Chem. 2005, 77, 3916–3924. [Google Scholar]
- Kalb, S.R.; Moura, H.; Boyer, A.E.; McWilliams, L.G.; Pirkle, J.L.; Barr, J.R. The use of Endopep-MS for the detection of botulinum toxins A, B, E, and F in serum and stool samples. Anal.Biochem. 2006, 351, 84–92. [Google Scholar]
- Kalb, S.R.; Lou, J.; Garcia-Rodriguez, C.; Geren, I.N.; Smith, T.J.; Moura, H.; Marks, J.D.; Smith, L.A.; Pirkle, J.L.; Barr, J.R. Extraction and inhibition of enzymatic activity of botulinum neurotoxins/A1, /A2, and /A3 by a panel of monoclonal anti-BoNT/A antibodies. PLoSOne 2009, 4, e5355. [Google Scholar]
- Boyer, A.E.; Quinn, C.P.; Hoffmaster, A.R.; Kozel, T.R.; Saile, E.; Marston, C.K.; Percival, A.; Plikaytis, B.D.; Woolfitt, A.R.; Gallegos, M.; Sabourin, P.; McWilliams, L.G.; Pirkle, J.L.; Barr, J.R. Kinetics of lethal factor and poly-D-glutamic acid antigenemia during inhalation anthrax in rhesus macaques. Infect. Immun. 2009, 77, 3432–3441. [Google Scholar] [CrossRef]
- Boyer, A.E.; Quinn, C.P.; Woolfitt, A.R.; Pirkle, J.L.; McWilliams, L.G.; Stamey, K.L.; Bagarozzi, D.A.; Hart, J.C., Jr.; Barr, J.R. Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal. Chem. 2007, 79, 8463–8470. [Google Scholar]
- Pellizzari, R.; Rossetto, O.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 259–268. [Google Scholar] [CrossRef]
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; Jackson, P.J.; Marks, J.D. Genetic diversity among Botulinum Neurotoxin-producing clostridial strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar]
- Moriishi, K.; Koura, M.; Abe, N.; Fujii, N.; Fujinaga, Y.; Inoue, K.; Ogumad, K. Mosaic structures of neurotoxins produced from Clostridium botulinum types C and D organisms. Biochim.Biophys. Acta 1996, 1307, 123–126. [Google Scholar]
- Moura, H.T.R.; Woolfitt, A.R.; Gallegos-Candela, M.; McWilliams, L.G.; Solano, M.I.; Pirkle, J.L.; Barr, J.R. Studies on Botulinum Neurotoxins Type /C1 and mosaic /DC Using Endopep-MS and Proteomics. FEMS Immunol. Microbiol. 2011, in press. [Google Scholar]
- Smith, T.J.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Tsai, R.; Laporte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; Marks, J.D. Sequence variation within botulinum neurotoxin serotypes impacts antibody binding and neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar]
- Mock, M.; Mignot, T. Anthrax toxins and the host: A story of intimacy. Cell Microbiol. 2003, 5, 15–23. [Google Scholar] [CrossRef]
- Turk, B.E. Manipulation of host signalling pathways by anthrax toxins. Biochem.J. 2007, 402, 405–417. [Google Scholar] [CrossRef]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar]
- Molloy, S.S.; Bresnahan, P.A.; Leppla, S.H.; Klimpel, K.R.; Thomas, G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 1992, 267, 16396–16402. [Google Scholar]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, II; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef]
- Warfel, J.M.; Steele, A.D.; D'Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Cui, X.; Solomon, S.B.; Vitberg, D.A.; Migone, T.S.; Scher, D.; Danner, R.L.; Natanson, C.; Subramanian, G.M.; Eichacker, P.Q. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J. Infect. Dis. 2010, 202, 1885–1896. [Google Scholar] [CrossRef]
- Boyer, A.E.; Quinn, C.P.; Hoffmaster, A.R.; Kozel, T.; Gallegos, M.; Saile, E.; Marston, C.; Beesley, C.; Percival, A.; Lins, R.; Woolfitt, A.; Pirkle, J.L.; Barr, J.R. Association of Early Lethal Factor Levels with Survival during Inhalation Anthrax in Rhesus Macaques. In Proceedings of ASM Biodefense and Emerging Diseases Research Meeting, Baltimore, MD, USA, 6-9 February 2010; American Society for Microbiology: Washington, DC, USA, 2010. [Google Scholar]
- Walsh, J.J.; Pesik, N.; Quinn, C.P.; Urdaneta, V.; Dykewicz, C.A.; Boyer, A.E.; Guarner, J.; Wilkins, P.; Norville, K.J.; Barr, J.R.; Zaki, S.R.; Patel, J.B.; Reagan, S.P.; Pirkle, J.L.; Treadwell, T.A.; Messonnier, N.R.; Rotz, L.D.; Meyer, R.F.; Stephens, D.S. A case of naturally acquired inhalation anthrax: Clinical care and analyses of anti-protective antigen immunoglobulin G and lethal factor. Clin.Infect. Dis. 2007, 44, 968–971. [Google Scholar]
- Ferreira, J.L.; Maslanka, S.; Johnson, E.; Goodnough, M. Detection of botulinal neurotoxins A, B, E, and F by amplified enzyme-linked immunosorbent assay: Collaborative study. J. AOAC Int. 2003, 86, 314–331. [Google Scholar]
- Ezzell, J.W., Jr.; Abshire, T.G. Serum protease cleavage of Bacillus anthracis protective antigen. J. Gen. Microbiol. 1992, 138, 543–549. [Google Scholar] [CrossRef]
- Panchal, R.G.; Halverson, K.M.; Ribot, W.; Lane, D.; Kenny, T.; Abshire, T.G.; Ezzell, J.W.; Hoover, T.A.; Powell, B.; Little, S.; Kasianowicz, J.J.; Bavari, S. Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J. Biol. Chem. 2005, 280, 10834–10839. [Google Scholar]
- Barr, J.R.; Maggio, V.L.; Patterson, D.G., Jr.; Cooper, G.R.; Henderson, L.O.; Turner, W.E.; Smith, S.J.; Hannon, W.H.; Needham, L.L.; Sampson, E.J. Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 1996, 42, 1676–1682. [Google Scholar]
- Williams, T.L.; Luna, L.; Guo, Z.; Cox, N.J.; Pirkle, J.L.; Donis, R.O.; Barr, J.R. Quantification of influenza virus hemagglutinins in complex mixtures using isotope dilution tandem mass spectrometry. Vaccine 2008, 26, 2510–2520. [Google Scholar]
- Kuklenyik, Z.; Boyer, A.E.; Lins, R.; Quinn, C.P.; Gallegos-Candela, M.; Woolfitt, A.R.; Pirkle, J.L.; Barr, J.R. Comparison of MALDI-TOF-MS and HPLC-ESI-MS/MS for Endopeptidase Activity Based Quantification of Anthrax Lethal Factor in Serum. Anal. Chem. 2011, in press. [Google Scholar]
- Lins, R.C.; Boyer, A.E.; Kuklenyik, Z.; Quinn, C.P.; Gallegos-Candela, M.; Leysath, C.E.; Chen, Z.; Chen, K.; Makiya, M.; Leppla, S.H.; Pirkle, J.L.; Barr, J.R. A High Sensitivity Adenylate Cyclase Assay for Anthrax Edema factor. In Proceedings of 58th ASMS Conference on Mass Spectrometry and Allied Topics, Salt Lake City, UT, USA, 23-27 May 2010; American Society for Mass Spectrometry: Santa Fe, NM, USA, 2010. [Google Scholar]
- Turk, B.E.; Wong, T.Y.; Schwarzenbacher, R.; Jarrell, E.T.; Leppla, S.H.; Collier, R.J.; Liddington, R.C.; Cantley, L.C. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 60–66. [Google Scholar]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; Vande Woude, G.F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 (Pt. 3), 739–745. [Google Scholar]
- Guo, Q.; Shen, Y.; Zhukovskaya, N.L.; Florian, J.; Tang, W.J. Structural and kinetic analyses of the interaction of anthrax adenylylcyclase toxin with reaction products cAMP and pyrophosphate. J. Biol. Chem. 2004, 279, 29427–29435. [Google Scholar]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef]
- Jank, T.; Giesemann, T.; Aktories, K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 2007, 17, 15R–22R. [Google Scholar] [CrossRef]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylatecyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef]
- Gallegos-Candela, M.; Boyer, A.E.; Quinn, C.P.; Woolfitt, A.R.; Barr, J.R. Validation of Rapid, Sensitive, Quantitative MALDI-TOF MS Assays for Anthrax Lethal Factor and Lethal Toxin, Bacillus-ACT 2009. In Proceedings of the International Bacillus Anthracis, B. Cereus, and B. Thuringiensis Conference, Santa Fe, NM, USA, 30 August-3 September, 2009; American Society for Microbiology: Washington, DC, USA, 2009. [Google Scholar]
- Kobiler, D.; Weiss, S.; Levy, H.; Fisher, M.; Mechaly, A.; Pass, A.; Altboum, Z. Protective antigen as a correlative marker for anthrax in animal models. Infect. Immun. 2006, 74, 5871–5876. [Google Scholar] [CrossRef]
- Mabry, R.; Brasky, K.; Geiger, R.; Carrion, R., Jr.; Hubbard, G.B.; Leppla, S.; Patterson, J.L.; Georgiou, G.; Iverson, B.L. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin.Vaccine Immunol. 2006, 13, 671–677. [Google Scholar] [CrossRef]
- Kalb, S.R.; Garcia-Rodriguez, C.; Lou, J.; Baudys, J.; Smith, T.J.; Marks, J.D.; Smith, L.A.; Pirkle, J.L.; Barr, J.R. Extraction of BoNT/A, /B, /E, and /F with a single, high affinity monoclonal antibody for detection of botulinum neurotoxin by Endopep-MS. PLoSOne 2010, 5, e12237. [Google Scholar]
- Tang, S.; Moayeri, M.; Chen, Z.; Harma, H.; Zhao, J.; Hu, H.; Purcell, R.H.; Leppla, S.H.; Hewlett, I.K. Detection of anthrax toxin by an ultrasensitive immunoassay using europium nanoparticles. Clin. Vaccine Immunol. 2009, 16, 408–413. [Google Scholar] [CrossRef]
- Boyer, A.E.; Quinn, C.P.; Hoffmaster, A.; Gallegos, M.; Saile, E.; Marston, C.; Kozel, T.; Meister, G.; Woolfitt, A.R.; Pirkle, J.L.; Barr, J.R. Kinetics of Toxemia of Inhalation Anthrax. In. In Proceedings of 6th Annual ASM Biodefense and Emerging Diseases Research Meeting, Baltimore, MD, USA, 24-27 February 2008; American Society for Microbiology: Washington, DC, USA, 2008. [Google Scholar]
- Kalb, S.R.; Goodnough, M.C.; Malizio, C.J.; Pirkle, J.L.; Barr, J.R. Detection of botulinum neurotoxin A in a spiked milk sample with subtype identification through toxin proteomics. Anal. Chem. 2005, 77, 6140–6146. [Google Scholar] [CrossRef]
- Barr, J.R.; Kalb, S.R.; Moura, H.; Pirkle, J.L. Biological Monitoring of Exposure to Botulinum Neurotoxins. Chem. Health Safety 2008, 15, 14–19. [Google Scholar]
- Belmares, J.; Gerding, D.N.; Tillotson, G.; Johnson, S. Measuring the severity of Clostridium difficile infection: implications for management and drug development. Expert Rev. Anti Infect. Ther. 2008, 6, 897–908. [Google Scholar] [CrossRef]
- Deshpande, A.; Pant, C.; Jain, A.; Fraser, T.G.; Rolston, D.D. Do fluoroquinolones predispose patients to Clostridium difficile associated disease? A review of the evidence. Curr.Med. Res. Opin. 2008, 24, 329–333. [Google Scholar] [CrossRef]
- Guris, D.; Strebel, P.M.; Bardenheier, B.; Brennan, M.; Tachdjian, R.; Finch, E.; Wharton, M.; Livengood, J.R. Changing epidemiology of pertussis in the United States: increasing reported incidence among adolescents and adults, 1990-1996. Clin. Infect. Dis. 1999, 28, 1230–1237. [Google Scholar] [CrossRef]
- King, A.J.; van Gorkom, T.; Pennings, J.L.; van der Heide, H.G.; He, Q.; Diavatopoulos, D.; Heuvelman, K.; van Gent, M.; van Leeuwen, K.; Mooi, F.R. Comparative genomic profiling of Dutch clinical Bordetellapertussis isolates using DNA microarrays: Identification of genes absent from epidemic strains. BMC Genomics 2008, 9, 311. [Google Scholar]
- Godfroid, F.; Denoel, P.; Poolman, J. Are vaccination programs and isolate polymorphism linked to pertussis re-emergence? Expert Rev. Vaccines 2005, 4, 757–778. [Google Scholar] [CrossRef]
- Kalb, S.R.; Baudys, J.; Egan, C.; Smith, T.J.; Smith, L.A.; Pirkle, J.L.; Barr, J.R. Clostridium baratii Type F Neurotoxin Cleaves Synaptobrevin-2 with Different Substrate Recognition Requirement than Clostridium botulinum Type F Neurotoxin. Appl. Environ. Microbiol. 2011, 77, 1301–1308. [Google Scholar] [CrossRef]
- Samples Availability: Samples of the compounds are commercially available. Anthrax toxins were acquired from List Biological Laboratories, USA and B. Pertussis adenylate cyclase toxins and ATP and cAMP are available from Sigma-Aldrich, USA.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boyer, A.E.; Gallegos-Candela, M.; Lins, R.C.; Kuklenyik, Z.; Woolfitt, A.; Moura, H.; Kalb, S.; Quinn, C.P.; Barr, J.R. Quantitative Mass Spectrometry for Bacterial Protein Toxins — A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis. Molecules 2011, 16, 2391-2413. https://doi.org/10.3390/molecules16032391
Boyer AE, Gallegos-Candela M, Lins RC, Kuklenyik Z, Woolfitt A, Moura H, Kalb S, Quinn CP, Barr JR. Quantitative Mass Spectrometry for Bacterial Protein Toxins — A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis. Molecules. 2011; 16(3):2391-2413. https://doi.org/10.3390/molecules16032391
Chicago/Turabian StyleBoyer, Anne E., Maribel Gallegos-Candela, Renato C. Lins, Zsuzsanna Kuklenyik, Adrian Woolfitt, Hercules Moura, Suzanne Kalb, Conrad P. Quinn, and John R. Barr. 2011. "Quantitative Mass Spectrometry for Bacterial Protein Toxins — A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis" Molecules 16, no. 3: 2391-2413. https://doi.org/10.3390/molecules16032391