3.1. Crocetin
The carotenoid crocetin has been recently tested as a photosensitizer for DSSC [
37].
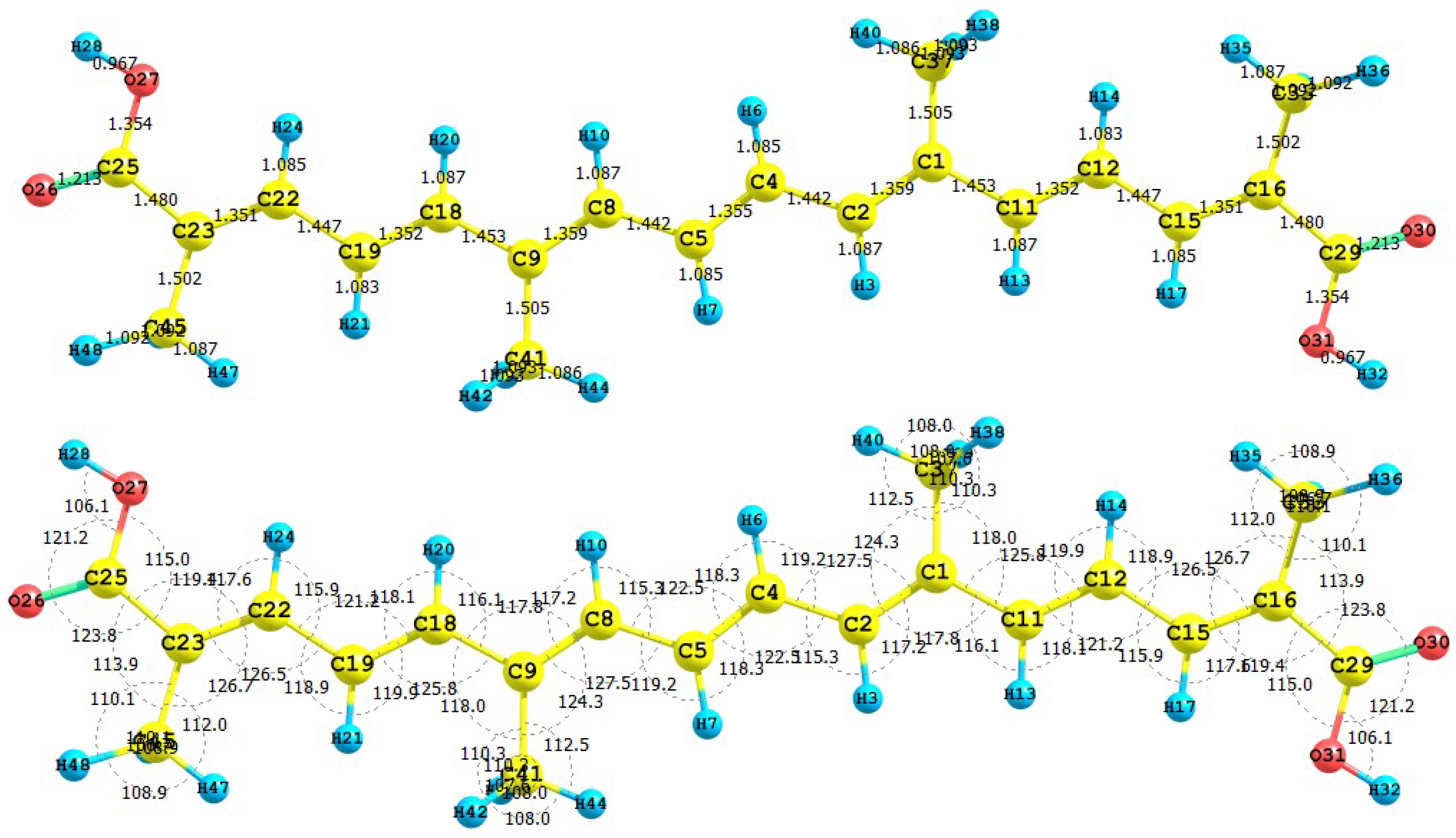
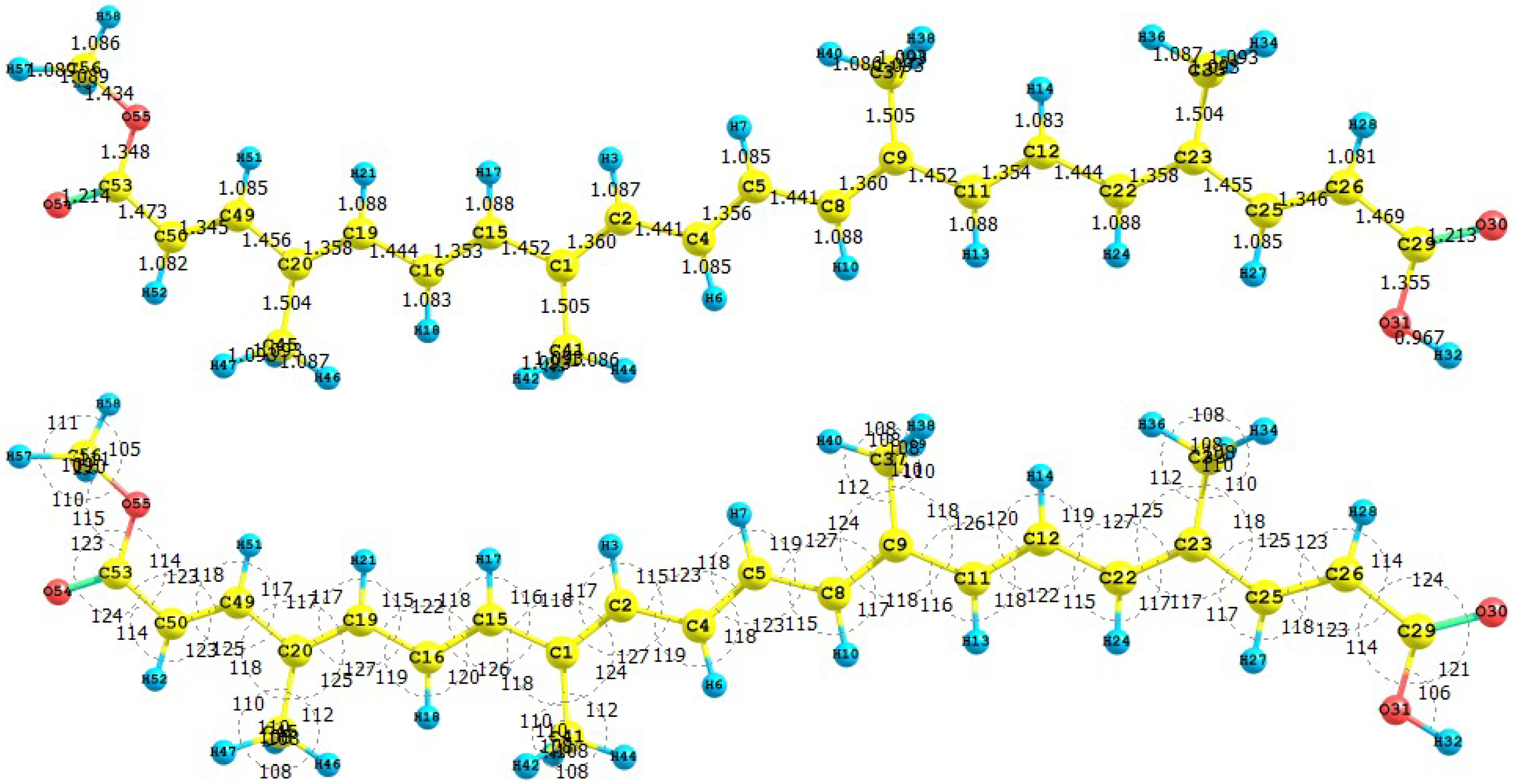
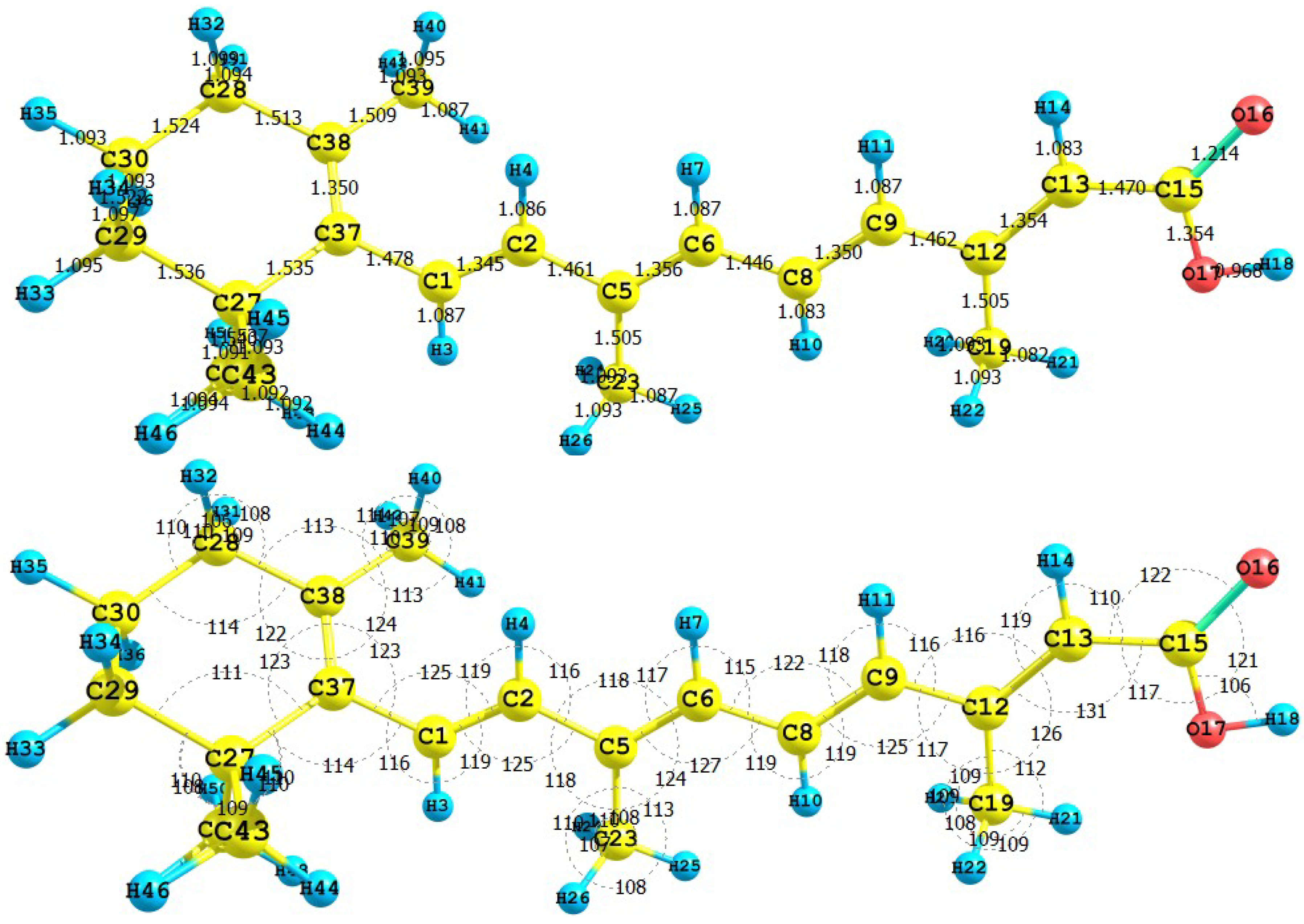
Figure 1 presents the results for the equilibrium conformation of the neutral molecule of crocetin through a representation of the molecular structure of the molecule showing the interatomic bond lengths and bond angles calculated at the M05-2X/6-31+G(d,p) level of theory.
Figure 1.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the crocetin molecule.
Figure 1.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the crocetin molecule.
The infrared spectrum (IR) for the crocetin molecule calculated at the M05-2X/6-31+G(d,p) level of theory is presented by displaying the most intense calculated frequencies together with an explanation of the transition assignments. The calculated spectrum is in a qualitative good agreement with the experimental results. The most intense calculated frequencies are: 542.5 cm-1 = O27-H28 rocking, 776.8 cm-1 = O31-H32 rocking, 1002.6 cm-1 = C33-methyl waving, 1020.4 cm-1 = C7-H8, C5-H6, C3-H4 waving, 1044.2 cm-1 = C1-O27 stretching, 1143.5 cm-1 = C41-methyl waving, 1204.2, 1222.1 and 1235.8 cm--1 = C-H rocking, 1353.4 cm-1 = C1-O-27-H28 twisting, 1394.8 cm-1 = C26-C31-H32 twisting, 1416.9 cm-1 = C21-C19 stretching, 1683.6 cm-1 = C25-C23 stretching, 1708.8 cm--1 = C2-C3 stretching, 1732.9 cm-1 = C26-O30 stretching, 1827.3 cm--1 = C1-O29 stretching, 3228.4 cm-1 = O27-H28 stretching, and 3848.1 cm-1 = O31-H32 stretching.
The ultraviolet spectrum (UV-Vis) of the crocetin molecule calculated with the M05-2X/6-31+G(d,p) level of theory is presented by showing the principal transitions in
Table 1. The wavelengths belonging to the HOMO-LUMO transition appear at 438.0 nm, while the experimental value from the spectrum taken in light petroleum is 450 nm, with a specific absorption coefficient of 4320 [
38].
From the present calculations, the total energy, the total dipole moment and the isotropic polarizability of the ground state of crocetin at the M05-2X/6-31+G(d,p) level of theory are -1077.368 au, 0.0033 Debye and 333.13 Bohr3, respectively. The calculated pKa related to H28 and H32 is 4.544. These results could be of interest as an indication of the solubility and chemical reactivity of the studied molecule.
Table 1.
Electronic transition states of crocetin (nm, eV, oscillator strengths (f), and transition assignments) as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
Table 1.
Electronic transition states of crocetin (nm, eV, oscillator strengths (f), and transition assignments) as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
| Number | Nm | eV | (f) | Assignment; H=HOMO, L=LUMO |
|---|
| 1 | 438.0 | 2.83 | 3.2313 | S H-0→L+0(+82%) |
| 2 | 245.6 | 5.05 | 0.2678 | S H-0→L+2(+32%) H-1→L+1(+25%) |
| | | | | H-2→L+0(+17%) |
| 3 | 241.9 | 5.13 | 0.0548 | S H-2→L+0(+55%) H-0→L+2(+40%) |
| 4 | 217.9 | 5.99 | 0.1235 | S H-1→L+1(+59%) H-0→L+2(17%) |
| H-2
→L+0(15%) |
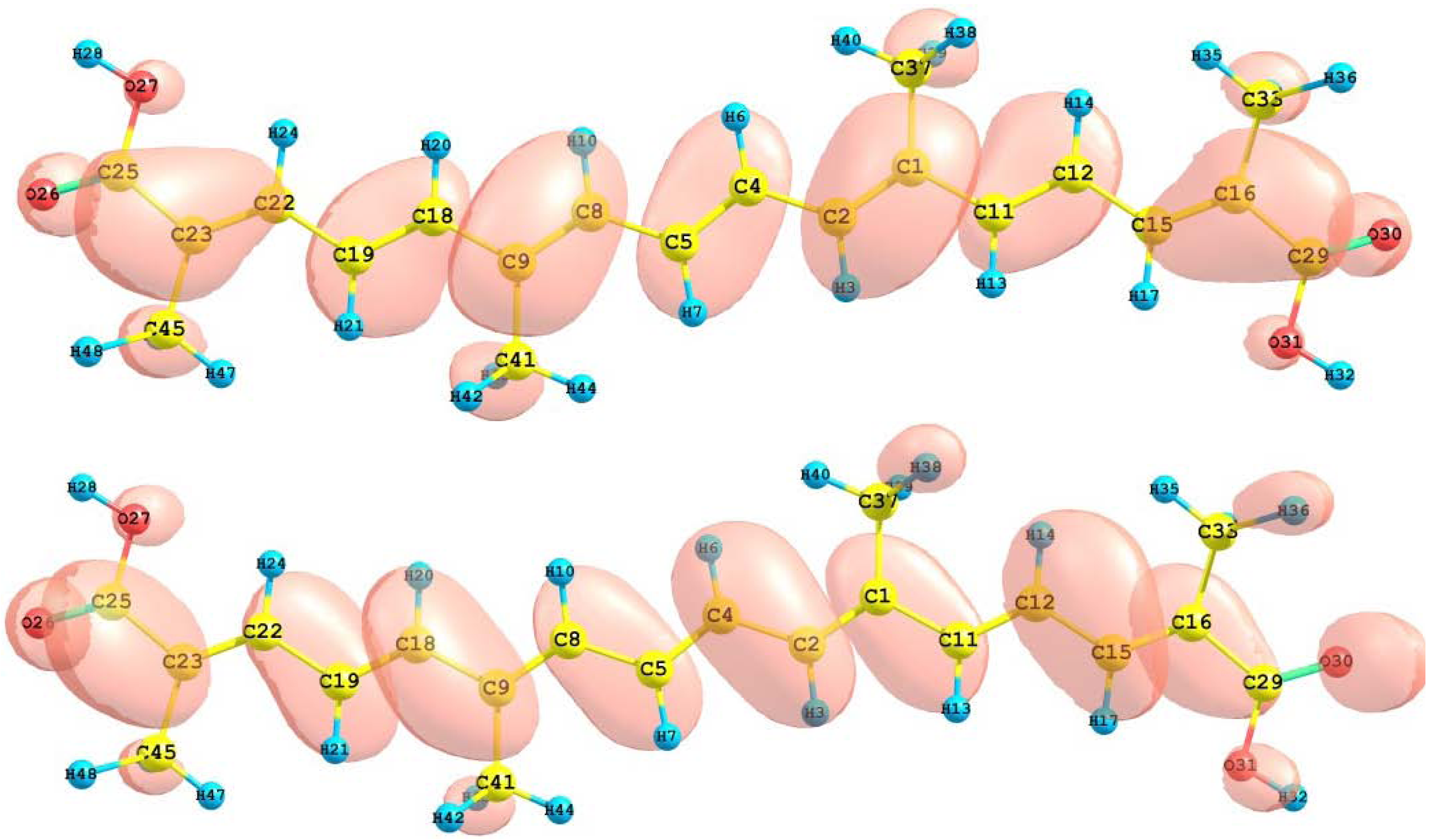
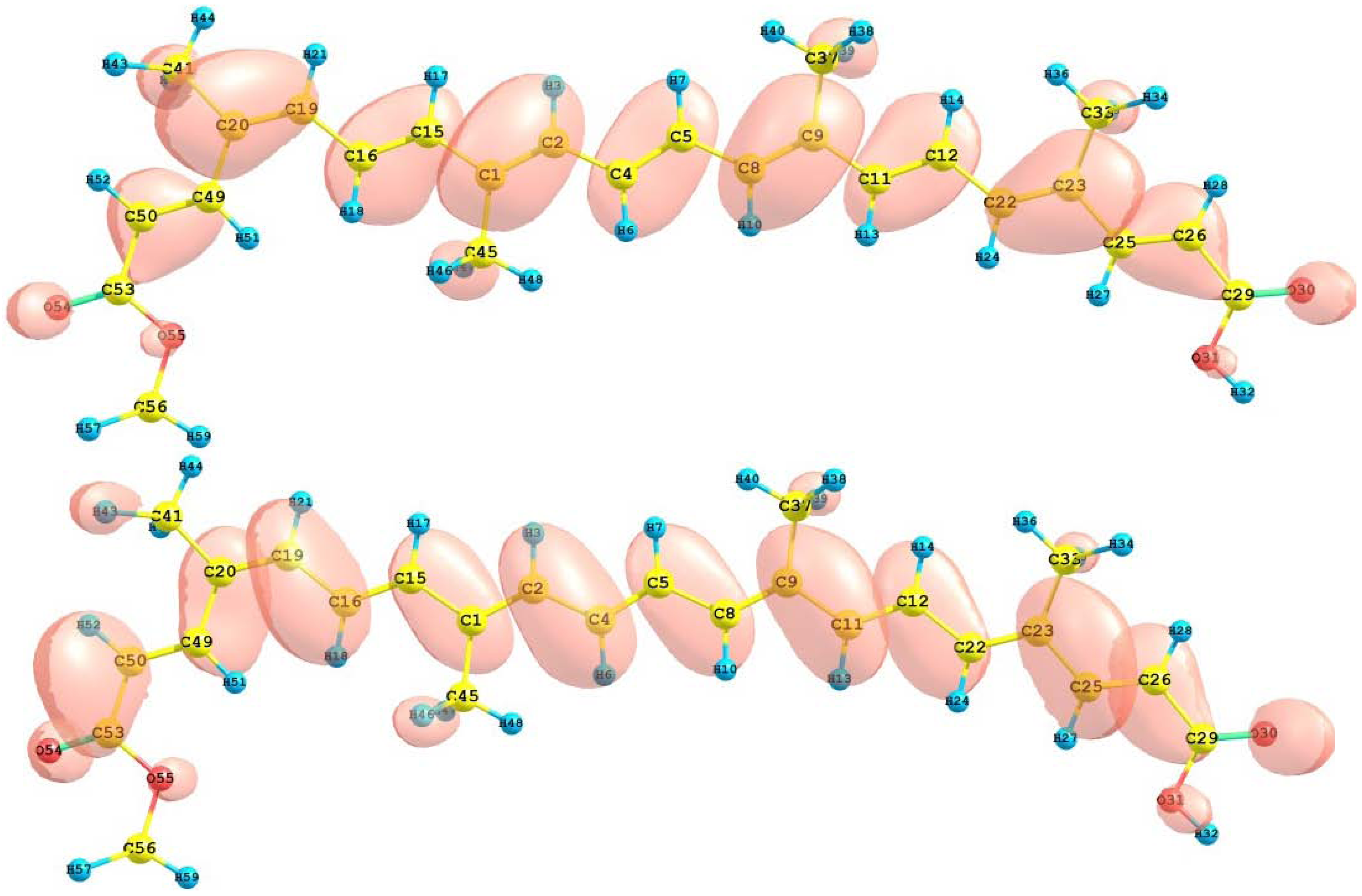
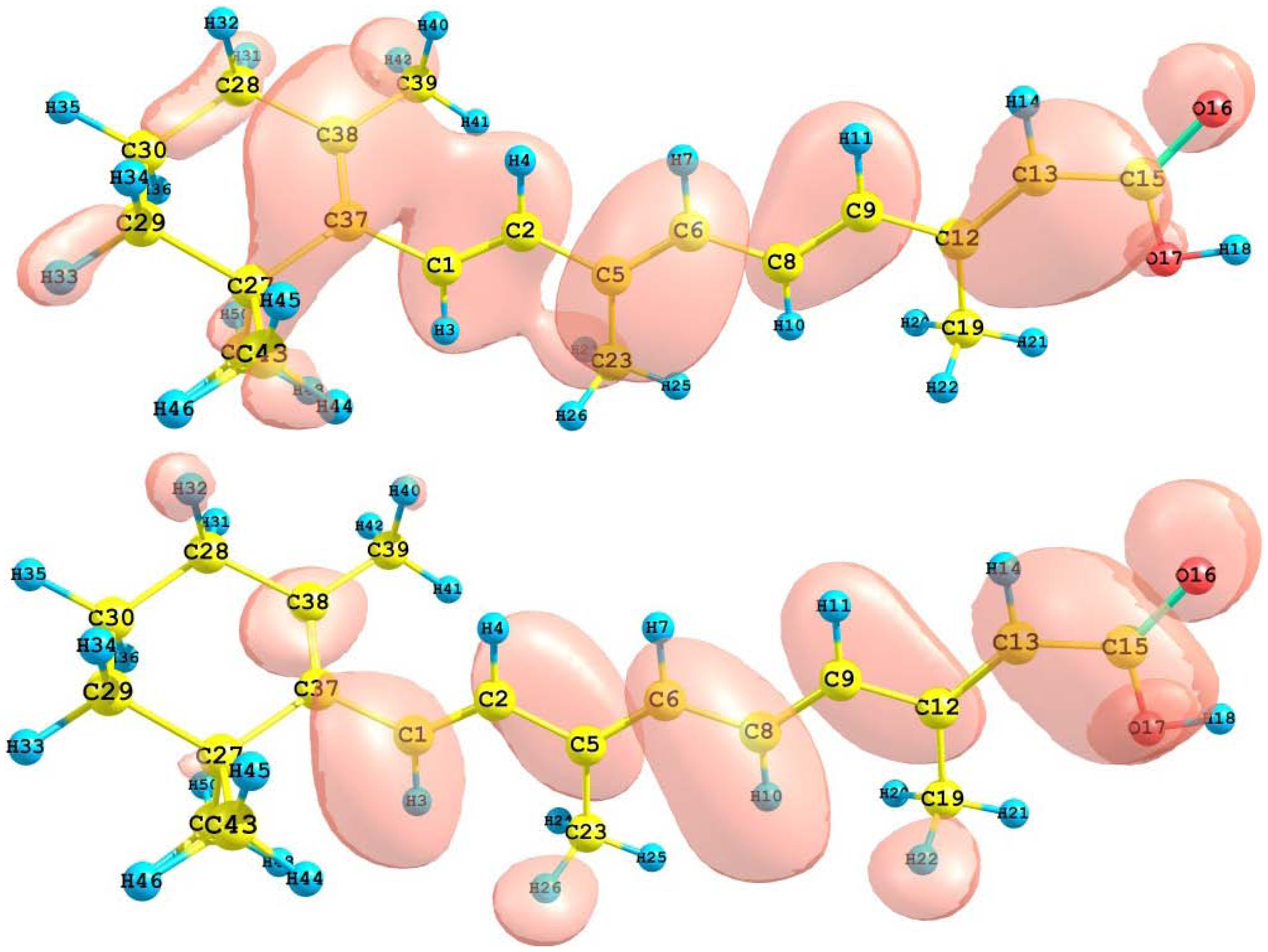
The HOMO and LUMO of the crocetin molecule calculated at the M05-2X/6-31+G(d,p) level of theory are displayed in
Figure 2. This can give us an idea of the reactivity of the molecule. The reactive sites can be identified through an analysis of the total and orbital densities. The representation of the calculated HOMO and LUMO densities in
Figure 2 show that the electrophilic attack would occur preferentially at the C=C double bonds and the nucleophilic attack at the C-C single bonds.
Figure 2.
HOMO and LUMO of the crocetin molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
Figure 2.
HOMO and LUMO of the crocetin molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
The sites for electrophilic attack will be those atoms bearing a negative charge and where the Fukui function fk- is a maximum. These values and those coming from the dual descriptor index confirm that the sites for the electrophilic attack are the C15 and C22 atoms. The sites for potential nucleophilic attack would depend on the values of fk+ on the atoms with a positive charge density. The calculated results from the Fukui functions and the dual descriptor index show that the sites for nucleophilic attack will be the C1 and C9 atoms.
The results for the vertical I and A of the crocetin molecule obtained through energy differences between the ionized and the neutral state, calculated at the geometry of the neutral molecule are I = 6.932 eV and A = 1.694 eV. The HOMO and LUMO energies are -6.589 eV and -2.075 eV, respectively. It can be seen that there is a good qualitative agreement between both results. The calculated values of the electronegativity, global hardness and global electrophilicity using the I and A are 𝜒 = 4.313 eV, η = 2.619 eV, and ω = 3.551 eV. Using the HOMO and LUMO energies, within the Koopmans’ theorem, the corresponding values are 𝜒 = 4.332 eV, η = 2.257 eV, and ω = 4.157 eV. Again, there is a good qualitative agreement for the reactivity parameters calculated through both procedures. It can be concluded that for the particular case of the crocetin molecule, the M05-2X/6-31+G(d,p) level of theory is able to predict the Conceptual DFT reactivity indices calculated through HOMO and LUMO energies as well as from the I and A obtained through energy differences with qualitative similar good accuracy.
3.2. Bixin, Norbixin and Transbixin
The apocarotenoids bixin (or
cis-bixin), norbixin and transbixin have also been recently tested as photosensitizers for DSSC [
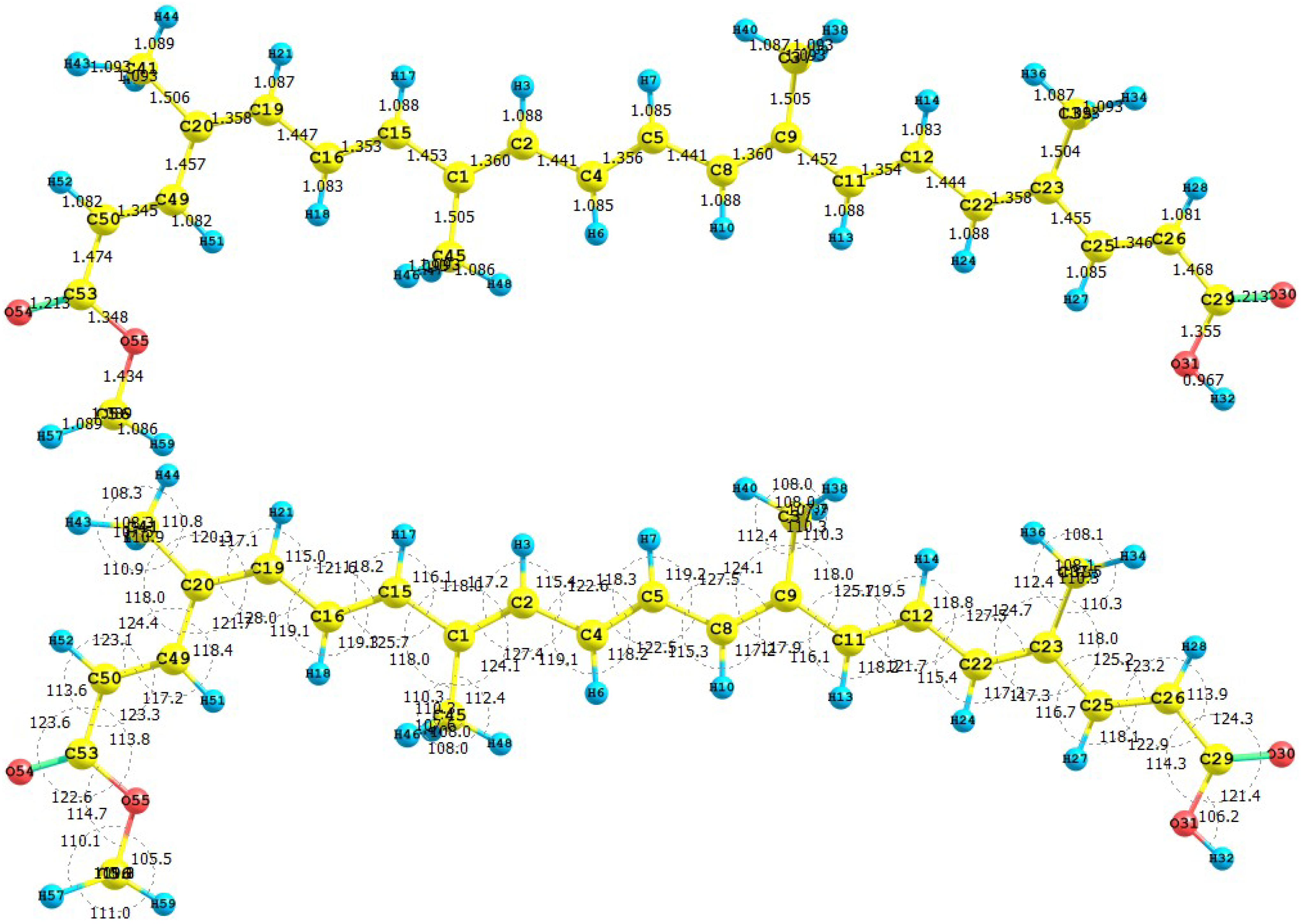
39]. The results for the equilibrium conformations of the neutral molecules of bixin, norbixin and transbixin calculated at the M05-2X/6-31+G(d,p) level of theory are presented through a representation of the molecular structure of the molecule showing the interatomic bond lengths and bond angles in
Figure 3,
Figure 4 and
Figure 5.
Figure 3.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the bixin molecule.
Figure 3.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the bixin molecule.
Figure 4.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the norbixin molecule.
Figure 4.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the norbixin molecule.
Figure 5.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the transbixin molecule.
Figure 5.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the transbixin molecule.
The infrared spectrum (IR) for the bixin molecule calculated with the M05-2X/6-31+G(d,p) level of theory is displayed by showing the most intense calculated frequencies and the corresponding assignments, which are: 611.0 cm-1 = O3-H4 rocking. 1040.4 cm-1 = methyl rocking, 1174.0 cm-1 = C1-O3-H4 waving, 1197.1 cm-1 = C-C stretching, 1214.1 cm-1 = C35-H36 rocking, 1330.2, 1370.7 and 1332.4 cm-1 = C-H rocking, 1462.2 cm-1 = C1-O3-H4 waving, 1662.8 cm-1 = C12-C14 and C26-C28 stretching, 1698.6 cm-1 = C30-C32 stretching, 1725.1 cm-1 = C5-C7 stretching, 1698.6 cm-1= C16-C17 and C23-C25 stretching, 1823.6 cm-1 = C33-C35 stretching, 1840.5 cm-1 = C1-O2 stretching, 3192.4 cm-1 = C-H stretching, 3228.0 cm-1 = methyl C-H asymmetric stretching, and 3622.2 cm-1 = O3-H4 stretching.
The most intense calculated frequencies in the infrared spectrum (IR) for the norbixin molecule calculated at the M05-2X/6-31+G(d,p) level of theory are: 556.9 cm-1 = O28-H29 rocking, 678.5 cm-1 = O49-H49 rocking, 1032.2 cm-1 = C-H rocking, 1034.6, 1078.6 and 1055.3 cm-1 = methyl waving, 1065.7 cm-1 = C47-O48 stretching, 1181.1 cm-1 = C1-O28 stretching, 1200.0 cm-1 = C-H waving, 1394.1 cm-1 = C1-C2 stretching, 1670.3 cm-1 = C20-C22 stretching, 1694.9 cm-1 = C23-C46 stretching, 1720.8 cm-1 = C11-C2 stretching, 1723.8 cm-1 = C25-C26 stretching, 1727.9 cm-1 = C2-C3 stretching, 1792.2 cm-1 = C47-O50 stretching, 1838.8 cm-1 = C1-O27 stretching, 3242.1 cm-1 = O28-H29 stretching, and 3855.3 cm-1 = O48-H49 stretching.
The most intense calculated frequencies in the infrared spectrum (IR) for the transbixin molecule calculated with the M05-2X/6-31+G(d,p) level of theory are: 650.7 cm-1 = O36-H37 rocking, 1014.3 cm-1 = C-H rocking, 1031.7 and 1035.0 cm-1 = methyl waving, 112.1 cm-1 = C1-O38 stretching, 1194.7 cm-1 = C33-H32 rocking, 1246.6, 1328.5, 1333.4 and 1343.6 cm-1 = C-H rocking, 1394.0 cm-1 = C1-O36-H37 scissoring, 1462.6 cm-1 = methyl waving, 1670.8 cm-1 = C9-C11 stretching, 1696.7 cm‑1 = C27-C29 stretching, 1721.5 cm-1 = C13-C14 stretching, 1724.6 cm-1 = C2-C4 stretching, 1822.5 cm-1 = C34-O38 stretching, 1838.6 cm-1 = C1-O35 stretching, and 3850.7 cm-1 = O36-H37 stretching.
The ultraviolet spectrum (UV-Vis) of the bixin, norbixin, and transbixin molecules calculated with the M05-2X/6-31+G(d,p) level of theory are presented by showing the principal transitions in
Table 2,
Table 3 and
Table 4. The wavelength corresponding to the HOMO-LUMO transition will appear at 471.6 nm for bixin, 476.9 nm for norbixin, and 476.2 nm for transbixin. The reported experimental absorptions occur at 456 nm for bixin in light petroleum, and at 474.0 nm for norbixin in chloroform [
35].
Table 2.
Electronic transition states of bixin (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
Table 2.
Electronic transition states of bixin (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
| Number | nm | eV | (f) | Assignment; H=HOMO, L=LUMO |
|---|
| 1 | 471.6 | 2.63 | 3.7551 | S H-0→L+0(+81%) H-1→L+1(+5%) |
| 2 | 346.1 | 3.58 | 0.1111 | S H-0→L+1(+67%) H-1→L+0(+12%) |
| 3 | 265.6 | 4.67 | 0.2404 | S H-2→L+0(+59%) H-0→L+2(14%) |
| 4 | 243.4 | 5.09 | 0.1220 | S H-1→L+1(+52%) H-2→L+0(+26%) |
| | | | | H-0→L+2(10%) |
| 5 | 235.1 | 5.27 | 0.1639 | S H-0→L+3(+40%) H-1→L+2(29%) |
| 6 | 227.1 | 5.46 | 0.0405 | S H-3→L+0(+45%) H-0→L+3(+21%) |
| | | | | H-2→L+1(+18%) |
Table 3.
Electronic transition states of norbixin (nm, eV, oscillator strengths (f), and transition assign-ments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
Table 3.
Electronic transition states of norbixin (nm, eV, oscillator strengths (f), and transition assign-ments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
| Number | nm | eV | (f) | Assignment; H=HOMO, L=LUMO |
|---|
| 1 | 476.9 | 2.60 | 3.9337 | S H-0→L+0(+82%) H-1→L+1(5%) |
| 2 | 276.0 | 4.49 | 0.3509 | S H-0→L+2(+46%) H-1→L+1(+25%) |
| 3 | 265.6 | 4.67 | 0.1728 | S H-2→L+0(+58%) H-0→L+2(+29%) |
| | | | | H-1→L+1(5%) |
| 4 | 243.3 | 4.63 | 0.1128 | S H-1→L+1(+53%) H-2→L+0(+23%) |
| | | | | H-0→L+2(14%) |
Table 4.
Electronic transition states of transbixin (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
Table 4.
Electronic transition states of transbixin (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31+G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
| Number | nm | eV | (f) | Assignment; H=HOMO, L=LUMO |
|---|
| 1 | 476.2 | 2.60 | 3.9436 | S H-0→L+0(+82%) H-1→L+1(5%) |
| 2 | 275.7 | 4.50 | 0.3689 | S H-0→L+2(+45%) H-1→L+1(+25%) |
| | | | | H-2→L+0(6%) |
| 3 | 265.5 | 4.67 | 0.1626 | S H-2→L+0(+59%) H-0→L+2(+29%) |
| 4 | 243.0 | 5.10 | 0.1156 | S H-1→L+1(+53%) H-2→L+0(+22%) |
| | | | | H-0→L+2(14%) |
The total energy, the total dipole moment, the isotropic polarizability of the ground state calculated at the M052-2X/6-31+G(d,p) level of theory and the pKa calculated with MOPAC 2009 and PM6 for the bixin, norbixin, and transbixin molecules are presented in
Table 5.
Table 5.
Total energy E, dipole moment μ, polarizability α, calculated with the M05-2X/6-31+G(d,p) level of theory, and pKa calculated with MOPAC 2009 and PM6.
Table 5.
Total energy E, dipole moment μ, polarizability α, calculated with the M05-2X/6-31+G(d,p) level of theory, and pKa calculated with MOPAC 2009 and PM6.
| Molecule | E (a.u.) | μ (Debye) | α (Bohr3) | pKa |
|---|
| Bixin | -1271.461 | 1.88 | 477.84 | 4.784 |
| Norbixin | -1232.061 | 1.43 | 466.17 | 4.768 |
| Transbixin | -1271.462 | 1.02 | 468.84 | 4.778 |
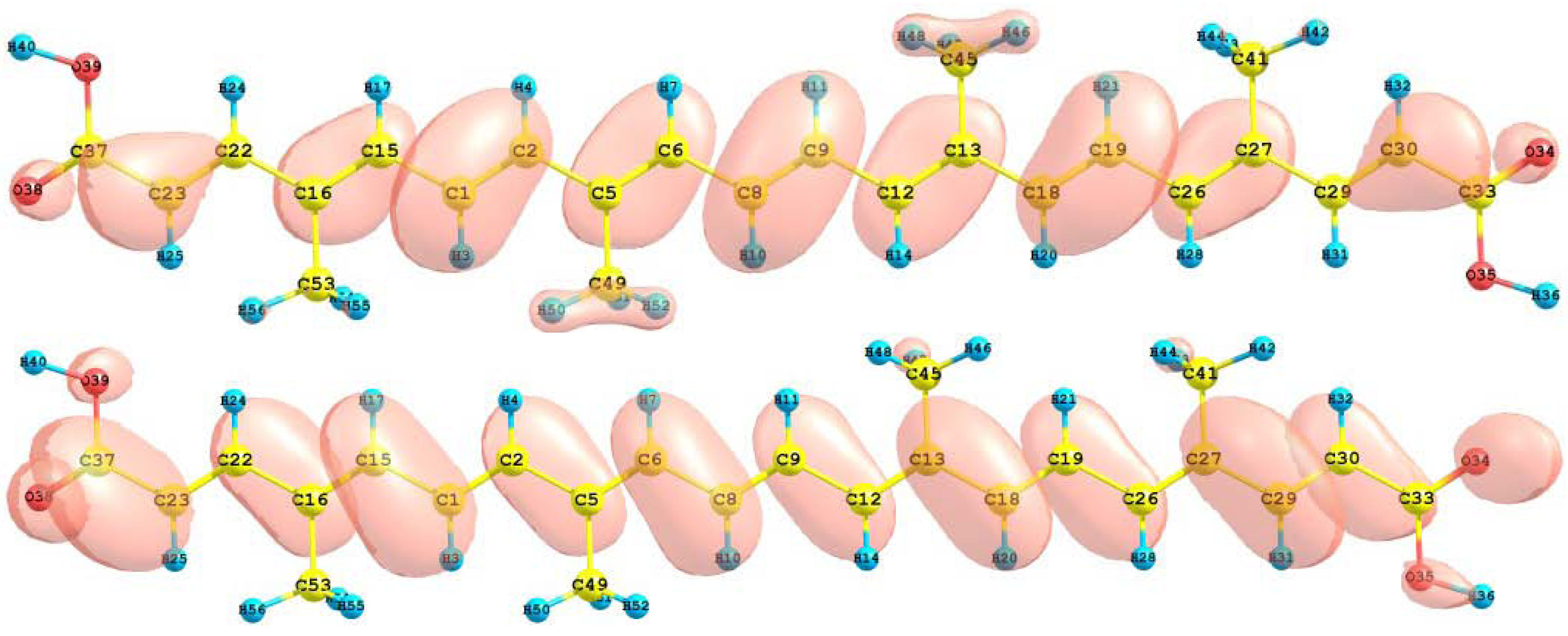
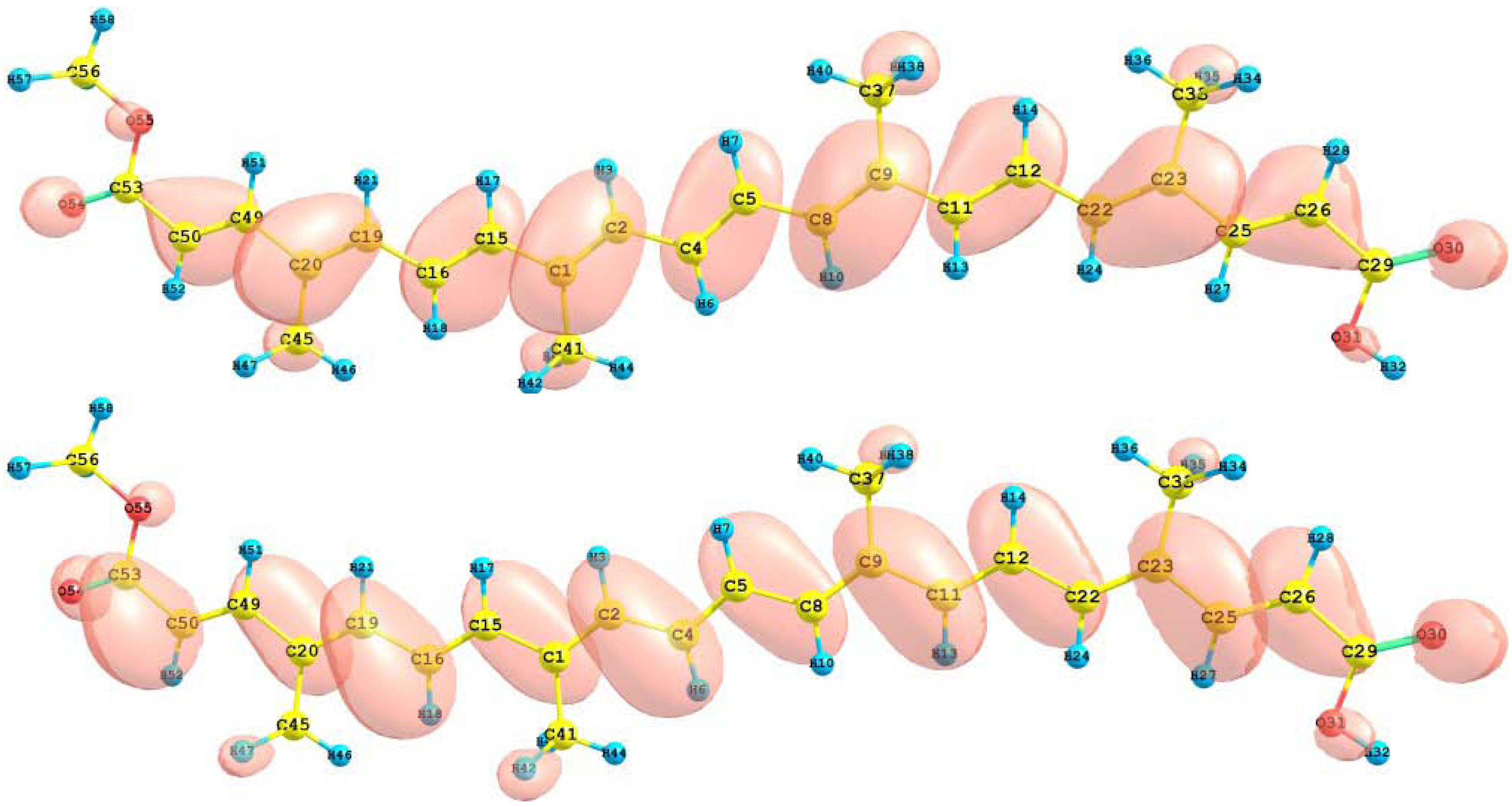
The HOMO and LUMO orbital of the bixin, norbixin and transbixin molecules calculated with the M05-2X/6-31+G(d,p) level of theory are displayed in
Figure 6,
Figure 7 and
Figure 8. The sites for electrophilic attack will be those atoms bearing a negative charge and where the Fukui function
fk- is a maximum, while the sites for potential nucleophilic attack would depend on the values of
fk+ on the atoms with a positive charge density. The calculated results are shown in
Table 6.
Figure 6.
HOMO and LUMO of the bixin molecule calculated at the M05-2X/6-31+G(d,p) level of theory.
Figure 6.
HOMO and LUMO of the bixin molecule calculated at the M05-2X/6-31+G(d,p) level of theory.
Figure 7.
HOMO and LUMO of the norbixin molecule calculated at the M05-2X/6-31+G(d,p) level of theory.
Figure 7.
HOMO and LUMO of the norbixin molecule calculated at the M05-2X/6-31+G(d,p) level of theory.
Figure 8.
HOMO and LUMO of the transbixin molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
Figure 8.
HOMO and LUMO of the transbixin molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
Table 6.
Electrophilic and nucleophilic attack sites for bixin, norbixin, and transbixin through Fukui functions and condensed dual descriptors calculated with the M05-2X/6-31+G(d,p) level of theory and the AOMix program. The value of the condensed dual descriptor (x 100) over the atom is shown in parentheses.
Table 6.
Electrophilic and nucleophilic attack sites for bixin, norbixin, and transbixin through Fukui functions and condensed dual descriptors calculated with the M05-2X/6-31+G(d,p) level of theory and the AOMix program. The value of the condensed dual descriptor (x 100) over the atom is shown in parentheses.
| Molecule | Bixin | Norbixin | Transbixin |
|---|
| Electrophilic attack site | C11 (-2.72) | C23, C29 (-4.61) | C22 (-2.33) |
| Nucleophilic attack site | C9 (3.04) | C5, C13 (5.77) | C9 (2.83) |
The results for the vertical IP and A of the bixin, norbixin and transbixin molecules obtained through energy differences between the ionized and the neutral state, calculated at the geometry of the neutral molecule, the HOMO and LUMO energies, and the calculated values of the electronegativity, global hardness and global electrofilicity using the I and A, and using the HOMO and LUMO energies, within the Koopmans’ theorem, are displayed in
table 7. The agreement between the results of both groups of Conceptual DFT reactivity descriptors is qualitatively correct for the three molecules, being the for bixin very good. The ionization potentials I are better described using the Koopmans’ theorem approximation than the electron affinities A, and this can be ascribed as the main source of discrepancies.
Table 7.
Ionization potential I, Electron affinity A, HOMO energy, LUMO energy, electronegativity, global hardness and global electrophilicity of bixin, norbixin and transbixin calculated with the M05-2X/6-31+G(d,p) level of theory and Eqns. 3, 4 and 5. The first group of Conceptual DFT reactivity descriptors corresponds to calculations based on energy differences, while the second group belongs to results based on HOMO and LUMO energies.
Table 7.
Ionization potential I, Electron affinity A, HOMO energy, LUMO energy, electronegativity, global hardness and global electrophilicity of bixin, norbixin and transbixin calculated with the M05-2X/6-31+G(d,p) level of theory and Eqns. 3, 4 and 5. The first group of Conceptual DFT reactivity descriptors corresponds to calculations based on energy differences, while the second group belongs to results based on HOMO and LUMO energies.
| Molecule | Bixin | Norbixin | Transbixin |
|---|
| I (eV) | 6.679 | 7.091 | 6.684 |
| A (eV) | 1.773 | 1.310 | 1.824 |
| ℇHOMO (eV) | -6.368 | -6.727 | -6.377 |
| ℇLUMO (eV) | -2.099 | -1.718 | -2.144 |
| 𝜒 (eV) | 4.226 | 4.201 | 4.254 |
| η(eV) | 2.453 | 2.891 | 2.430 |
| ω(eV) | 3.640 | 3.052 | 3.724 |
| 𝜒 (eV) | 4.234 | 4.223 | 4.261 |
| η (eV) | 2.135 | 2.505 | 2.117 |
| ω(eV) | 4.198 | 3.560 | 4.288 |
3.3. Retinoic acid
Retinol, also known as vitamin A, is essential for life. Retinol in the human body is produced by a series of metabolic processes that begin with dietary intake of
β-carotene and retinyl esters. Retinoic acid (RA), the oxidized form of Vitamin A, is one of the most potent physiological metabolites of retinol. Retinoic acid has also been tried as a sensitizer for DSSC [
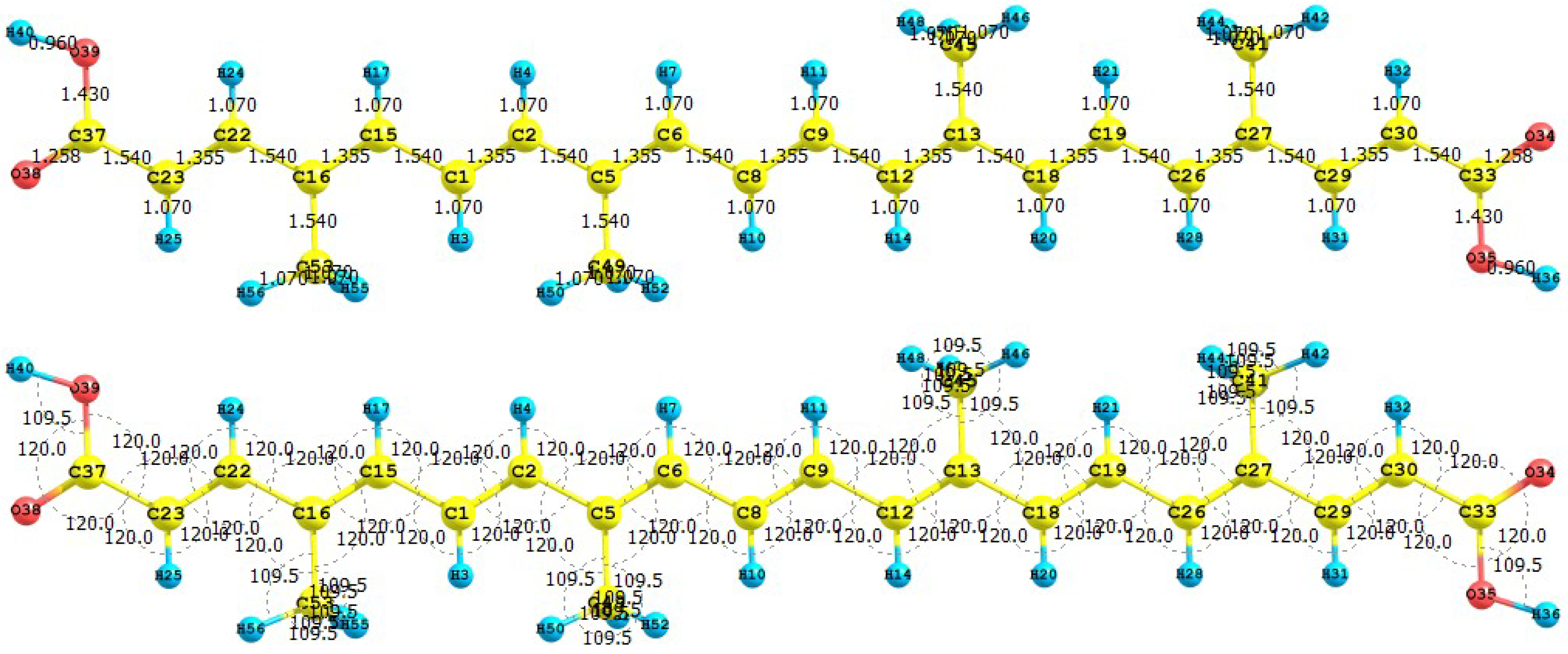
40]. The results for the equilibrium conformation of the neutral molecule of retinoic acid calculated with the M05-2X/6-31+G(d,p) level of theory through a representation of the molecular structure of the molecule showing the interatomic bond lengths and bond angles are presented in
Figure 9.
Figure 9.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the retinoic acid molecule.
Figure 9.
Interatomic bond distances (
![Molecules 15 04490 i024]()
) and bond angles (deg) for the retinoic acid molecule.
The infrared spectrum (IR) for the retinoic acid molecule calculated with the M05-2X/6-31+G(d,p) level of theory are presented by showing the most intense calculated frequencies and their assignments, which are: 609.3 cm-1 = O41-H42 rocking, 1021.9 cm-1 = C-H rocking, 1193.1 cm-1 = C39-O41 stretching, 1210.6 cm-1 = C-C stretching, 1263.9 and 1404.8 cm-1 = C-H rocking, 1702.5 and 1725.3 cm-1 = C-C asymmetric stretching, 1824.0 cm-1 = C39-O40 stretching, and 3833.9 cm-1 = O41-H42 stretching.
The ultraviolet spectrum (UV-Vis) of the retinoic acid molecule calculated with the M05-2X/6-31+G(d,p) level of theory is presented by showing the principal transitions in
Table 8. The wavelength belonging to the HOMO-LUMO transition will appear at 360.7 nm.
Table 8.
Electronic transition states of retinoic acid (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
Table 8.
Electronic transition states of retinoic acid (nm, eV, oscillator strengths (f), and transition assignments as calculated with TD-DFT and the M05-2X/6-31G(d,p) level of theory. Only the transition states with f > 0.02 are shown.
| Number | nm | eV | (f) | Assignment; H=HOMO, L=LUMO |
|---|
| 1 | 360.7 | 3.44 | 1.7642 | S H-0→L+0(+83%) |
| 2 | 264.3 | 4.69 | 0.1850 | S H-1→L+0(+76%) H-0→L+1(+6%) |
| 3 | 237.6 | 5.22 | 0.1527 | S H-0→L+1(+80%) H-1→L+0(8%) |
| 4 | 221.1 | 5.61 | 0.0325 | S H-2→L+0(+73%) |
| 5 | 202.5 | 6.12 | 0.0224 | S H-0→L+3(+40%) H-3→L+0(11%) |
| 6 | 198.0 | 6.26 | 0.1169 | S H-1→L+1(+22%) H-0→L+7(17%) |
From the present calculations, the total energy, the total dipole moment and the isotropic polarizability of the ground state of retinoic acid with the M052-2X/6-31+G(d,p) level of theory are ‑929.349 au, 4.790 Debye and 176.53 Bohr3, respectively. The calculated pKa related to the H18 is 6.690. These results could be of interest as an indication of the solubility and chemical reactivity of the studied molecule.
The HOMO and LUMO of the retinoic acid molecule calculated with the M05-2X/6-31+G(d,p) level of theory are displayed in
Figure 10. This can give us an idea of the reactivity of the molecule. The sites for electrophilic attack will be those atoms bearing a negative charge and where the Fukui function
fk- is a maximum. These values as well as the results from the condensed dual descriptor confirm that the site for the electrophilic attack is the C12 atom. The site for potential nucleophilic attack would depend on the values of
fk+ on the atoms with a positive charge density and where the condensed dual descriptor has the largest positive value. The calculated results show that the site for nucleophilic attack will be the C38 atom.
Figure 10.
HOMO and LUMO of the retinoic acid molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
Figure 10.
HOMO and LUMO of the retinoic acid molecule calculated with the M05-2X/6-31+G(d,p) level of theory.
The results for the vertical I and A of the retinoic acid molecule obtained through energy differences between the ionized and the neutral state, calculated at the geometry of the neutral molecule are I = 7.221 eV and A = 0.942 eV. The HOMO and LUMO energies are -7.986 eV and -1.445 eV, respectively. There is a good qualitative agreement between both results. The calculated values of the electronegativity, global hardness and global electrophilicity using the I and A are 𝜒 = 4.082 eV, η = 3.140 eV, and ω = 3.303 eV. Using the HOMO and LUMO energies, within the Koopmans’ theorem, the corresponding values are 𝜒 = 4.716 eV, η = 3.271 eV, and ω = 3.399 eV. Again, there is a good qualitative agreement for the reactivity parameters calculated through both procedures. It can be concluded that also for the particular case of the retinoic acid molecule, the M05-2X/6-31+G(d,p) level of theory is able to predict the Conceptual DFT reactivity indices calculated through HOMO and LUMO energies with the same degree of accuracy as the results from energy differences.
3.4. Zinc oxide (ZnO)
In its simplest configuration, the dye-sensitized solar cell (DSSC) is comprised of a transparent conducting glass electrode coated with porous nanocrystalline TiO
2, dye molecules attached to the surface of the TiO
2, an electrolyte containing a reduction-oxidation couple such as I
-/ I
-3 and a catalyst coated counter-electrode. Upon illumination, the cell produces an overvoltage and current through an external load connected to the electrodes. Due to the energy level positioning in the system, the cell is capable of producing voltage between its electrodes and across the external load. The maximum theoretical value for the photovoltage at an open circuit condition is determined by the potential difference between the conduction band edge of the TiO
2 and the redox potential of the I
-/I
-3 pair in the electrolyte. The operation of the cell is regenerative in nature, since no chemical substances are neither consumed nor produced during the working cycle [
41].
At the heart of the system is a mesoporous oxide layer composed of nanometer-sized particles which have been sintered together to allow for electronic conduction to take place. The material of choice has traditionally been TiO
2 (anatase), although alternative wide band gap oxides such as ZnO, and Nb
2O
5 have also been investigated [
42].
Zinc oxide (ZnO) has a large application potential owing to its diverse physical properties and the fine-tuning in the preparation process. Historically, ZnO has been used for a long time as pigment and protective coatings on metal surfaces. Its wide band gap of 3.2 eV at room temperature has rendered the use as protective UV-absorbing additive in everything from skin creams to advanced plastic and rubber composites. ZnO is an attractive material for nanoscale optoelectronic devices, as it is a wide-band gap semiconductor with good carrier mobility and can be doped both n and p-type. The electron mobility is much higher in ZnO than in TiO
2, while the conduction band edge of both materials is located at approximately the same level [
43].
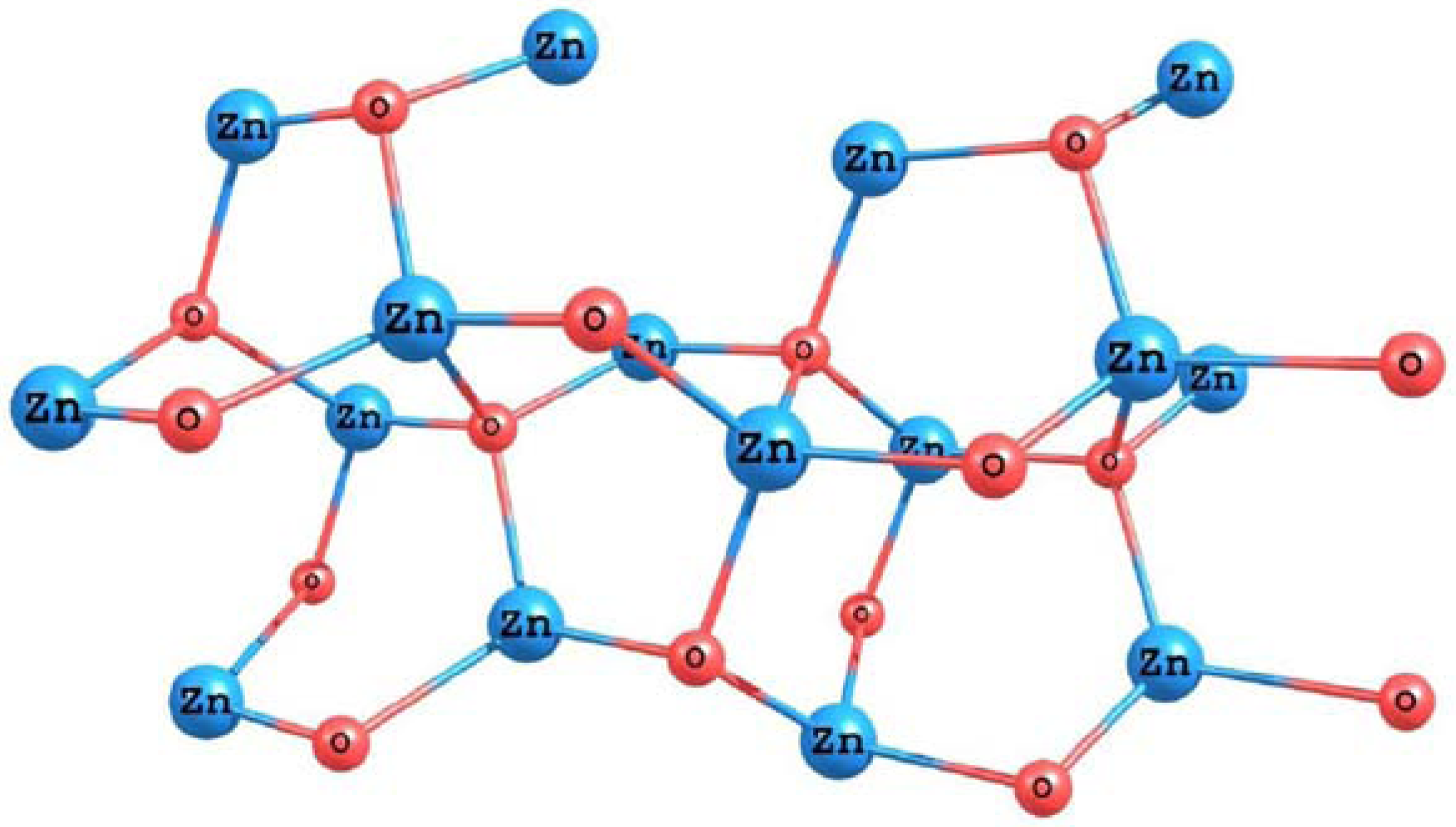
In order to simulate the ZnO surface, we have optimized the structure of a small cluster based on the (101) face of a zincite crystal. The results for the optimized structure of the system obtained using the M05-2X density functional and the LANL2DZ basis set are presented in
Figure 11.
The results for the vertical I and A of the studied ZnO cluster obtained through energy differences between the ionized and the neutral state, calculated at the geometry of the neutral systems are I = 6.941 eV and A = 1.759 eV. The HOMO and LUMO energies are -6.286 eV and -2.340 eV, respectively. The calculated values of the electronegativity, global hardness and global electrophilicity using the I and A are 𝜒 = 4.350 eV, η = 2.591 eV, and ω = 3.652 eV. Using the HOMO and LUMO energies, within the Koopmans’ theorem, the corresponding values are 𝜒 = 4.313 eV, η = 1.973 eV, and ω = 4.714 eV. The calculated band gap is 3.95 eV. Indeed, this value is different from the experimental band gap not only due to the quality of the density functional employed, but also because we are considering a small cluster instead of the bulk system.
Figure 11.
Molecular structure of the ZnO surface calculated with the M05-2X/LANL2DZ level of theory.
Figure 11.
Molecular structure of the ZnO surface calculated with the M05-2X/LANL2DZ level of theory.
3.5. Photovoltaic properties
The conversion efficiency of the solar cell η is defined as the ratio of the generated maximum electric output power to the total power of the incident light P
in [
44]:
where I
sc is the short-circuit current, V
oc is the open-circuit voltage, and FF is the fill factor of the solar cell which is defined as:
The photovoltaic parameters are evaluated under standard test conditions: the air mass (AM) 1.5 spectrum with an incident power density of 1,000 W/m
2 and a temperature of 25
oC. In order to improve the efficiency, it is necessary to maximize all the three photovoltaic parameters, such as V
oc, I
sc and FF [
44].
The aim of device modeling is to develop a link between materials’ properties and the electrical device characteristics of a nanostructured solar cell. The goal of device modeling is to simulate the I-V curve of a nanostructured solar cell, both in dark and under illumination. From this, the main photovoltaic parameters of the solar cell are deduced [
45]. However, this procedure is very involved both from a theoretical and computational point of view. Thus, we prefer to perform materials modeling, where materials parameters are studied and theoretically modeled based on physical and chemical phenomena and interactions.
For example, it is well known that there exists a relationship between the V
oc and the interfacial band gap, which is defined as the difference between the HOMO of the donor (the absorbing dye) and the LUMO of the acceptor (the nanostructured metallic oxide). For a series of similar compounds, knowing the experimental efficiency, it should be possible to estimate the proportionality constant. However, the data for the carotenoids in our work have taken from different sources [
37,
39,
40], with experiments performed in different conditions, and for this reason, it is not possible to do quantitative comparisons. On the contrary, some qualitative comparisons can be performed. For a given acceptor (ZnO in this case), the interfacial band gap will be larger when the HOMO of donor (the carotenoid) become larger. The results for the HOMO for the five carotenoids studied in this work are:
From this, it can be concluded that the retinoic acid caroteonid will be the more efficient for a DSSC based on nanostructured ZnO. A similar analysis, based on the calculated electronegativity
𝜒, total hardness
η and global elec-trophilicity
ω, gives, respectively:
It can be concluded that if retinoic acid is the carotenoid which will render the best efficiency in a ZnO based DSSC according the results of the Voc related to the interfacial band gap, then the calculated values of electronegativity 𝜒,total hardness η and global electrophilicity ω could also give an idea of the efficiency of the solar cell. That is, the larger the value of the Conceptual DFT reactivity parameters, the larger the efficiency of the DSSC. However, the opposite holds for the case of the global electrophilicity. Thus, it could be that the smaller the global electrophilicity, the larger the efficiency of the solar cell, and this deserves experimental confirmation.
Indeed, it must be recognized that a series of another factor could also be important, and that in our analysis we are not considering the variation of Isc with the calculated values. This suggests that a good theoretical relationship between the Isc and the electronic descriptors must be found. Moreover, the results of our work suggest that a Quantitative Structure-Property Relationship (QSPR) equation could be constructed in order to estimate the efficiency of the DSSC in terms of the HOMO, LUMO, and the conceptual DFT reactivity parameters.



 is the electronegativity. The global hardness η can be seen as the resistance to charge transfer:
is the electronegativity. The global hardness η can be seen as the resistance to charge transfer:



 and
and  represent the energies of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO), respectively.
represent the energies of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO), respectively.
 (for nucleophilic attack),
(for nucleophilic attack),  (for electrophilic attack), and
(for electrophilic attack), and  (for radical attack), where
(for radical attack), where  is the gross charge of atom
is the gross charge of atom  in the molecule.
in the molecule. 

 =1 and
=1 and  =[
=[  +
+  ]/2.
]/2.
 , then the site is favored for a nucleophilic attack, whereas if
, then the site is favored for a nucleophilic attack, whereas if  then the site may be favored for an electrophilic attack. Through a similar procedure, one finds that for the condensed dual descriptor [36]:
then the site may be favored for an electrophilic attack. Through a similar procedure, one finds that for the condensed dual descriptor [36]:

 ) and bond angles (deg) for the crocetin molecule.
) and bond angles (deg) for the crocetin molecule.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











