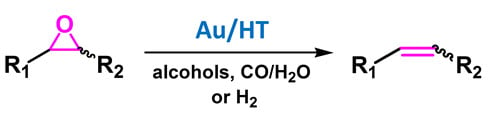
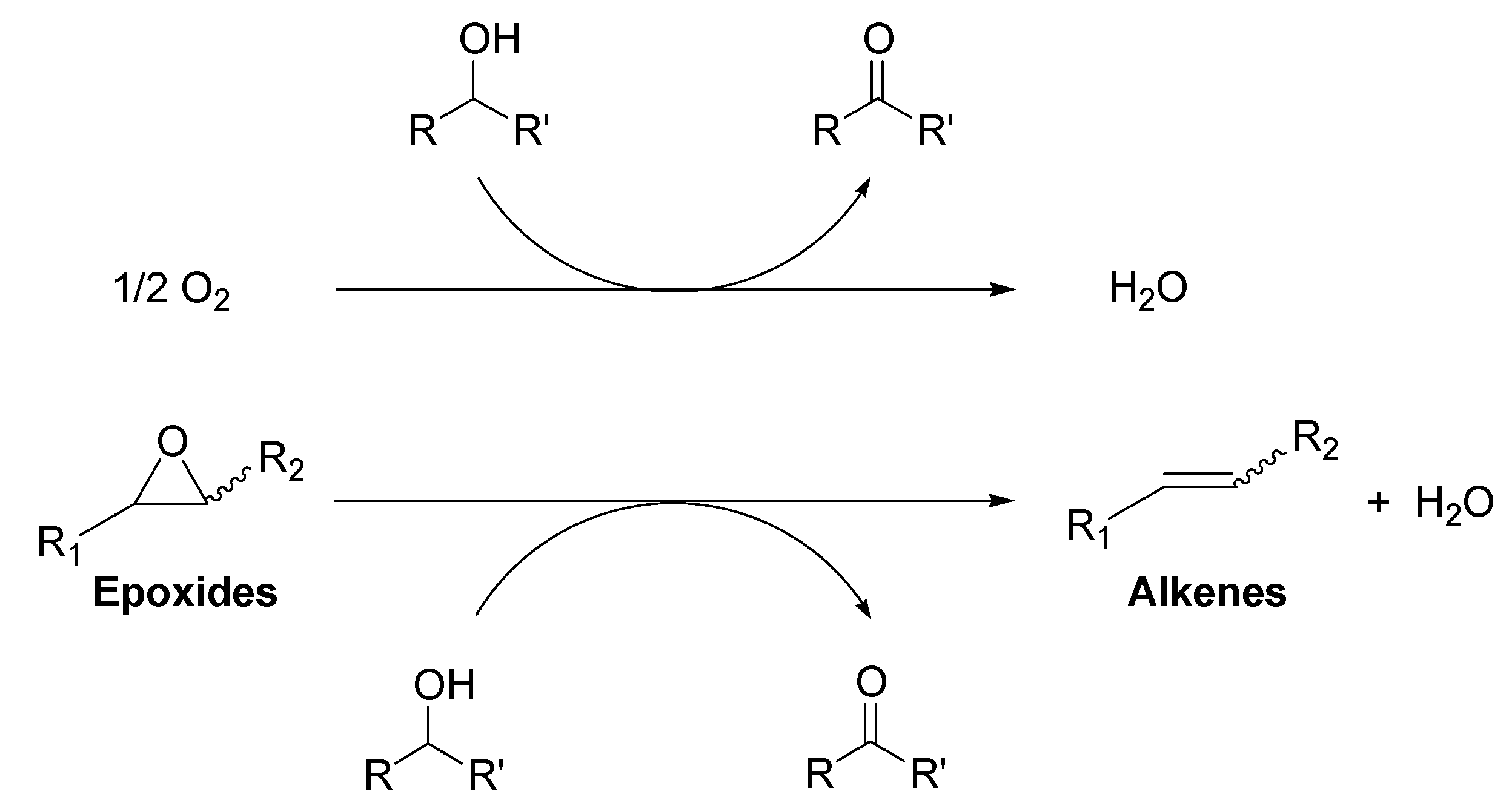
2.2.1. Au/HT-Catalyzed Deoxygenation of Epoxides Using Alcohols as a Reductant
Recently, we have found that Au/HT could catalyze the highly efficient aerobic oxidation of alcohols [
40] and the lactonization of diols [
41]. These results allowed us to predict that if epoxides could be employed as hydrogen acceptors in place of molecular oxygen under the alcohol oxidation conditions, a green catalytic deoxygenation of epoxides with alcohols could be developed (
Scheme 3) [
36].
Scheme 3.
The oxidation of alcohols using O2 vs. deoxygenation of epoxides using alcohols.
Scheme 3.
The oxidation of alcohols using O2 vs. deoxygenation of epoxides using alcohols.
To demonstrate the above hypothesis, we carried out the deoxygenation of
trans-stilbene oxide (
1) using Au/HT with 2-propanol in toluene as the solvent at 110 °C under Ar atmosphere for 4 h.
1 was successfully deoxygenated to give the corresponding alkene
trans-stilbene (
2) in quantitative yield. Notably, no by-products such as 1,2-diphenylethanol or 1,2-diphenylethane resulting from the hydrogenation of
1 or
2 were formed (
Table 1, Entry 1). During the deoxygenation of
1, the amounts of acetone and water generated were almost equivalent to that of
2. Among the alcohols tested, 1-phenylethanol and benzyl alcohol could also function as reductants (Entries 2 and 3), while the use of an aliphatic primary alcohol such as 1-octanol resulted in lower yield (Entry 4). Next, the effects of inorganic supports of Au NPs were investigated. Au NPs on basic supports of Al
2O
3 and MgO afforded good to moderate yields of
2 (Entries 5 and 6), whereas non-basic supports like TiO
2 and SiO
2 were not effective (Entries 7 and 8). Other Au compounds like HAuCl
4, Au
2O
3 and bulk Au metal did not promote the deoxygenation (Entries 9–11). These results indicate that the combination of Au NPs and a basic support is necessary to achieve the high catalytic activity for the deoxygenation.
Table 1.
Deoxygenation of
trans-stilbene oxide
a.
![Molecules 16 08209 i001]()
Table 1.
Deoxygenation of trans-stilbene oxide a. ![Molecules 16 08209 i001]()
| Entry | Catalyst | Alcohol | Conv. b (%) | Sel. b (%) |
|---|
| 1 | Au/HT | 2-propanol | 99 | >99 |
| 2 | Au/HT | 1-phenylethanol | 99 | >99 |
| 3 | Au/HT | benzyl alcohol | 91 | >99 |
| 4 | Au/HT | 1-octanol | 37 | >99 |
| 5 | Au/Al2O3 | 2-propanol | 60 | >99 |
| 6 | Au/MgO | 2-propanol | 43 | >99 |
| 7 | Au/TiO2 | 2-propanol | 19 | >99 |
| 8 | Au/SiO2 | 2-propanol | 5 | >99 |
| 9 | HAuCl4 | 2-propanol | <1 | - |
| 10 | Au2O3 | 2-propanol | <1 | - |
| 11 | bulk Au metal | 2-propanol | 0 | - |
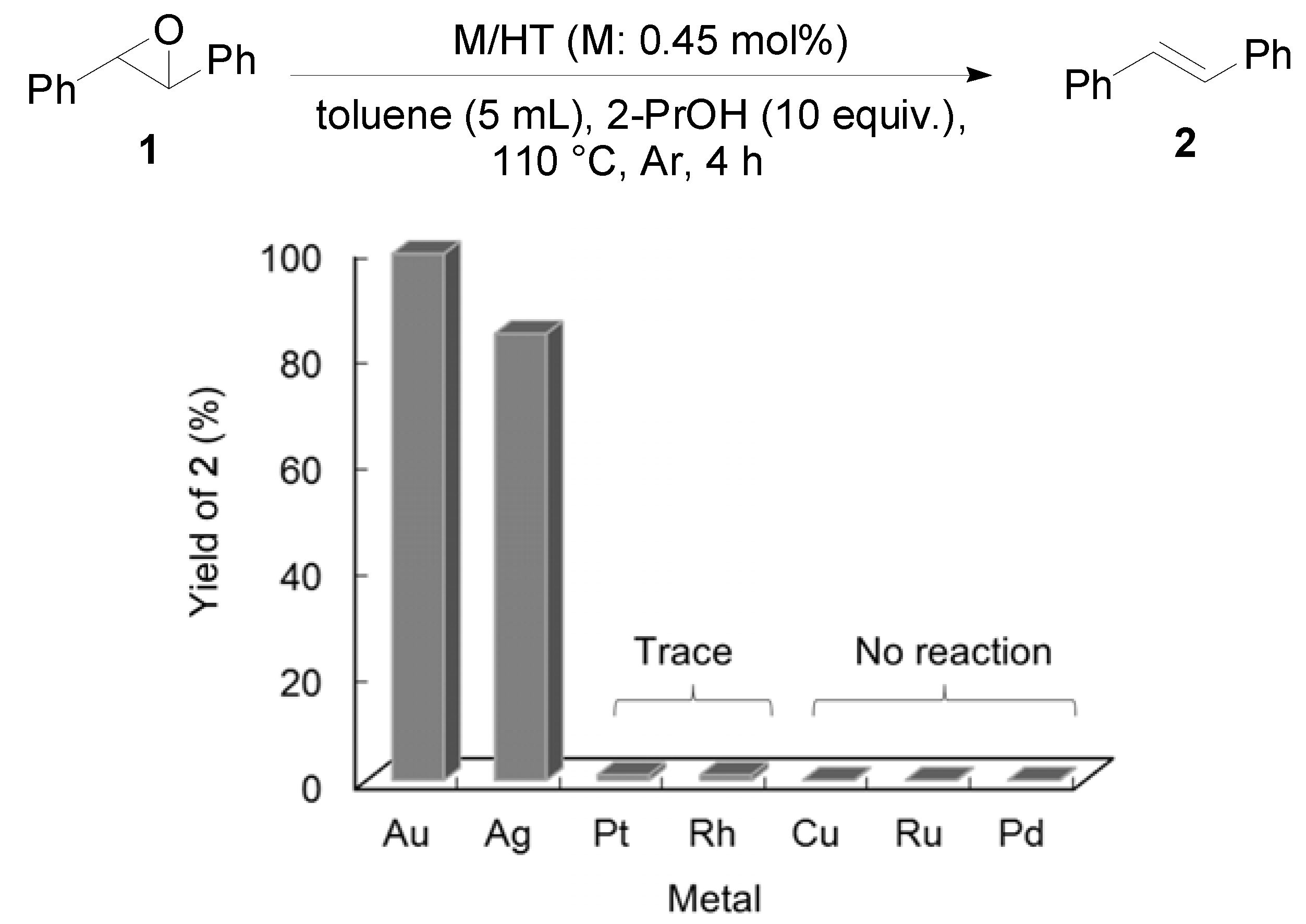
Other metal NPs on HT were examined for the deoxygenation of
1 (
Figure 2). Among the catalysts tested, Ag/HT also showed excellent catalytic activity for deoxygenation, while other metal NPs did not function as catalysts.
Figure 2.
Deoxygenation of trans-stilbene oxide using HT-supported various metal NPs.
Figure 2.
Deoxygenation of trans-stilbene oxide using HT-supported various metal NPs.
To investigate the possibility of the leaching of active metal species from Au/HT into the reaction mixtures, Au/HT was filtered from the reaction mixture at 50% conversion of 1, and treatment of the filtrate with additional stirring under similar conditions did not give any product. Furthermore, inductively coupled plasma atomic emission spectral (ICP-AES) analysis revealed no Au species in the filtrates (detection limit: 0.1 ppm). These results clearly proved that no leaching occurred and the deoxygenation proceeded on the Au NPs on HT.
The outstanding catalytic activity of Au NPs encouraged us to investigate the scope of epoxides in the deoxygenation (
Table 2). Various epoxides were efficiently converted into the corresponding alkenes with over 99% selectivity. Both aromatic and aliphatic epoxides could be deoxygenated. Epoxides having ether and hydroxyl groups were also successfully employed as substrates (Entries 9 and 16). Notably, the reducible C=O bonds of epoxyketones were tolerated in the deoxygenation (Entries 14 and 15).
cis-Stilbene oxide and
cis-2,3-epoxyoctane gave (
Z)/(
E)-alkene stereoisomers. The selectivities for
cis-alkenes were 60% and 50%, respectively (Entries 4 and 13).
Table 2.
Deoxygenation of various epoxides using Au/HT
a .
![Molecules 16 08209 i002]()
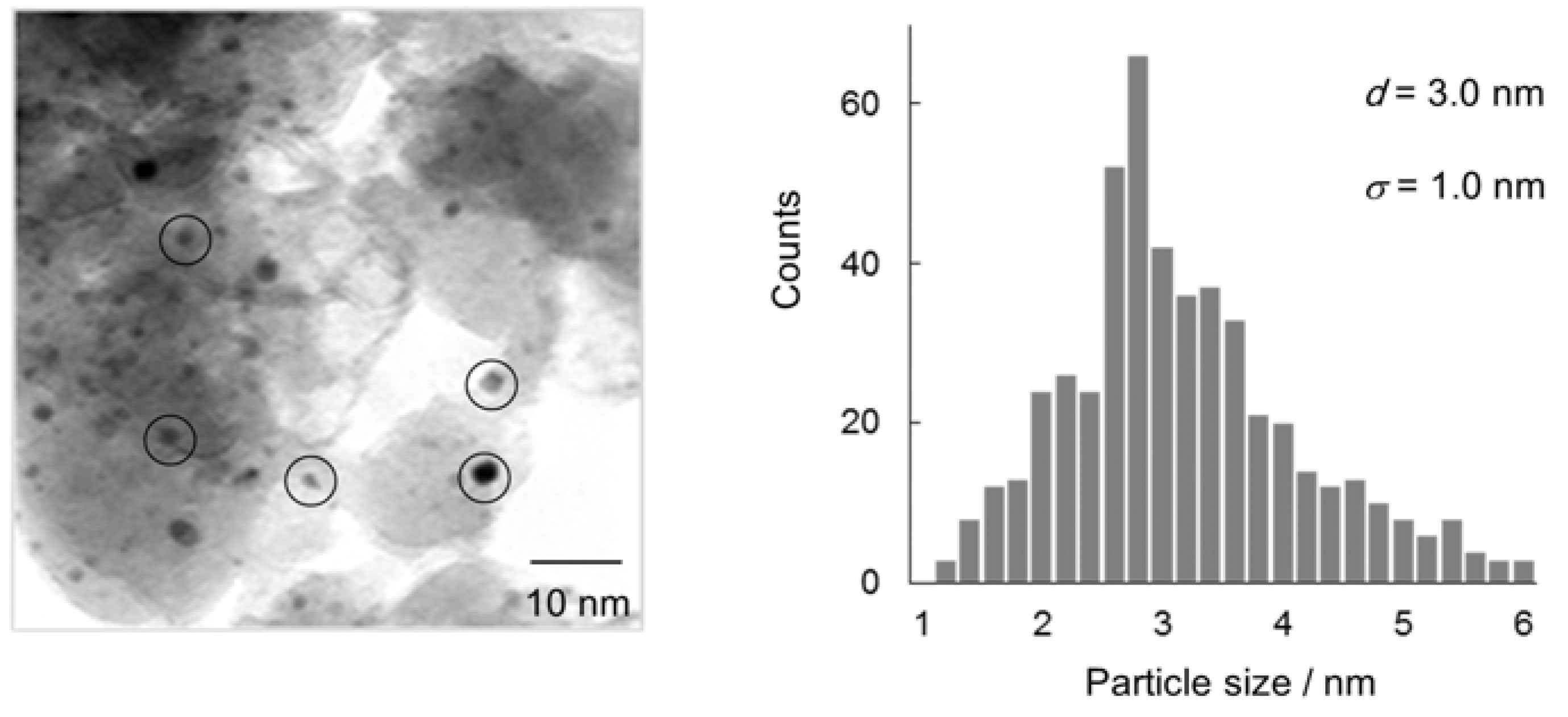
After the deoxygenation of
1, the solid Au/HT catalyst could be easily separated from the reaction mixtures and reused with retention of its performance (Entries 2 and 3). TEM images showed that the Au NPs on HT after reuse were similar to fresh Au/HT in average diameter and size distribution and no aggregation of the used Au NPs was observed (
Figure 3).
Figure 3.
TEM image and size distribution of Au NPs of Au/HT after reuse experiments.
Figure 3.
TEM image and size distribution of Au NPs of Au/HT after reuse experiments.
Atomic-scale analysis using Au L-edge EXAFS of Au/HT showed that the intensity of the FT peak derived from the Au-Au shell at 2.8 Å was not changed, supporting the observation that the Au NPs after reuse were of the same size as the originals. These results are consistent with the high durability of Au/HT in the recycling experiments.
Au/HT was also applicable in a preparative scale reaction (
Scheme 4). Thus, the deoxygenation of 20 mmol of
1 successfully proceeded to afford
2 with 95% isolated yield in 2-propanol as solvent after 72 h, where the TON and TOF reached 20,000 and 270 h
−1, respectively. These values are three orders of magnitude greater than those of previously reported catalytic systems such as Tp’ReO
3-PPh
3 (TON = 19) [
24], polystyrene-supported Re-PPh
3 (TON = 18, TOF = 12 h
−1) [
26], CH
3ReO
3-PPh
3 (TON = 8, TOF = 0.4) [
25], [Fe
4S
4(SC
6H
5)
4]
3−-NaBH
4 (TON = 4, TOF = 1 h
−1) [
27], and Co complex-Na (Hg) (TON = 5, TOF = 0.8 h
−1) [
28].
Scheme 4.
Preparative deoxygenation of trans-stilbene oxide using Au/HT.
Scheme 4.
Preparative deoxygenation of trans-stilbene oxide using Au/HT.
In separate experiments, the use of
d-benzhydrol [C
6H
5CD(OD)C
6H
5] as a reductant for the deoxygenation of
1 afforded
2 and D
2O with all hydrogen atoms in the alkene product retained. From both these results and the positive effect of basic supports as shown in
Table 1 (Entries 1, 5 and 6), we propose the following mechanism as shown in
Scheme 5.
Scheme 5.
A plausible reaction mechanism for the Au/HT-catalyzed deoxygenation of an epoxide through the cooperation of the Au NPs with basic sites (BS) of HT.
Scheme 5.
A plausible reaction mechanism for the Au/HT-catalyzed deoxygenation of an epoxide through the cooperation of the Au NPs with basic sites (BS) of HT.
A basic site (denoted as BS) of HT abstracts the H
+ from the hydroxyl group of the alcohol to promote the formation of an Au-alcoholate species which subsequently forms an Au-hydride species and the corresponding carbonyl compound through β-hydride elimination [
42]. The Au-hydride species and H
+ attack an epoxide, providing an alkene and H
2O. The distinguished deoxygenation activities of Au NPs from those of other metal NPs should be attributed to the reactivity toward an epoxides (III→I) because the other metal NPs of Cu, Ru, Pd and Rh, which can form metal-hydride species from the reaction with the alcohol (I→III), do not deoxygenate the epoxide.
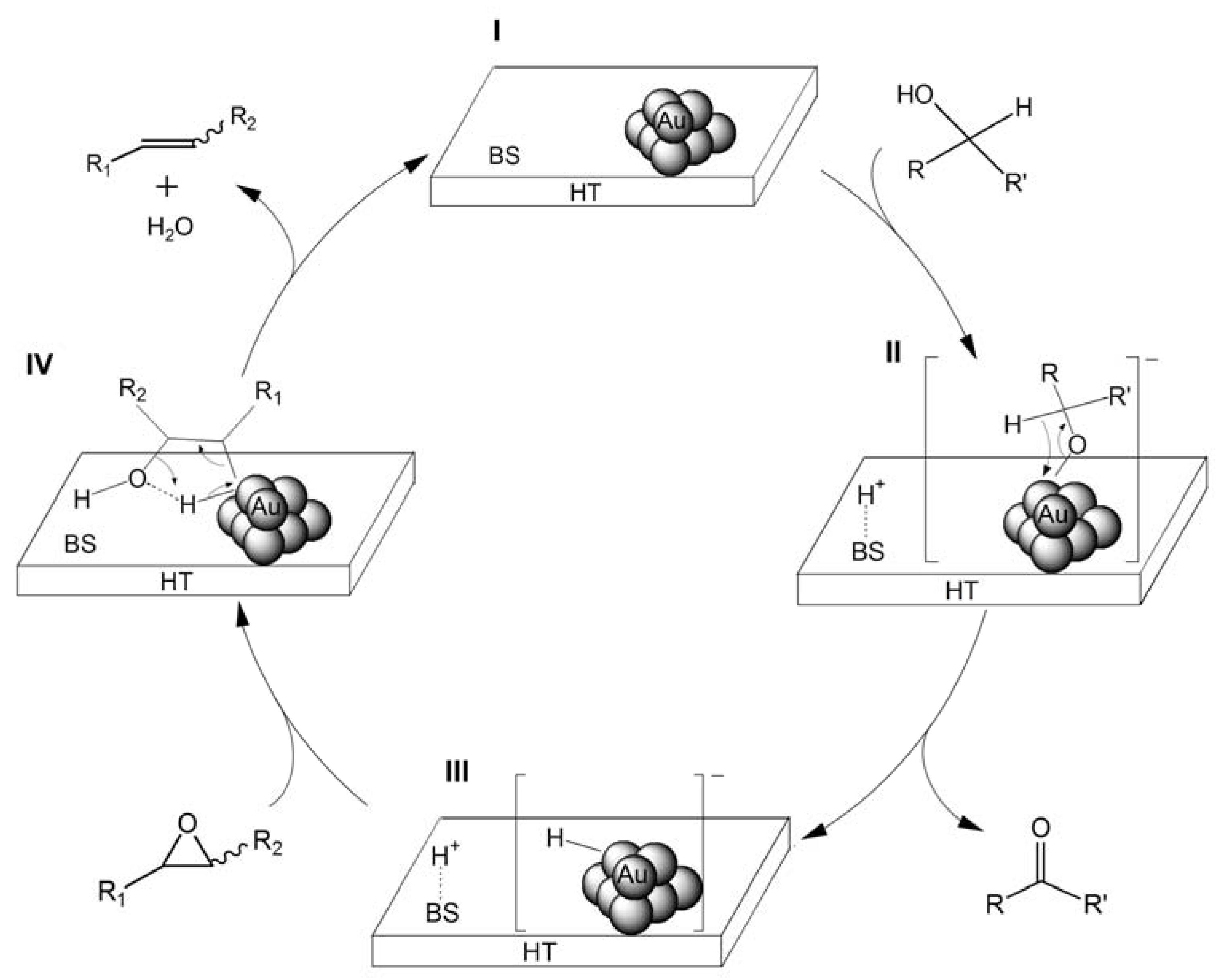
2.2.2. Deoxygenation of Epoxides with CO/H2O
We have previously reported that Rh carbonyl species can deoxygenate nitro compounds in the presence of amines as bases under water-gas shift reaction conditions (CO + H
2O → CO
2 + H
2) [
43]. In this reaction, CO and H
2O react with the Rh carbonyl species and an amine to form a Rh-hydride species that is active for the reduction of nitro compounds. The formation of the Rh-hydride species in cooperation with amines under water-gas shift reaction conditions inspired us to develop an alternative catalytic deoxygenation system using Au/HT. Namely, we proposed that an active Au-hydride species for the deoxygenation of epoxides can be formed through the cooperative effect of HT as a base under water-gas shift reaction conditions (
Scheme 6). Thus, the attack of H
2O on the basic sites of HT to CO adsorbed on Au NPs generates [Au-COOH]
−, followed by the elimination of CO
2 to give the Au-hydride species and H
+, which then act in concert to deoxygenate epoxides to alkenes [
37].
Scheme 6.
Concerted effects between HT and Au NPs for the formation of Au-H species (BS represents base site of HT).
Scheme 6.
Concerted effects between HT and Au NPs for the formation of Au-H species (BS represents base site of HT).
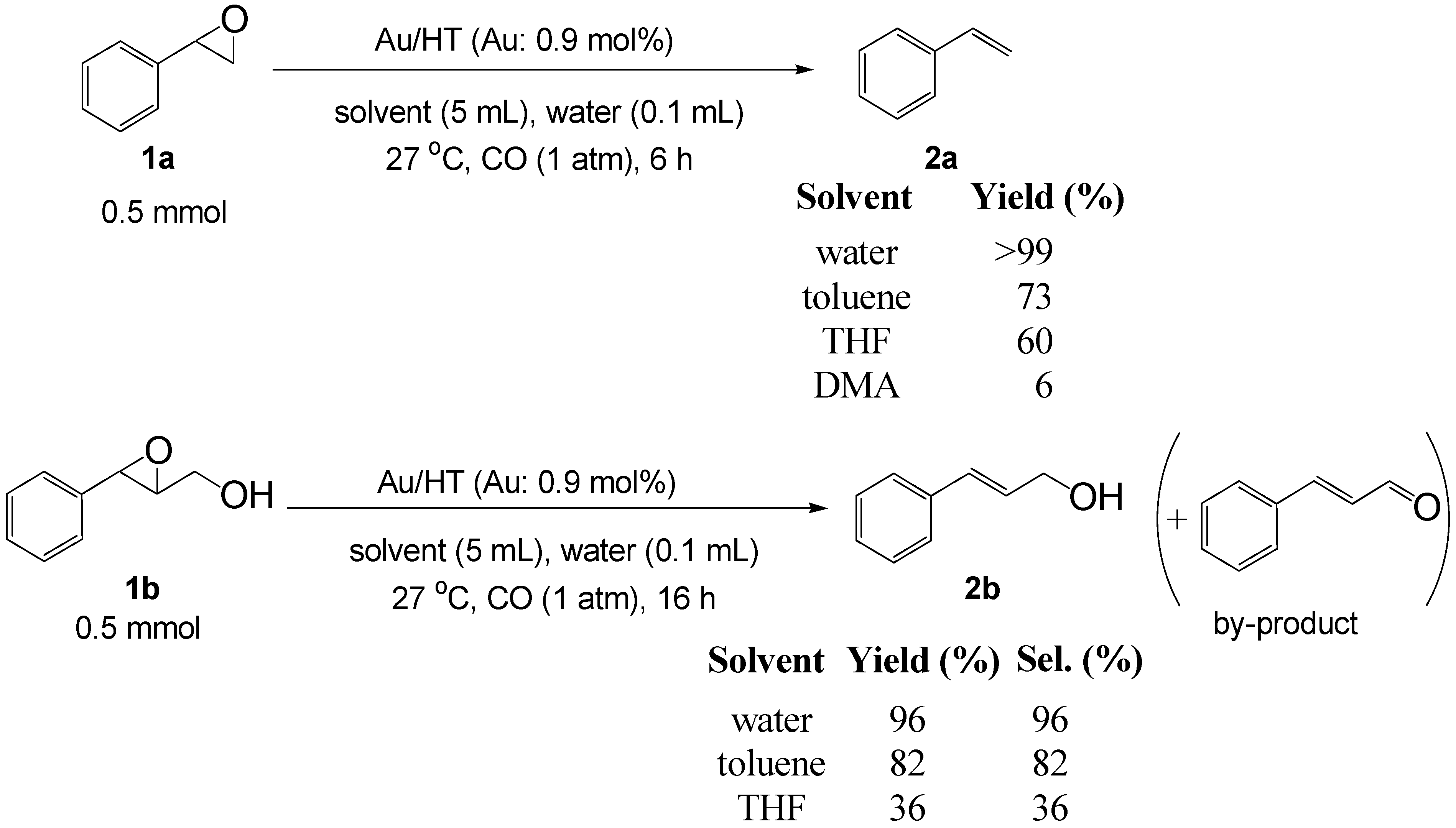
Based on the above hypothesis, we carried out the deoxygenation of styrene oxide (
1a) under water-gas shift reaction conditions in the presence of Au/HT. Styrene (
2a) was quantitatively obtained as the sole product under atmospheric pressure CO in water at room temperature (
Table 3, Entry 1). Various epoxides tested in the Au/HT-2-propanol system were also reactive under water-gas shift conditions (
Table 4). Compared with the Au/HT-2-propanol system, this Au/HT-CO/H
2O system can promote the deoxygenation of epoxides under mild and convenient reaction conditions, e.g., at room temperature, 1 atm of CO, and in the absence of organic solvents.
Table 3.
Deoxygenation of styrene oxide using CO/H
2O
a.
![Molecules 16 08209 i033]()
Table 3.
Deoxygenation of styrene oxide using CO/H2O a. ![Molecules 16 08209 i033]()
| Entry | Catalyst | Yield b (%) | Sel. b (%) |
|---|
| 1 | Au/HT | >99 | >99 |
| 2 c | Au/HT | >99 | >99 |
| 3 d | Au/HT | 97 | >99 |
| 4 | Au/Al2O3 | 79 | >99 |
| 5 | Au/MgO | 36 | >99 |
| 6 | Au/TiO2 | 18 | >99 |
| 7 e | Au/TiO2 + Na2CO3 | 57 | >99 |
| 8 | Au/SiO2 | 3 | >99 |
Table 4.
Deoxygenation of various epoxides with CO/H
2O using Au/HT
a.
![Molecules 16 08209 i034]()
Scheme 7.
Solvent effect on the deoxygenation of epoxides.
Scheme 7.
Solvent effect on the deoxygenation of epoxides.
The solvent effect on the deoxygenation is shown in
Scheme 7. Notably, water was found to provide the highest yield among all the solvents tested despite the water-insoluble nature of
1a. In the case of the epoxy alcohol 2,3-epoxy-3-phenyl-1-propanol (
1b), the highest yield and selectivity of cinnamyl alcohol (
2b) were obtained in water. After the deoxygenation of
1a,
2a was easily extracted from the reaction mixture by
n-hexane and the recovered aqueous phase containing solid Au/HT could be recycled with no decrease in catalytic activity (
Table 3, Entries 2 and 3).
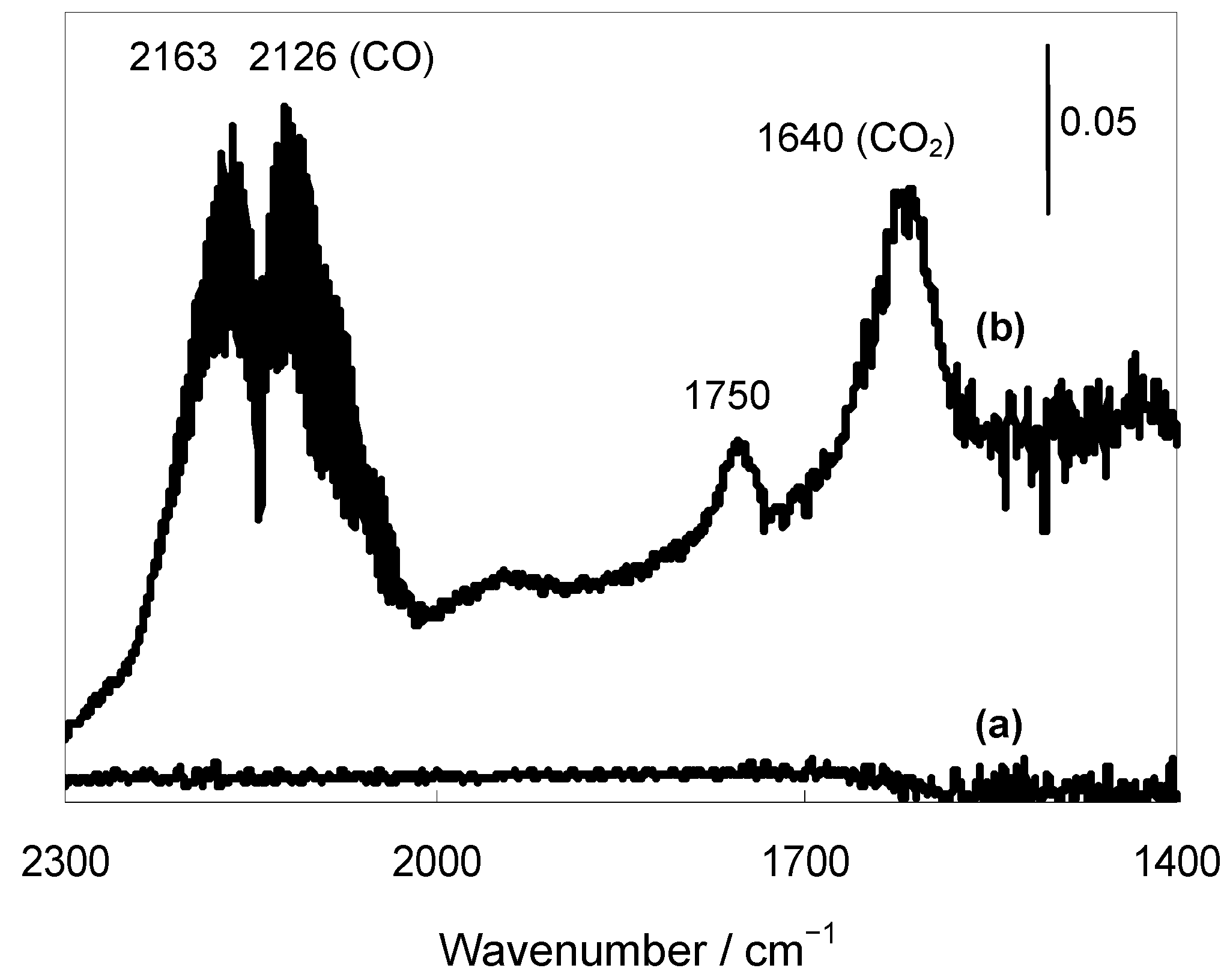
Figure 4.
FTIR spectra of (a) Au/HT and (b) after CO/H2O adsorption at 298 K.
Figure 4.
FTIR spectra of (a) Au/HT and (b) after CO/H2O adsorption at 298 K.
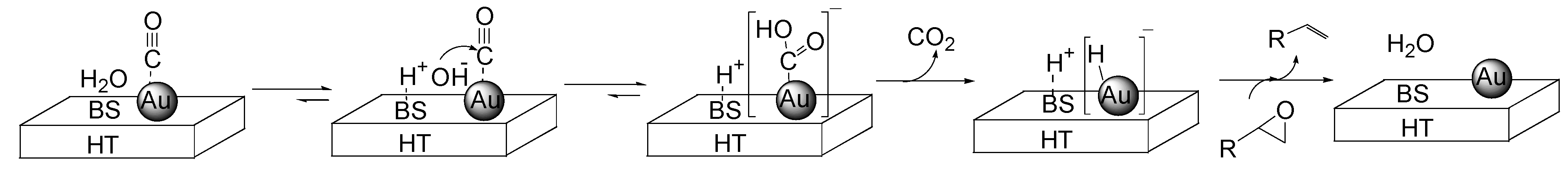
To gain more insight into the deoxygenation under the water-gas shift conditions, the following control experments were carried out. When the reaction was conducted in the absence of
1a under otherwise identical conditions, H
2 was not detected. The use of D
2O in place of H
2O significantly affected the reaction rate for the deoxygenation of
1a with a
kH/kD value of 3.9. These results rule out the participation of H
2 in the Au/HT-catalyzed deoxygenation reaction, while indicating that not only CO functions as a sole reductant, but also water takes part in the deoxygenation. An additional experiment using
trans-2-octenal in place of
1a revealed that chemoselective reduction occurred to give
trans-2-octen-1-ol as the sole product while retaining the C=C double bond of the starting material. From the above results, we believe that an active Au-hydride species is generated in situ from the reaction of H
2O with CO during the deoxygenation of epoxides. According to the proposed reaction mechanism (
Scheme 6), a basic site of HT facilitates the formation of the Au-hydride species through the nucleophilic attack of OH
− on the Au-CO species followed by a decarboxylation, which is well evidenced by the positive effect of the additional base of Na
2CO
3 to the Au NPs on the non-basic material of TiO
2 (
Table 3, Entry 6
vs. 7). Finally, to confirm the generation of the Au-H species, Fourier transform infrared (FT-IR) studies of Au/HT were conducted in the presence of CO and H
2O. When Au/HT was treated with CO and H
2O vapor at 298 K, a new band attributed to the generation of Au-H species was detected around 1750 cm
−1 (
Figure 4). Next, this treated Au/HT was exposed to the vapor of
1a, and the band attributed to the Au-H species gradually disappeared. The detection of the Au-H species agreed with recent IR and theoretical studies on Au–H species that predicted a band around 1800 cm
−1 [
44,
45]. These above control experiments are consistent with the proposed reaction mechanism as shown in
Scheme 6. The heterolytic H
+ and Au-hydride species generated in situ on Au/HT deoxygenate the epoxide to form the corresponding alkene and water.
After the publication of our Au/HT-CO/H
2O catalyst system, Cao
et al. reported a deoxygenation method using Au/TiO
2-VS (very small gold NPs on TiO
2) under water-gas shift reaction conditions [
46]. Au/TiO
2-VS showed high catalytic activity (TON = 9,600, TOF = 400 h
−1) in the deoxygenation of styrene oxide in the mixed solvent of acetone with H
2O under a high pressure CO atmosphere (10 atm).
2.2.3. Selective Deoxygenation of Epoxides Using Molecular Hydrogen
The ideal green methodology for the catalytic deoxygenation of epoxides is to utilize molecular hydrogen (H
2) as a reductant due to the formation of non-toxic water as a by-product. However, the use of H
2 often causes non-selective reduction of epoxides to yield alcohols and alkanes as byproducts through hydrogenation of the epoxides and overhydrogenation of the desired alkenes, respectively. Although there are a few successful reports on the selective deoxygenation of epoxides using H
2, high selectivity for alkenes is restricted to low conversion levels [
46] or a limited range of substrates [
47]. Therefore, the development of an efficient catalytic system for the selective deoxygenation of epoxides to the corresponding alkenes using H
2 is a challenging task.
With supported Au NPs in hand, the deoxygenation conditions were optimized [
38]. When the deoxygenation of
1a using Au/HT was carried out at 80 °C under 1 atm of H
2 for 6 h,
1a was converted to
2a in 97% yield accompanied by the formation of ethylbenzene (
3a) as a byproduct through the hydrogenation of the desired product
2a (
Table 5, Entry 1). Next, Au NPs on other supports were investigated in the deoxygenation of
1a under similar reaction conditions. Au/CeO
2 and Au/Al
2O
3 had lower selectivities for
2a, which caused hydrogenation of
2a (Entries 4 and 5) [
48]. Interestingly, Au/TiO
2 gave
2a with over 99% selectivity, though the conversion of
1a was much lower than that of Au/HT (Entry 6). Au/SiO
2 did not exhibit any catalytic activity toward this reaction (Entry 7). Remarkably, when the reaction was carried out at 60 °C for 8 h, Au/HT produced
2a in over 99% yield without formation of any side products (Entry 2). Moreover, the C=C bond of
2a was completely intact when the reaction time was prolonged (Entry 3). The Au NP catalysis exhibited different activity from other metal NPs. Ag/HT, Rh/HT, Ru/HT, and Cu/HT did not function as catalysts for this reaction (Entries 10-13). On the other hand, Pd/HT and Pt/HT afforded undesired
4a with over 99% selectivity through the hydrogenation of
1a (Entries 8 and 9).
Table 5.
Deoxygenation of styrene oxide using H
2 a.
![Molecules 16 08209 i049]()
Table 5.
Deoxygenation of styrene oxide using H2 a. ![Molecules 16 08209 i049]()
| Entry | Catalyst | Temp. (°C) | Time (h) | Conv. b (%) | Sel. for 2a b (%) | Sel. for 3a b (%) | Sel. for 4a b (%) |
|---|
| 1 | Au/HT | 80 | 6 | 97 | 97 | 3 | 0 |
| 2 | Au/HT | 60 | 8 | >99 | >99 | 0 | 0 |
| 3 | Au/HT | 60 | 24 | >99 | >99 | 0 | 0 |
| 4 | Au/CeO2 | 80 | 6 | 64 | 81 | 19 | 0 |
| 5 | Au/Al2O3 | 80 | 6 | 82 | 36 | 64 | 0 |
| 6 | Au/TiO2 | 80 | 6 | 26 | >99 | <1 | 0 |
| 7 | Au/SiO2 | 80 | 6 | <1 | - | - | - |
| 8 | Pd/HT | 80 | 6 | >99 | 0 | 0 | >99 |
| 9 | Pt/HT | 80 | 6 | 87 | 0 | <1 | >99 |
| 10 | Ag/HT | 80 | 6 | <1 | - | - | - |
| 11 | Rh/HT | 80 | 6 | <1 | - | - | - |
| 12 | Ru/HT | 80 | 6 | <1 | - | - | - |
| 13 | Cu/HT | 80 | 6 | <1 | - | - | - |
Next, we conducted further studies on Au/HT and Au/TiO
2 in the hydrogenation of
2a in the presence or absence of
p-methylstyrene oxide (
1c) (
Scheme 8). In the absence of
1c, Au/TiO
2 hydrogenated
2a into
3a. Surprisingly, Au/HT did not show any activity toward the hydrogenation of
2a. Neither Au/HT nor Au/TiO
2 hydrogenated
2a in the presence of
1c. These sharply contrasting results indicate that the hydrogen species generated on Au/HT are active for the deoxygenation of epoxides, but are completely inactive for the hydrogenation of C=C bonds. On the other hand, the high selectivity of Au/TiO
2 for alkenes in the deoxygenation of epoxides (
Table 5, Entry 6) is attributable to the preferential adsorption of epoxides over alkenes, which is a similar phenomenon to the previous report that the nitro group in 3-nitrostyrene was reduced by Au/TiO
2 catalyst while retaining C=C bonds [
49].
Scheme 8.
Hydrogenation of 2a in the presence or absence of p-methylstyrene oxide.
Scheme 8.
Hydrogenation of 2a in the presence or absence of p-methylstyrene oxide.
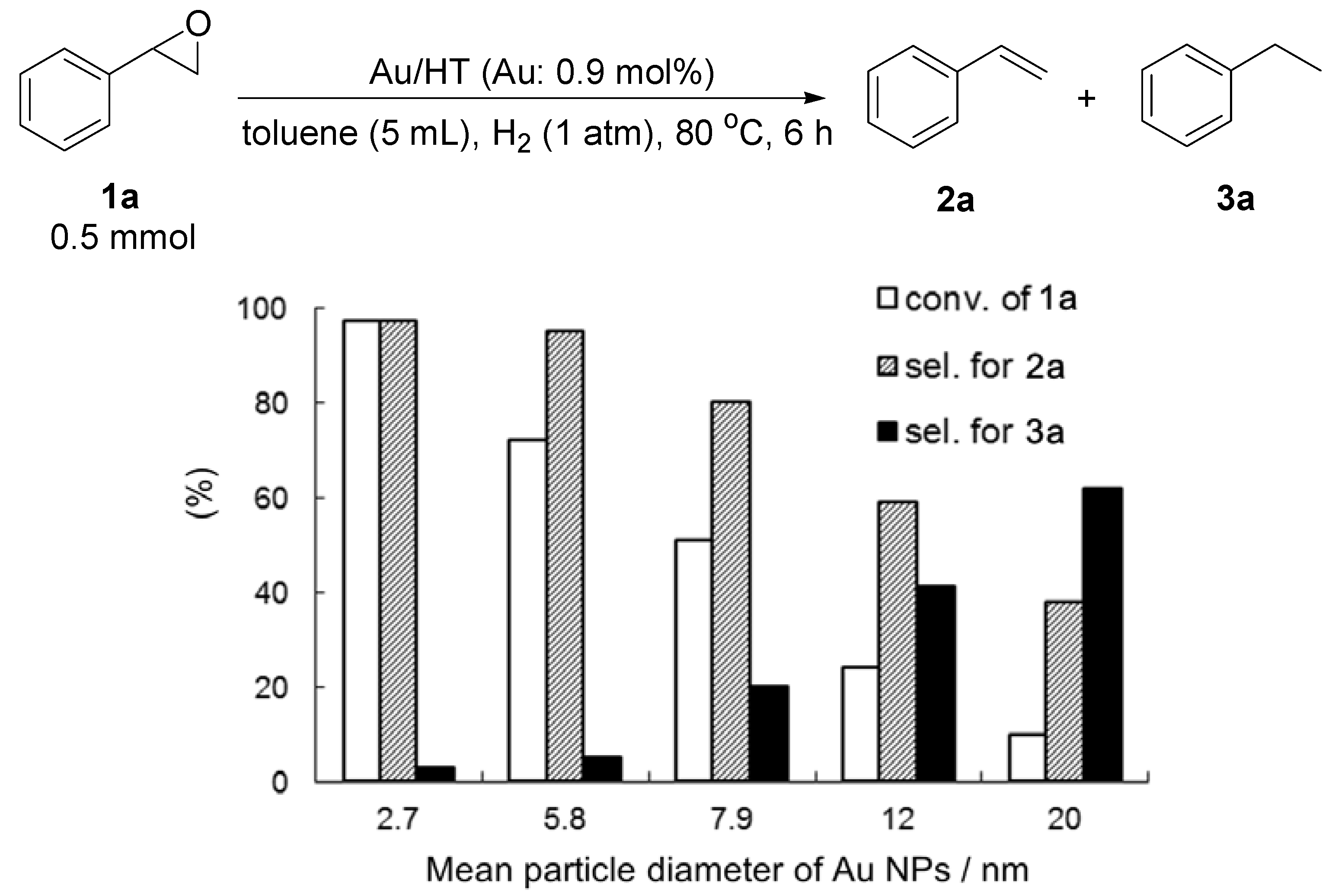
Various Au/HTs with different mean diameters of Au NPs were tested for the deoxygenation of
1a with H
2 (
Figure 5). Different sized Au/HTs could be prepared by varying the concentration of HAuCl
4 solution [
38]. Interestingly, larger Au NPs (>3 nm) showed lower catalytic activity and selectivity for the deoxygenation due to the hydrogenation of the C=C bond. The selectivity and yield of
2a increased with decreasing mean diameter of supported Au NPs. From these results, it can be said that immobilizing small Au NPs (<3 nm) is the key to promoting the selective deoxygenation of epoxides to alkenes. The lower selectivity of larger Au NPs indicates that non-polar hydrogen species active for the hydrogenation of C=C bond were easily generated on the surface of larger Au NPs through the homolytic dissociation of H
2. Au/HT with a mean diameter of 2.7 nm showed high catalytic activity and selectivity for the H
2-driven deoxygenation of both aromatic and aliphatic epoxides to alkenes (
Table 6). After the reaction, Au/HT could be reused with no loss of its catalytic efficiency (Entries 2 and 3).
Figure 5.
Size effect on the deoxygenation of 1a.
Figure 5.
Size effect on the deoxygenation of 1a.
Table 6.
Deoxygenation of various epoxides using Au/HT
a.
![Molecules 16 08209 i050]()
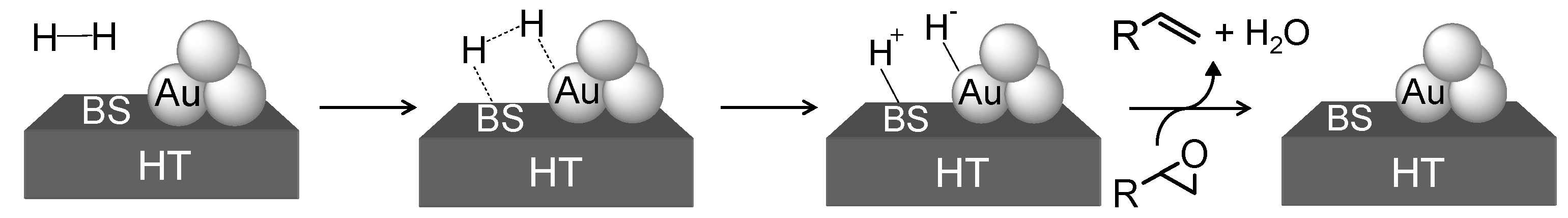
Bearing in mind that basic ligands of transition-metal complexes promote heterolytic cleavage of H
2 to give metal-hydride species, we propose a concerted effect between the basic sites of HT and Au NPs. Namely, heterolytic cleavage of H
2 occurs to give [Au-H]
− and [HT-H]
+ species at the interface between Au NPs and HT, which then react with an epoxide to afford an alkene and H
2O (
Scheme 9). The formation of [Au-H]
− and [HT-H]
+ was confirmed by FT-IR studies. When Au/HT was treated with 50 mbar of H
2, IR bands attributed to [Au-H]
− and [HT-H]
+ appeared around 1,748 cm
−1 and 3,200 cm
−1, respectively. The exclusive formation of the heterolytically cleaved hydrogen species of Au-hydride and H
+ on HT would selectively deoxygenate epoxides to alkenes.
Scheme 9.
Heterolytic dissociation of H2 at the interface between Au NPs and basic sites (BS) of HT in the deoxygenation of epoxides.
Scheme 9.
Heterolytic dissociation of H2 at the interface between Au NPs and basic sites (BS) of HT in the deoxygenation of epoxides.

































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
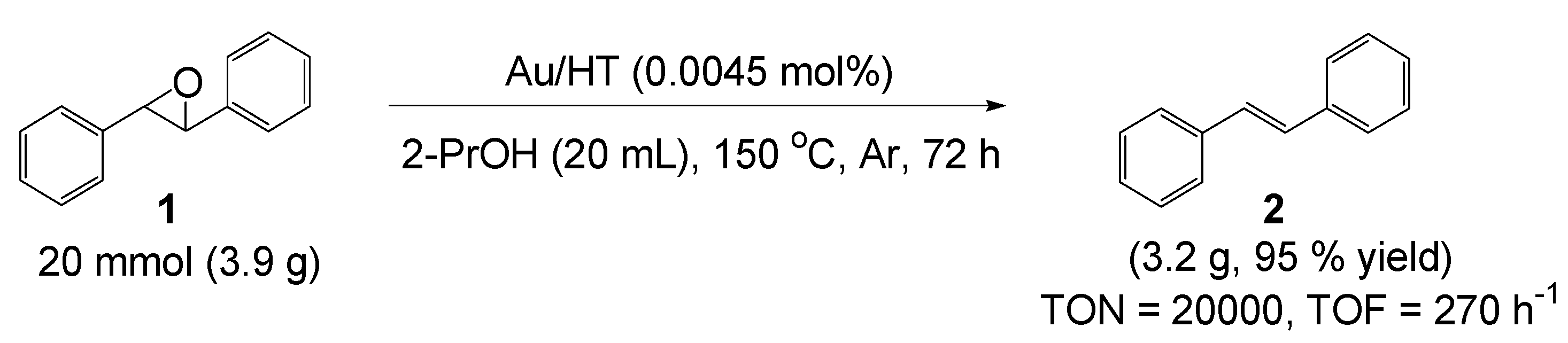{kind=link}
{kind=link}
{kind=link}
{kind=link}
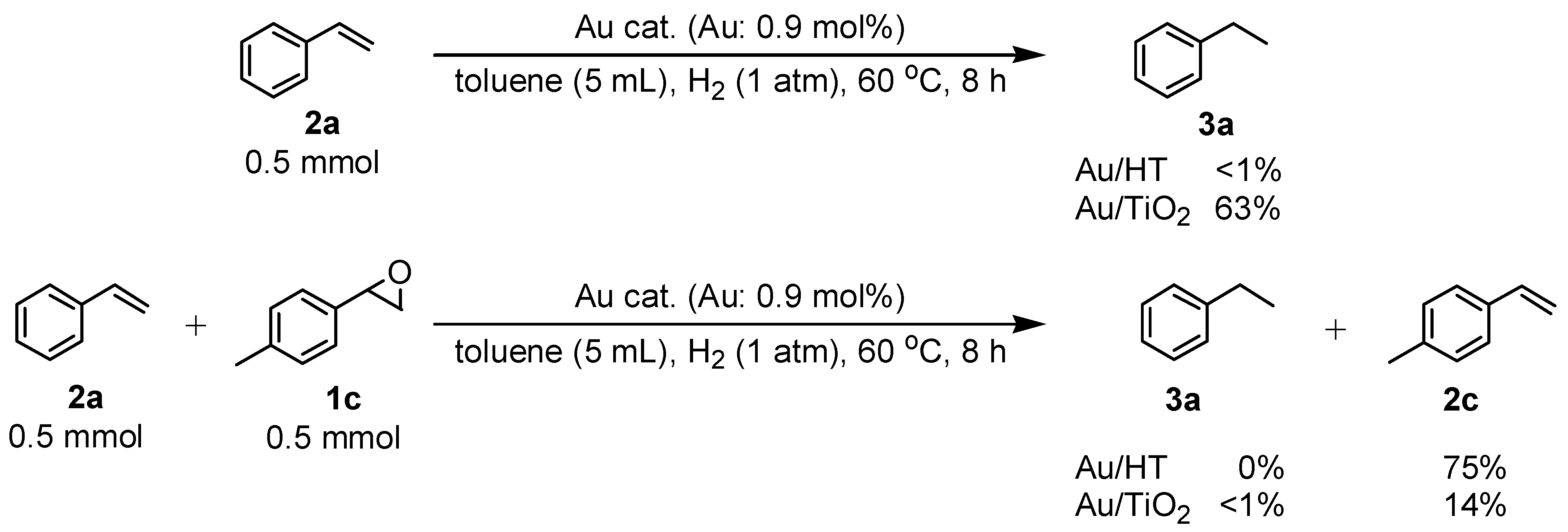{kind=link}
{kind=link}


















