Recent Advances in the Synthesis of Ammonium-Based Rotaxanes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
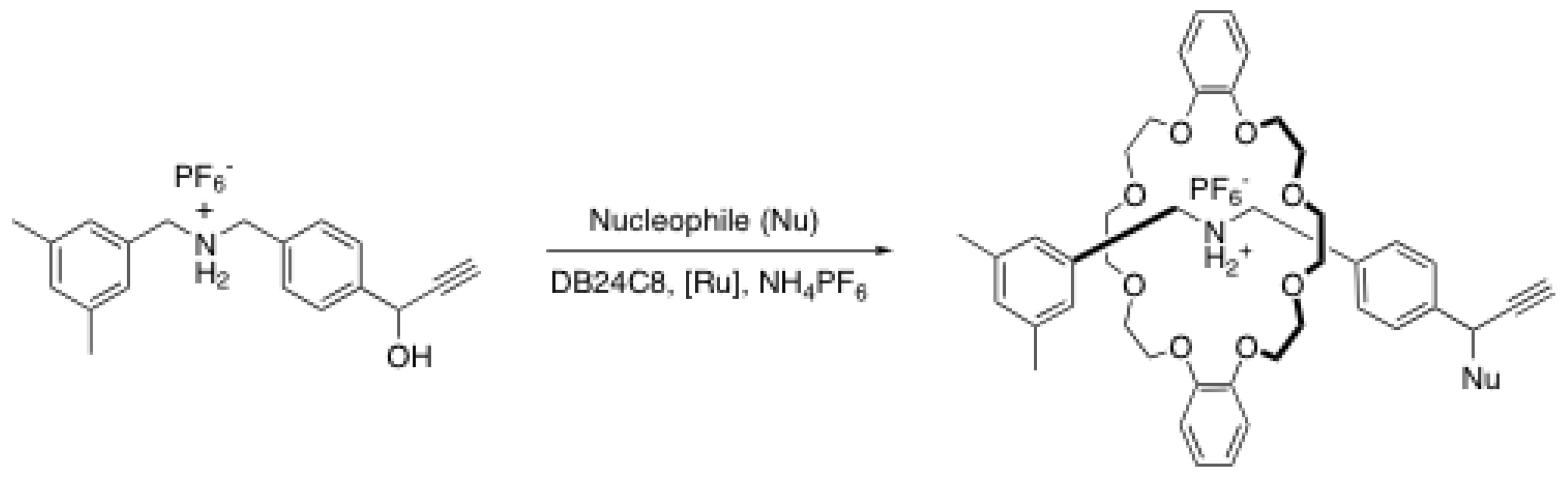{kind=link}
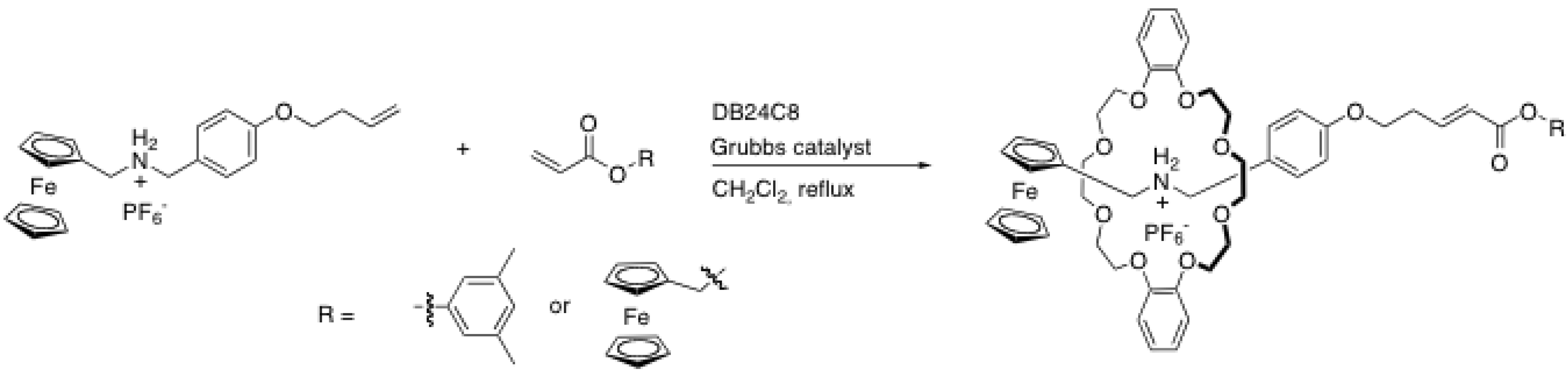{kind=link}
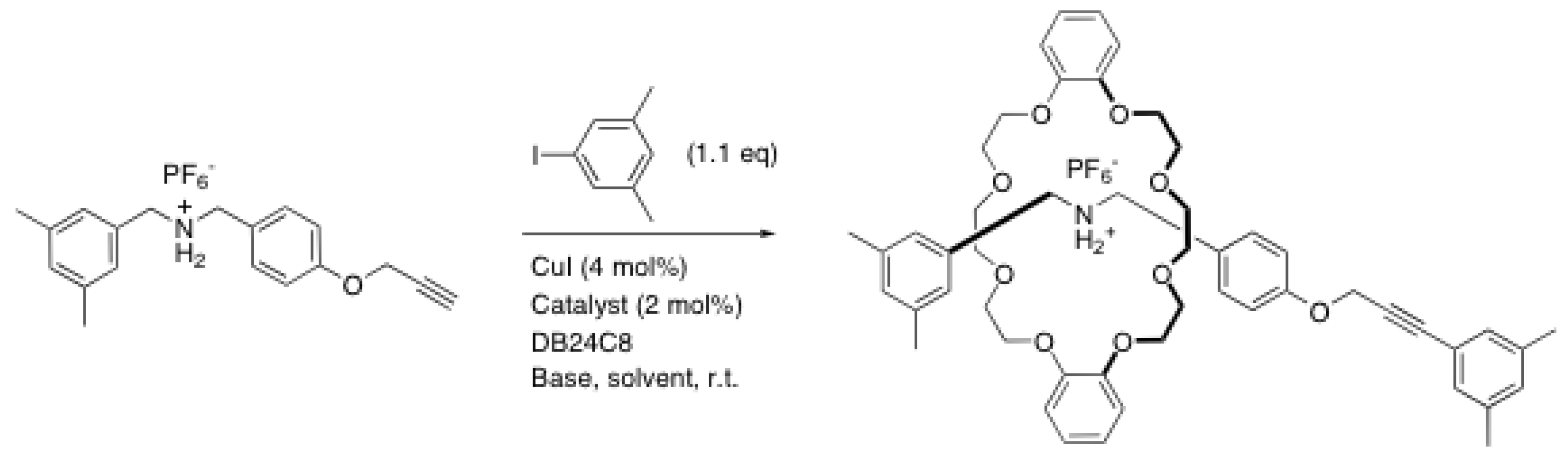{kind=link}
{kind=link}
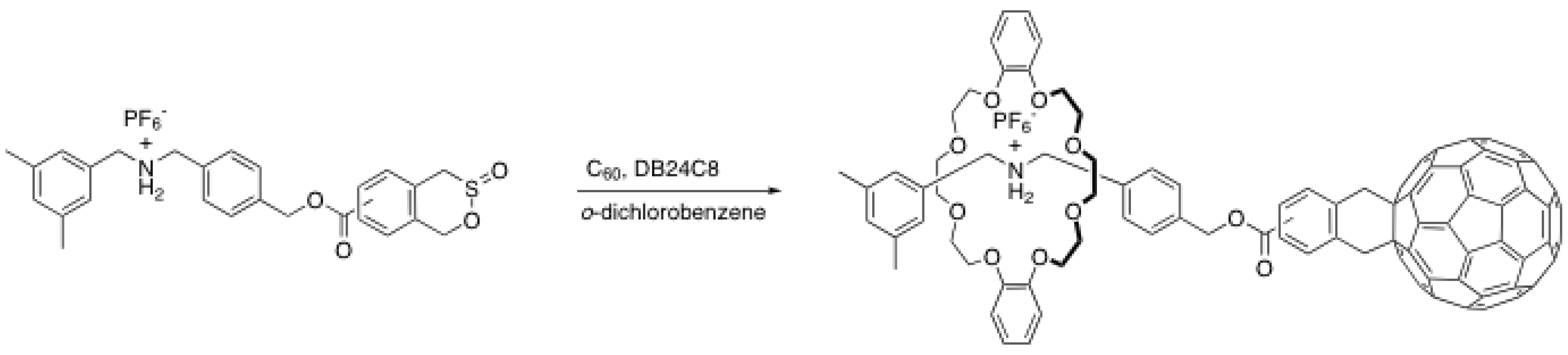{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

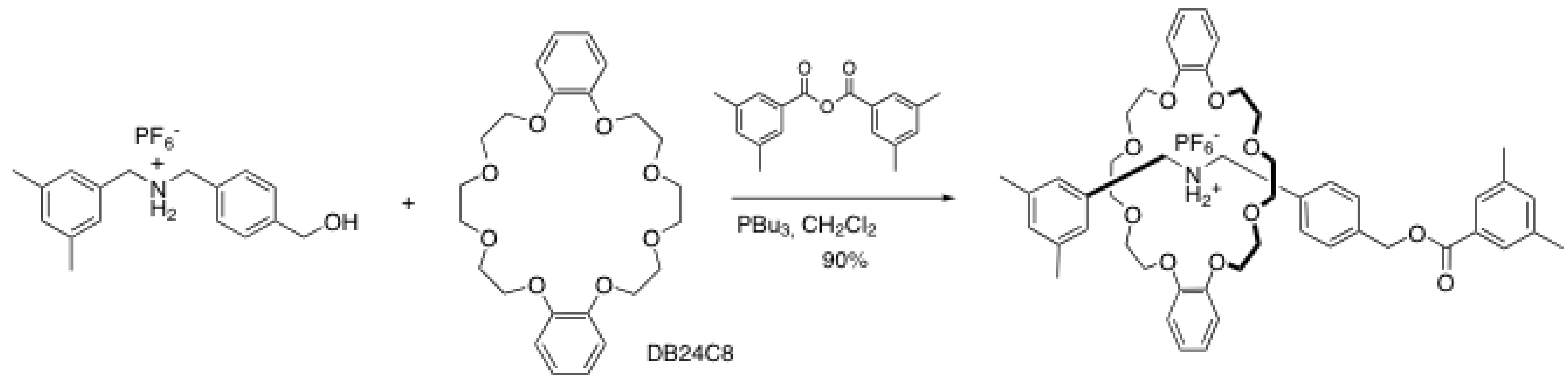
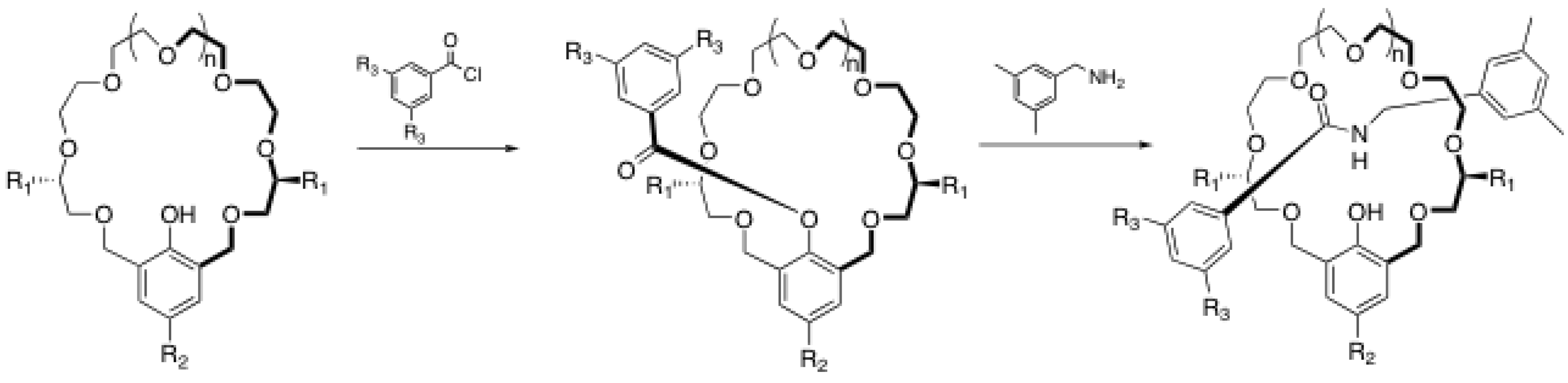
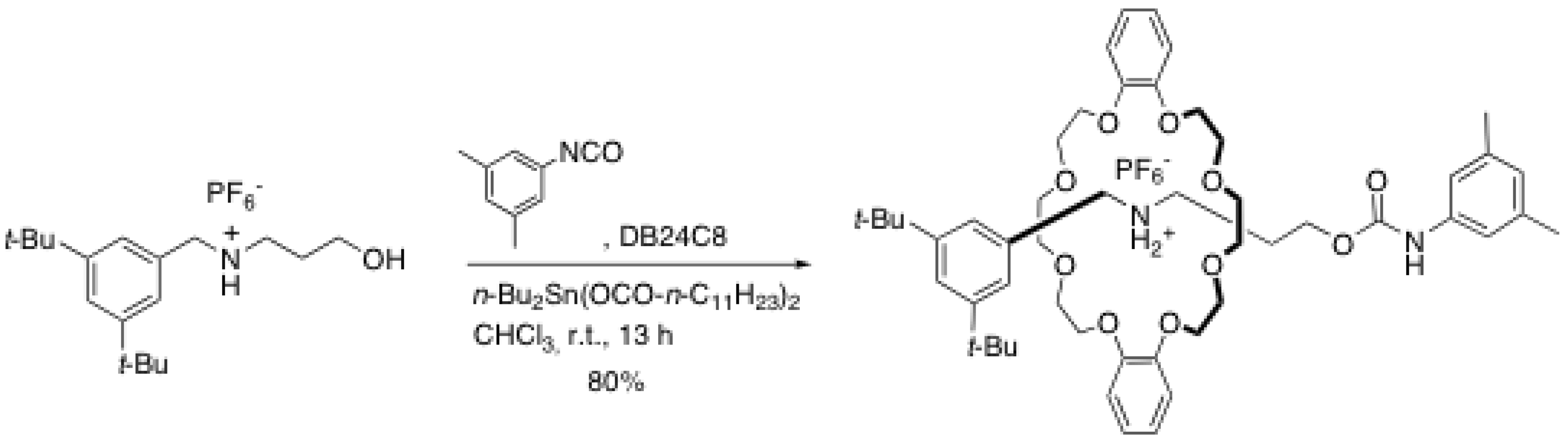
2. Rotaxane Formation through End-Capping Reactions
2.1. Esterification reactions




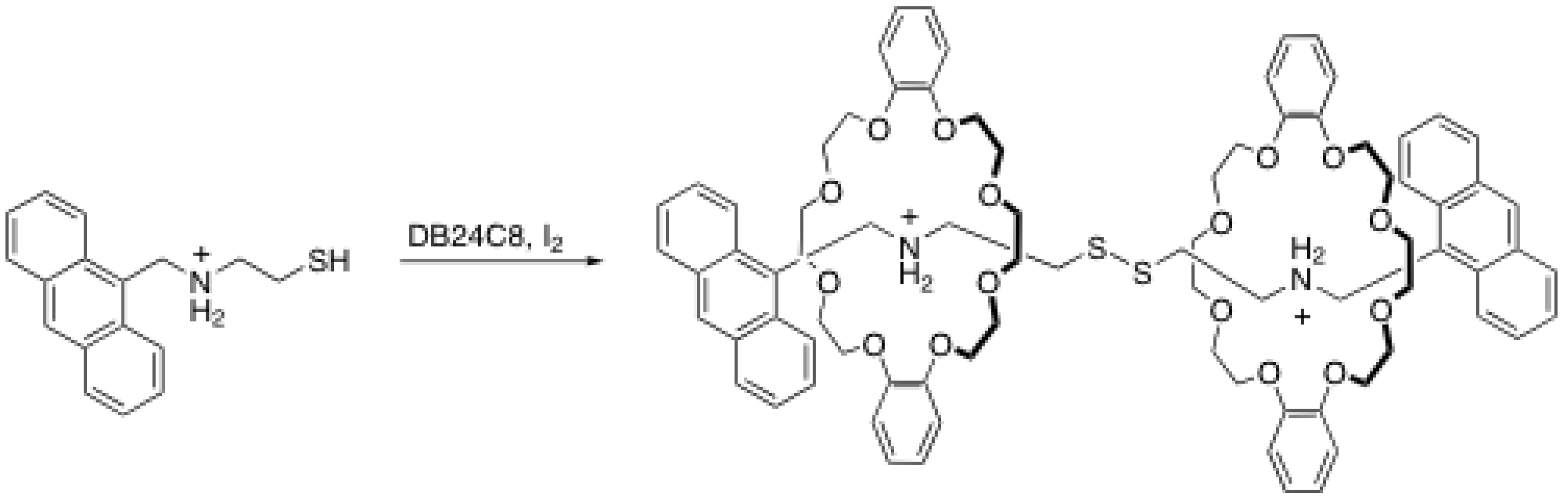
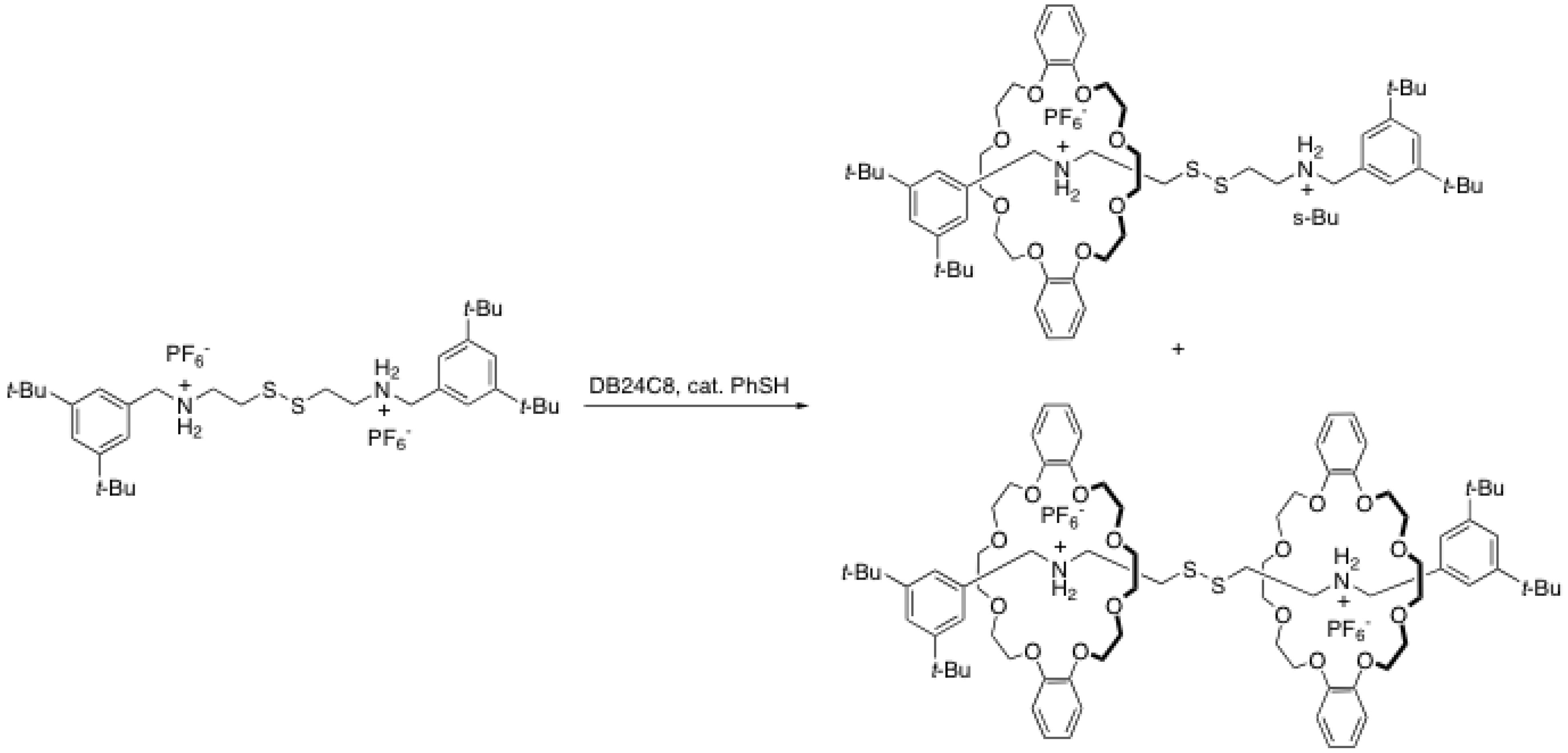
2.2. Reversible disulfide and imine formation


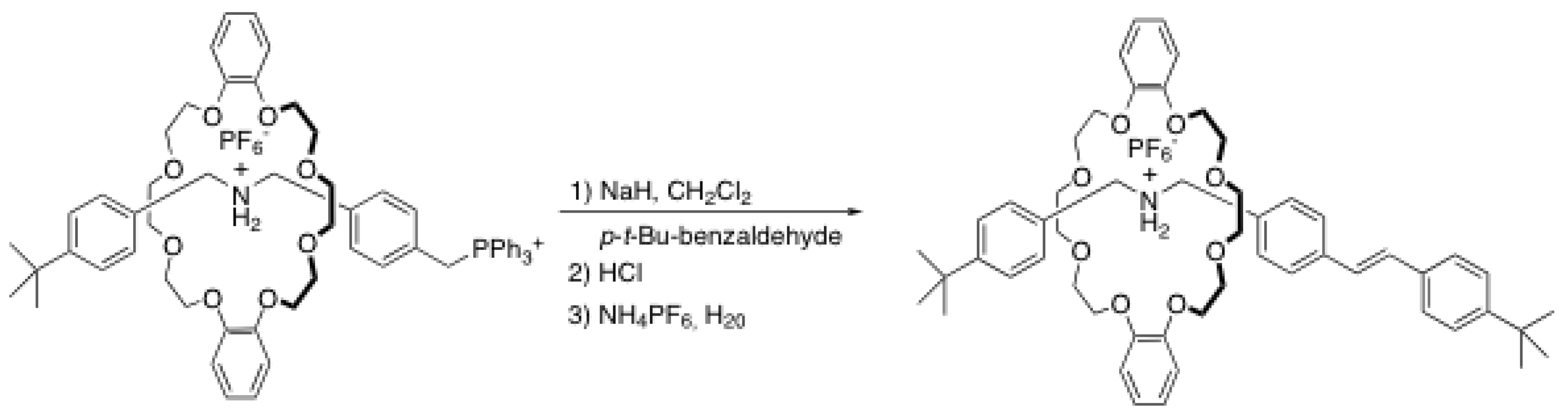
2.3. Wittig reaction

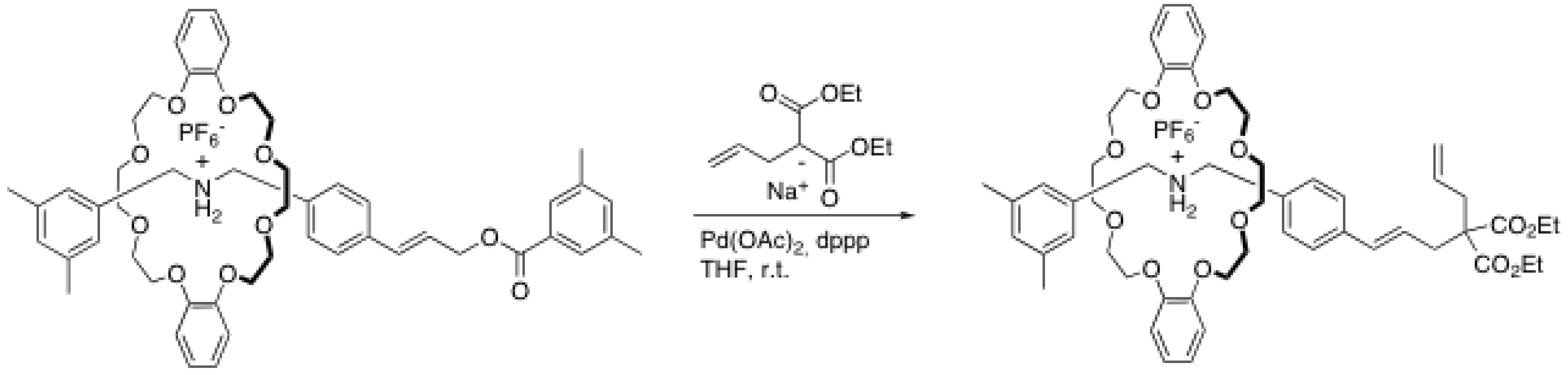
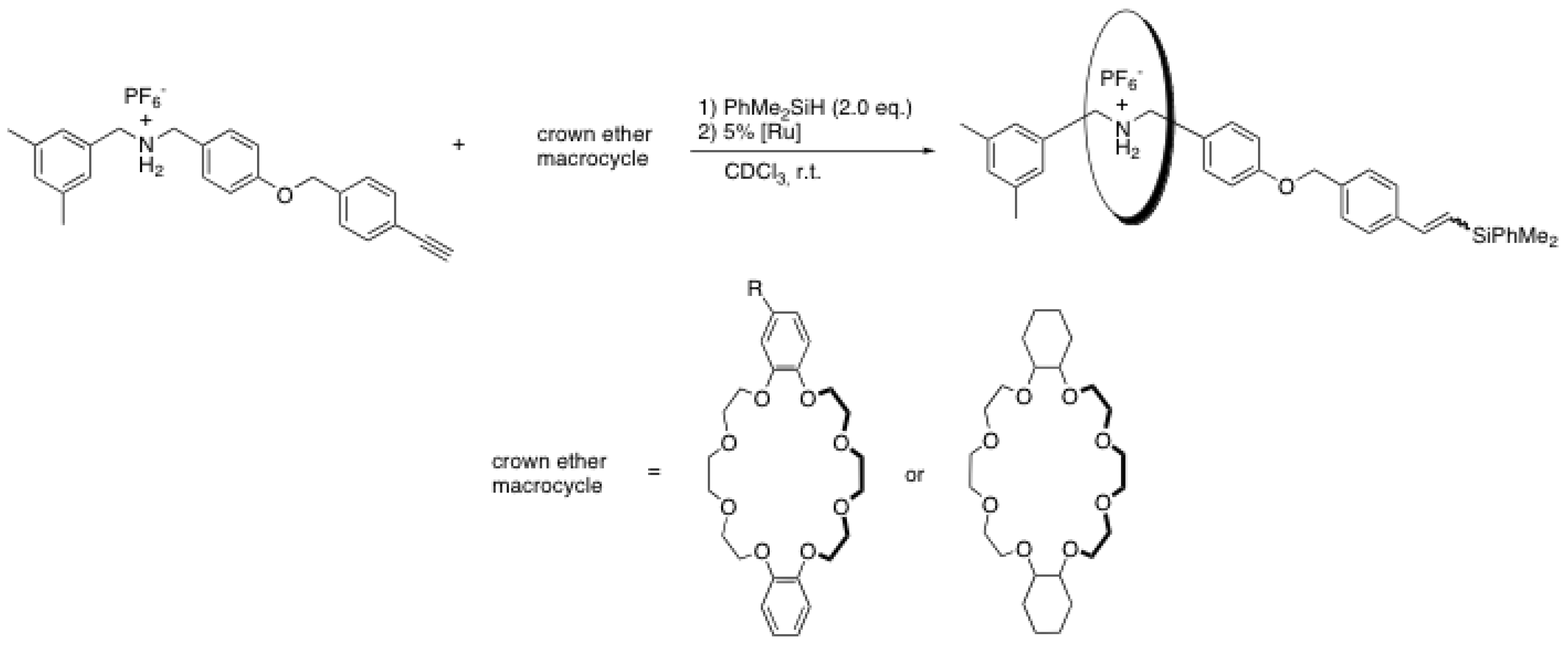
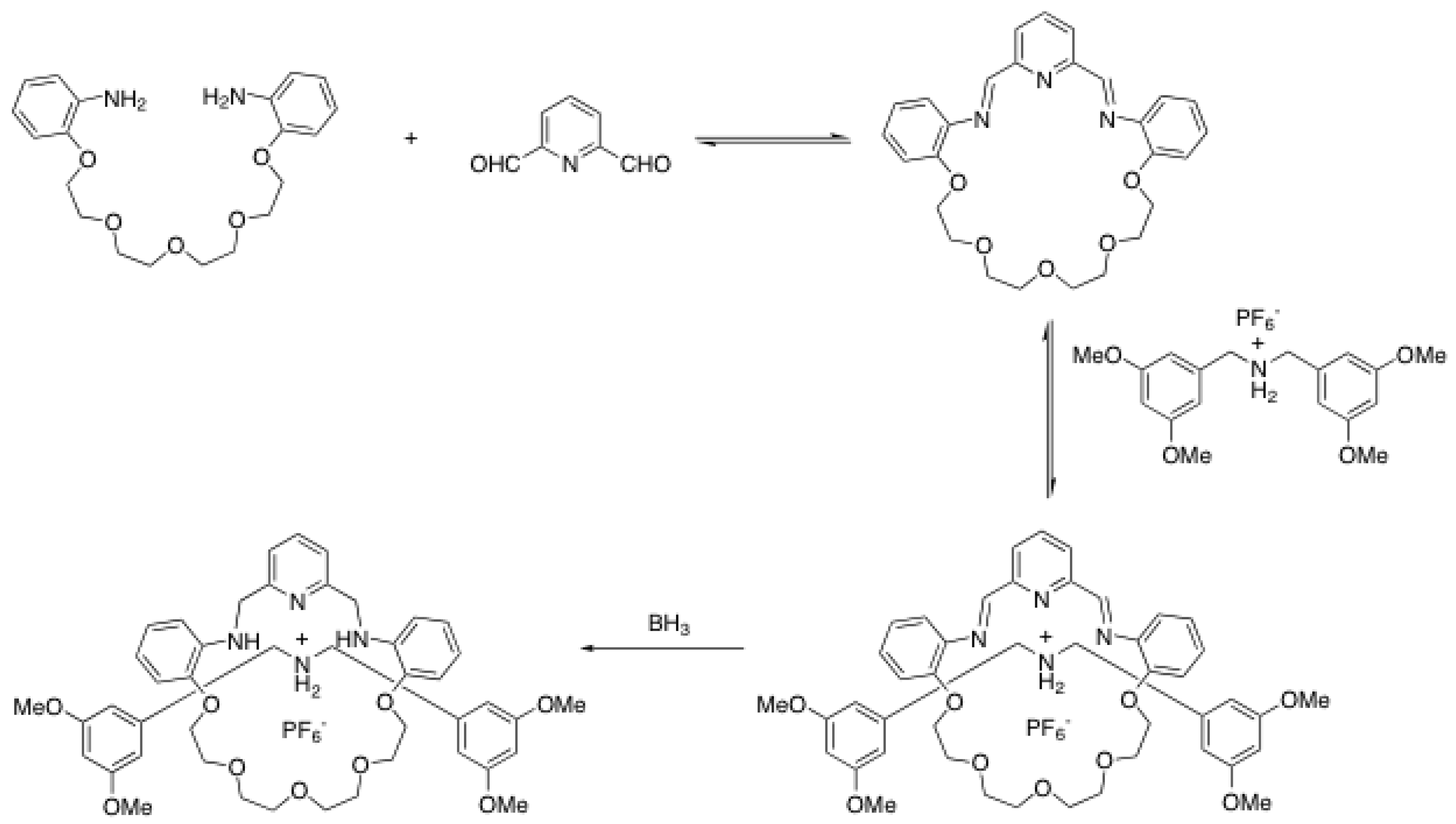
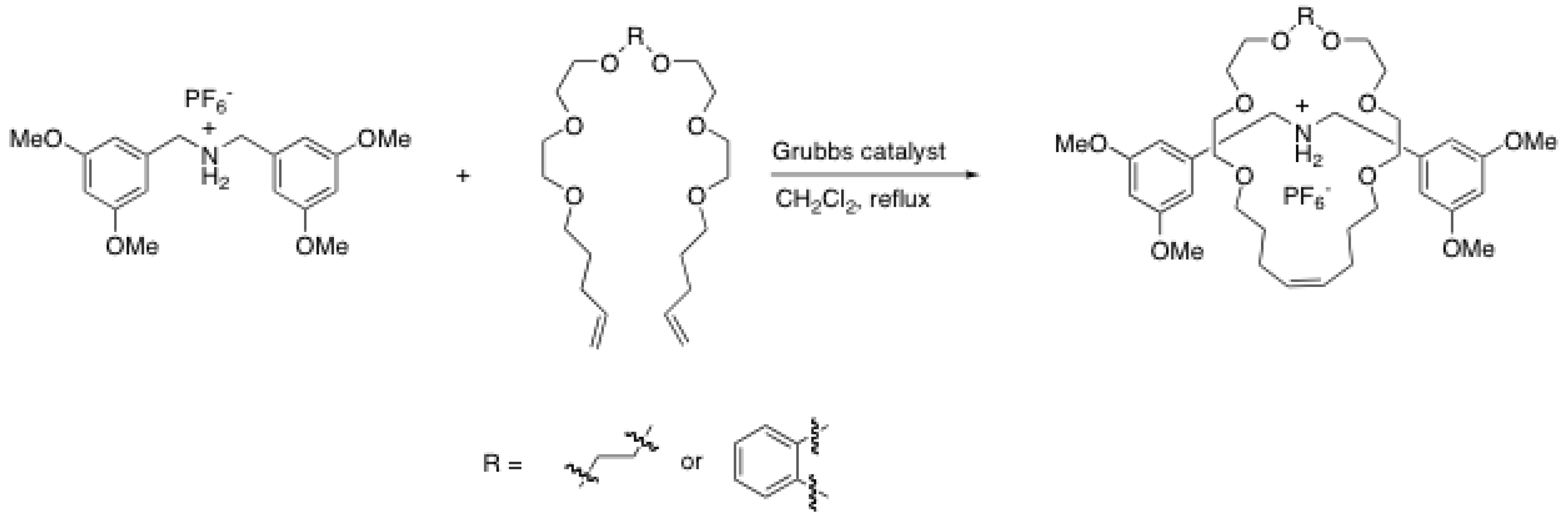
2.4. Metal-catalyzed reactions





2.5. Cycloaddition reactions


2.6. Solvent-free reactions
2.7. Miscellaneous reactions
3. Rotaxane Formation through Macrocycle Ring-Closing Reactions


4. Functionalization of Ammonium Ion in Rotaxanes
5. Synthesis of Functional Rotaxanes
5.1. Bioactive rotaxanes
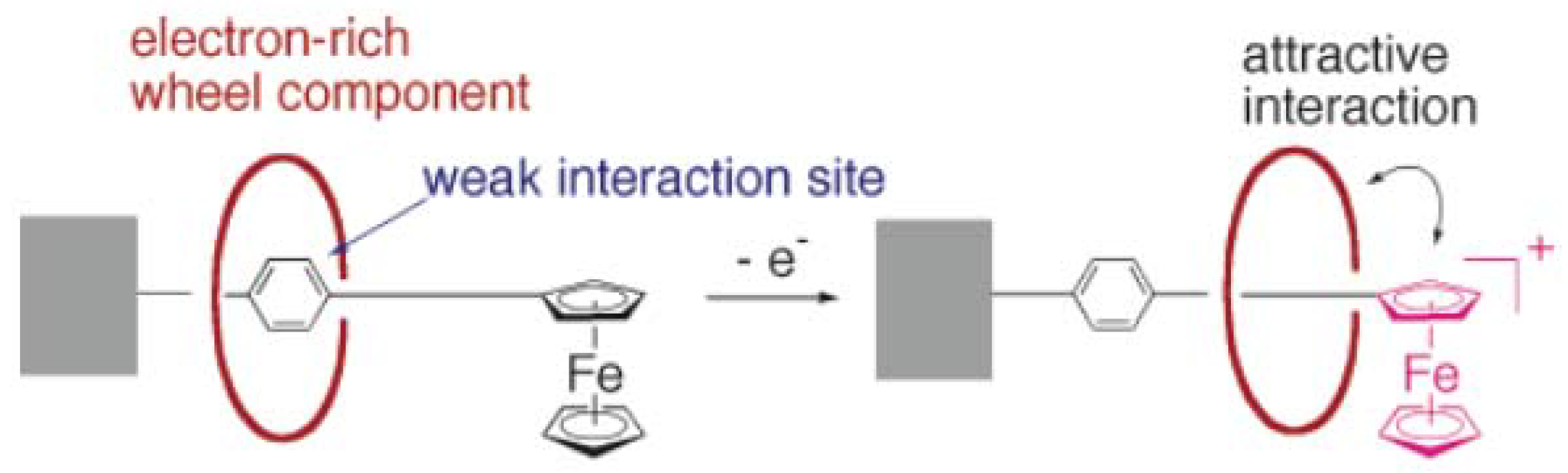
5.2. Photo- and electroactive rotaxanes



6. Conclusions
Acknowledgements
References and Notes
- Kawasaki, H.; Kihara, N.; Takata, T. High yielding and practical synthesis of rotaxanes by acetylative end-capping catalyzed by tributylphosphine. Chem. Lett. 1999, 1015–1016. [Google Scholar]
- Tachibana, Y.; Kawasaki, H.; Kihara, N.; Takata, T. Sequential O- and N-acylation protocol for high-yield preparation and modification of rotaxanes: synthesis, functionalization, structure, and intercomponent interaction of rotaxanes. J. Org. Chem. 2006, 71, 5093–5104. [Google Scholar] [CrossRef]
- Nakazono, K.; Kuwata, S.; Takata, T. Crown ether-tert-ammonium salt complex fixed as rotaxane and its derivation to nonionic rotaxane. Tetrahedron Lett. 2008, 49, 2397–2401. [Google Scholar] [CrossRef]
- Tachibana, Y.; Kihara, N.; Furusho, Y.; Takata, T. Is the tert-butyl group bulky enough to end-cap a pseudorotaxane with a 24-crown-8-ether wheel? Org. Lett. 2004, 6, 4507–4509. [Google Scholar]
- Watanabe, N.; Yagi, T.; Kihara, N.; Takata, T. Highly efficient synthesis of [3] and [5]-rotaxanes consisting of crown ether and a sec-ammonium salt. Chem. Comm. 2002, 2720–2721. [Google Scholar]
- Sato, T.; Takata, T. Rotaxane as an effective scaffold: synthesis of functionalized [3]rotaxane and connection of the wheel components arranged on the axle. TetrahedronLett. 2007, 48, 2797–2801. [Google Scholar] [CrossRef]
- Zhang, C.; Li, S.; Zhang, J.; Zhu, K.; Li, N.; Huang, F. Benzo-21-crown-7/secondary dialkylammonium salt [2]pseudorotaxane- and [2]rotaxane-type threaded structures. Org. Lett. 2007, 9, 5553–5556. [Google Scholar] [CrossRef]
- Jiang, W.; Winkler, H.D.F.; Schalley, C.A. Integrative self-sorting construction of a cascade-stoppered hetero[3]rotaxane. J. Am. Chem. Soc. 2008, 130, 13852–13853. [Google Scholar]
- Zhang, C.; Zhu, K.; Li, S.; Zhang, J.; Wang, F.; Liu, M.; Li, N.; Huang, F. Binding of secondary dialkylammonium salts by pyrido-21-crown-7. Tetrahedron Lett. 2008, 49, 6917–6920. [Google Scholar]
- Kihara, N.; Nakakoji, N.; Takata, T. Tributylphosphine-catalyzed acylation of alcohol by active ester directed toward effective end-capping of pseudorotaxane consisting of ammonium group and crown ether. Chem. Lett. 2002, 924–925. [Google Scholar]
- Tokunaga, Y.; Kakuchi, S.; Akasaka, K.; Nishikawa, N.; Shimomura, Y.; Isa, K.; Seo, T. A high-yielding and convenient synthesis of rotaxane based on an ester forming capping methodology. Chem. Lett. 2002, 810–811. [Google Scholar]
- Hirose, K.; Nishihara, K.; Harada, N.; Nakamura, Y.; Masuda, D.; Araki, M.; Tobe, Y. Highly selective and high-yielding rotaxane synthesis via aminolysis of prerotaxanes consisting of a ring component and a stopper unit. Org. Lett. 2007, 9, 2969–2972. [Google Scholar]
- Furusho, Y.; Sasabe, H.; Narsui, D.; Murakawa, K.; Takata, T.; Harada, T. Synthesis and [2]- and [3]rotaxanes by an end-capping approach utilizing urethane formation. Bull. Chem. Soc. Jpn. 2004, 77, 179–185. [Google Scholar] [CrossRef]
- Sandanayaka, A.S.D.; Sasabe, H.; Araki, Y.; Furusho, Y.; Ito, O.; Takata, T. Photoinduced electron-transfer processes between [C60]fullerene and triphenylamine moieties tethered by rotaxane structures. Through-space electron tranfer via excited triplet states of [60]fullerene. J. Phys. Chem. A 2004, 108, 5145–5155. [Google Scholar]
- Sasabe, H.; Ikeshita, K.; Rajkumar, G.A.; Watanabe, N.; Kihara, N.; Furusho, Y.; Mizuno, K.; Ogawa, A.; Takata, T. Synthesis of [60]fullerene-functionalized rotaxanes. Tetrahedron 2006, 62, 1988–1997. [Google Scholar]
- Rajkumar, G.A.; Sandanayaka, A.S.D.; Ikeshita, K.; Araki, Y.; Furusho, Y.; Takata, T.; Ito, O. Prolongation of the lifetime of the charge-separated state at low temperature in a photoinduced electron-transfer system of [60]fullerene and ferrocene moieties tethered by rotaxane structures. J. Phys. Chem. B 2006, 110, 6516–6525. [Google Scholar]
- Sasabe, H.; Inomoto, N.; Kihara, N.; Suzuki, Y.; Ogawa, A.; Takata, T. Synthesis of poly[2]rotaxane by Sonogashira polycondensation. J. Polym. Sci. Part A 2007, 45, 4154–4160. [Google Scholar]
- Marois, J.-S.; Cantin, K.; Desmarais, A.; Morin, J.-F. [3]Rotaxane-porphyrin conjugate as a novel supramolecular host for fullerenes. Org. Lett. 2008, 10, 33–36. [Google Scholar]
- Marois, J.-S.; Morin, J.-F. Synthesis and surface self-assembly of [3]rotaxane-porphyrin conjugates: toward the development of a supramolecular surface tweezer for C60. Langmuir 2008, 24, 10865–10873. [Google Scholar] [CrossRef]
- Kolchinski, A.G.; Alcock, N.W.; Roesner, R.A.; Busch, D.H. Molecular riveting: high yield preparation of a [3]rotaxane. Chem. Comm. 1998, 1437–1438. [Google Scholar]
- Furusho, Y.; Hasegawa, T.; Tsuboi, A.; Kihara, N.; Takata, T. “Unlock-lock” approach to[2] and [3]rotaxanes: entering of a ring through disulfide linkage that is unlocked by thiol “key”. Chem. Lett. 2000, 18–19. [Google Scholar]
- Furusho, Y.; Oku, T.; Hasegawa, T.; Tsuboi, A.; Kihara, N.; Takata, T. Dynamic covalent approach to [2]- and [3]rotaxane by utilizing reversible thiol-disulfide interchange reaction. Chem. Eur. J. 2003, 8, 2895–2903. [Google Scholar]
- Oku, T.; Furusho, Y.; Takata, T. Rotaxane-stabilized thiophosphonium salt from disulfide and phosphine. Org. Lett. 2003, 5, 4923–4925. [Google Scholar] [CrossRef]
- Oku, T.; Furusho, Y.; Takata, T. First poly[3]rotaxane synthesized through the noncovalent step-groeth polymerization of homoditopic dumbbell compound and a macrocycle with a reversible thiol-disulfide interchange reaction. J. Polym. Sci. Part A 2003, 41, 119–123. [Google Scholar] [CrossRef]
- Cantrill, S.J.; Rowan, S.J.; Stoddart, J.F. Rotaxane formation under thermodynamic control. Org. Lett. 1999, 1, 1363–1366. [Google Scholar] [CrossRef]
- Leigh, D.A.; Thomson, A.R. An ammonium/bis-ammonium switchable shuttle. Tetrahedron 2008, 64, 8411–8416. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Stoddart, J.F. Triphenylphosphoniumstoppered [2]rotaxanes. Org. Lett. 1999, 1, 129–132. [Google Scholar]
- Rowan, S.J.; Stoddart, J.F. Precision molecular grafting: exchanging surrogate stoppers in [2]rotaxanes. J. Am. Chem. Soc. 2000, 122, 164–165. [Google Scholar]
- Chang, T.; Heiss, A.M.; Cantrill, S.J.; Fyfe, M.C.T.; Pease, A.R.; Rowan, S.J.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Ammonium ion binding with pyridine-containing crown ethers. Org. Lett. 2000, 2, 2947–2950. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Toward daisy chain polymers: “Wittig exchange” of stoppers in [2]rotaxane monomers. Org. Lett. 2000, 2, 759–762. [Google Scholar]
- Chiu, S.-H.; Stoddart, J.F. Reversing a rotaxane recognition motif: threading oligoethylene glycol derivatives through a dicationiccyclophane. J. Am. Chem. Soc. 2002, 124, 4174–4175. [Google Scholar] [CrossRef]
- Chiu, S.-H.; Elizarov, A.M.; Glink, P.T.; Stoddart, J.F. Translational Isomerism in a [3] Catenane and a [3] Rotaxane. Org. Lett. 2002, 4, 3561–3564. [Google Scholar] [CrossRef]
- Elizarov, A.M.; Chiu, S.-H.; Glink, P.T.; Stoddart, J.F. Dendrimer with rotaxane-like mechanical branching. Org. Lett. 2002, 4, 679–682. [Google Scholar]
- Kihara, N.; Motoda, S.; Yokozawa, T.; Takata, T. End-cap exchange of rotaxane by the Tsuji-Trost allylation reaction. Org. Lett. 2005, 7, 1199–1202. [Google Scholar] [CrossRef]
- Sasabe, H.; Kihara, N.; Mizuno, K.; Ogawa, K.; Takata, T. Efficient synthesis of [2]- and higher order rotaxanes via the transition metal-catalyzed hydrosilylation of alkyne. Tetrahedron Lett. 2005, 46, 3851–3853. [Google Scholar]
- Tokunaga, Y.; Kawai, N.; Shimomura, Y. Using ruthenium-catalysedpropargylic substitutions for the efficient syntheses of rotaxanes. Tetrahedron Lett. 2007, 48, 4995–4998. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The development of L2X2Ru=CHR olefin metathesis catalysts: an organometallic success story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef]
- Suzaki, Y.; Osakada, K. End-cappind of pseudo[2]rotaxane composed of alkyl(ferrocenylmethyl)ammonium and dibenzo[24]crown-8 via cross metathesis reactions. Chem. Lett. 2006, 35, 374–375. [Google Scholar] [CrossRef]
- Suzaki, Y.; Osakada, K. Ferrocene-containing |2]- and [3]rotaxanes. Preparation via an end-capping cross-metathesis reaction and electrochemical properties. Dalton Trans. 2007, 2376–2383. [Google Scholar] [CrossRef]
- Giguère, J.-B.; Thibeault, D.; Cronier, F.; Marois, J.-S.; Auger, M.; Morin, J.-F. Synthesis of [2]- and [3]rotaxanes through Sonogashira Coupling. Tetrahedron Lett. 2009, 50, 5497–5500. [Google Scholar]
- Cao, J.; Fyfe, M.C.T.; Stoddart, J.F.; Cousins, G.R.L.; Glink, P.T. Molecular shuttles by the protecting group approach. J. Org. Chem. 2000, 65, 1937–1946. [Google Scholar]
- Coutrot, F.; Busseron, E. Controlling the chair conformation of a mannopyranose in a large-amplitude [2]rotaxane molecular machine. Chem. Eur. J. 2009, 15, 5186–5190. [Google Scholar] [CrossRef]
- Coutrot, F.; Busseron, E. A New Glycorotaxane Machine Based on an Anilinium and a Triazolium Station. Chem. Eur. J. 2008, 14, 4784–4787. [Google Scholar] [CrossRef]
- Coutrot, F.; Romuald, C.; Busseron, E. A new pH-switchable dimannosyl[c2]daisy chain molecular machine. Org. Lett. 2008, 10, 3741–3744. [Google Scholar]
- Sasabe, H.; Kihara, N.; Furusho, Y.; Mizuno, K.; Ogawa, A.; Takata, T. End-capping of a pseudorotaxane via Diels-Alder reaction for the construction of C60-terminated [2]rotaxane. Org. Lett. 2004, 6, 3957–3960. [Google Scholar] [CrossRef]
- Hsueh, S.-Y.; Cheng, K.-W.; Lai, C.-C.; Chiu, S.-H. Efficient solvent-free syntheses of [2]- and [4]rotaxanes. Angew. Chem. Int. Ed. 2008, 47, 4436–4439. [Google Scholar]
- Hsu, C.-C.; Chen, N.-C.; Lai, C.-C.; Liu, Y.-H.; Peng, S.-M.; Chiu, S.-H. Solvent-free synthesis of the smallest rotaxane prepared to date. Angew. Chem. Int. Ed. 2008, 47, 7475–7478. [Google Scholar]
- Hsu, C.-C.; Lai, C.-C.; Chiu, S.-H. Using Diels-Alder reactions to synthesize [2]rotaxanes under solvent-free conditions. Tetrahedron 2009, 65, 2824–2829. [Google Scholar] [CrossRef]
- Asakawa, M.; Ikeda, T.; Yui, N.; Shimizu, T. Preparation of porphyrin-stopperedrotaxane aiming at immobilization on substrate. Chem. Lett. 2002, 174–175. [Google Scholar]
- Coutrot, F.; Busseron, E.; Montero, J.-L. A very efficient synthesis of a mannosyl orthoester [2]rotaxane and mannosidic [2]rotaxanes. Org. Lett. 2008, 10, 753–756. [Google Scholar] [CrossRef]
- Zehnder II, D.W.; Smithrud, D.B. Facile synthesis of rotaxanes through condensation reactions of DCC-[2]rotaxanes. Org. Lett. 2001, 3, 2485–2487. [Google Scholar] [CrossRef]
- Furusho, Y.; Rajkumar, G.A.; Oku, T.; Takata, T. Synthesis of [2]rotaxanes by tritylativeendcapping of in situ formed pseudorotaxanes having thiol or hydroxyl functionality on the axle termini. Tetrahedron 2002, 58, 6609–6613. [Google Scholar]
- Hung, W.-C.; Liao, K.-S.; Liu, Y.-H.; Peng, S.-M.; Chiu, S.-H. Mild and high yielding syntheses of diethyl phosphoramidate-stoppered [2]rotaxanes. Org. Lett. 2004, 6, 4183–4186. [Google Scholar]
- Tokunaga, Y.; Nakamura, T.; Yoshioka, M.; Shimomura, Y. A molecular switch based on acid and base promoted, cation governed binding in a crown ether threaded rotaxane. Tetrahedron Lett. 2006, 47, 5901–5904. [Google Scholar] [CrossRef]
- Suzaki, Y.; Osakada, K. Formation, dynamic behavior, and chemical transformation of Pt complexes with a rotaxane-like structure. Chem. Asian J. 2006, 1, 331–343. [Google Scholar] [CrossRef]
- Chiu, C.-W.; lai, C.-C.; Chiu, S.-H. “Threading-followed-by-swelling”: a new protocol for rotaxane synthesis. J. Am. Chem. Soc. 2007, 129, 3500–3501. [Google Scholar] [CrossRef]
- Nakazono, K.; Oku, T.; Takata, T. Synthesis of rotaxanes consisting of crown ether wheel and sec-ammonium axle under basic condition. Tetrahedron Lett. 2007, 48, 3409–3411. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Ito, T.; Sugawara, H.; Nakata, R. Dynamic covalent chemistry of a boronylammonium ion and a crown ether: formation of a C3-symmetric [4]rotaxane. Tetrahedron Lett. 2008, 49, 3449–3452. [Google Scholar] [CrossRef]
- Chuang, C.-J.; Li, W.-S.; Lai, C.-C.; Liu, Y.-H.; Peng, S.-M.; Chao, I.; Chiu, S.-H. A molecular cage-based [2]rotaxane that behaves as a molecular muscle. Org. Lett. 2009, 11, 385–388. [Google Scholar]
- Glink, P.T.; Oliva, A.I.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Template-directed synthesis of a [2]rotaxane by the clipping under thermodynamic control of a crown ether like macrocycle around a dialkylammonium ion. Angew. Chem. Int. Ed. 2001, 40, 1870–1874. [Google Scholar]
- Leung, K.C.-F.; Arico, F.; Cantrill, S.J.; Stoddart, J.F. Template-directed dynamic synthesis of mechanically interlocked dendrimers. J. Am. Chem. Soc. 2005, 127, 5808–5810. [Google Scholar] [CrossRef]
- Wu, J.; Leung, K.C.-F.; Stoddart, J.F. Efficient production of [n]rotaxanes by using template-directed clipping reactions. Proc. Natl. Acad. Sci. USA 2007, 104, 17266–17271. [Google Scholar]
- Haussmann, P.C.; Khan, S.I.; Stoddart, J.F. Equilibrating dynamic [2]rotaxanes. J. Org. Chem. 2007, 72, 6708–6713. [Google Scholar] [CrossRef]
- Kilbinger, A.F.M.; Cantrill, S.J.; Waltman, A.W.; Day, M.W.; Grubbs, R.H. Magic ring rotaxanes by olefin metathesis. Angew. Chem. Int. Ed. 2003, 42, 3281–3285. [Google Scholar] [CrossRef]
- Yoon, I.; Narita, M.; Shimizu, T.; Asakawa, M. Threading-followed-by-shrinking protocol for the synthesis of a [2]rotaxane incorporating a Pd(II)-salophen moiety. J. Am. Chem. Soc. 2004, 126, 16740–16741. [Google Scholar] [CrossRef]
- Yoon, I.; Narita, M.; Goto, M.; Shimizu, T.; Asakawa, M. Synthesis of a [2]rotaxane incorporating a Ni(II)-salen moiety: evidence of ring-opening-and-closing protocol. Org. Lett. 2006, 8, 2341–2344. [Google Scholar] [CrossRef]
- Umemiya, T.; Takeuchi, D.; Osakada, K. Synthesis of macrocyclicpolyethers via Ru complex-catalyzed metathesis cyclization and their use as the ring component of rotaxanes. J. Organomet. Chem. 2006, 691, 5260–5266. [Google Scholar] [CrossRef]
- Kihara, N.; Tachibana, Y.; Kawasaki, H.; Takata, T. unusually lowered acidity of ammonium group surrounded by crown ether in a rotaxane system and its acylative neutralization. Chem. Lett. 2000, 506–507. [Google Scholar]
- Makita, Y.; Kihara, N.; Takata, T. Quantitative active transport in [2]rotaxane using a one-shot acylation reaction toward the linear molecular motor. J. Org. Chem. 2008, 73, 9245–9250. [Google Scholar] [CrossRef]
- Tachibana, Y.; Kihara, N.; Takata, T. Asymmetric benzoin condensation catalyzed by chiralrotaxanes tethering a thiazolium salt moiety via the cooperation of the component: can rotaxane be an effective reaction field? J. Am. Chem. Soc. 2004, 126, 3438–3439. [Google Scholar]
- Badjic, J.D.; Ronconi, C.M.; Stoddart, J.F.; Balzani, V.; Silvi, S.; Credi, A. Operating molecular elevators. J. Am. Chem. Soc. 2006, 128, 1489–1499. [Google Scholar]
- Balzani, V.; Credi, A.; Raymo, F.M.; Stoddart, J.F. Artificial Molecular Machines. Angew. Chem. Int. Ed. 2000, 39, 3348–3391. [Google Scholar] [CrossRef]
- Smukste, I.; Smithrud, D.B. Structure-function relationship of amino acid-[2]rotaxanes. J. Org. Chem. 2003, 68, 2547–2558. [Google Scholar] [CrossRef]
- Smukste, I.; House, B.E.; Smithrud, D.B. Host-[2]rotaxane: advantage of converging functional groups for guest recognition. J. Org. Chem. 2003, 68, 2559–2571. [Google Scholar] [CrossRef]
- Dvornikovs, V.; House, B.E.; Kaetzel, M.; Dedman, J.R.; Smithrud, D.B. Host-[2]rotaxanes as cellular transport agents. J. Am. Chem. Soc. 2003, 125, 8290–8301. [Google Scholar]
- Bao, X.; Isaacsohn, I.; Drew, A.F.; Smithrud, D.B. Determining the binding and intracellular transporting ability of a host-[3]rotaxane. J. Org. Chem. 2007, 72, 3988–4000. [Google Scholar] [CrossRef]
- Zhu, J.; House, B.E.; Fleck, E.; Isaacsohn, I.; Drew, A.F.; Smithrud, D.B. A host-rotaxane derivatized with carboxylic acids efficiently delivers a highly cationic fluoresceinated peptide. Bioorg. Med. Chem. Lett. 2007, 17, 5058–5062. [Google Scholar]
- Zhu, J.; McFarland-Mancini, M.; Drew, A.F.; Smithrud, D.B. Host-rotaxanes with oligomeric axles are intracellular transport agents. Bioorg. Med. Chem. Lett. 2009, 19, 520–523. [Google Scholar] [CrossRef]
- Kihara, N.; Hashimoto, M.; Takata, T. Redox behavior of ferrocene-containing rotaxane: transport of the rotaxane wheel by redox reaction of a ferrocene moiety tethered at the end of the axle. Org. Lett. 2004, 6, 1693–1696. [Google Scholar] [CrossRef]
- Rajkumar, G.A.; Sandanayaka, A.S.D.; Ikeshita, K.; Araki, Y.; Furusho, Y.; Takata, T.; Ito, O. Prolongation of the lifetime of the charge-separated state at low temperatures in a photoinduced electron-transfer system of [60]fullerene and ferrocene moieties tethered by rotaxane structures. J. Phys. Chem. B 2006, 110, 6516–6525. [Google Scholar]
- Sasabe, H.; Ikeshita, K.; Rajkumar, G. A.; Watanabe, N.; Kihara, N.; Furusho, Y.; Mizuno, K.; Ogawa, A.; Takata, T. Synthesis of [60]fullerene-functionalized rotaxanes. Tetrahedron 2006, 62, 1988–1997. [Google Scholar] [CrossRef]
- Illescas, B.M.; Santos, J.; Diaz, M.C.; Martin, N.; Atienza, C.M.; Guldi, D.M. Supramolecular threaded complexes from fullerene-crown ether and π-extended TTF derivatives. Eur. J. Org. Chem. 2007, 5027–5037. [Google Scholar]
- Wang, J.-Y.; Han, J.-M.; Yan, J.; Ma, Y.; Pei, J. A mechanically interlocked [3]rotaxane as a light-harvesting antenna: synthesis, characterization, and intramolecular energy transfer. Chem. Eur. J. 2009, 15, 3585–3594. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Thibeault, D.; Morin, J.-F. Recent Advances in the Synthesis of Ammonium-Based Rotaxanes. Molecules 2010, 15, 3709-3730. https://doi.org/10.3390/molecules15053709
Thibeault D, Morin J-F. Recent Advances in the Synthesis of Ammonium-Based Rotaxanes. Molecules. 2010; 15(5):3709-3730. https://doi.org/10.3390/molecules15053709
Chicago/Turabian StyleThibeault, Dominic, and Jean-François Morin. 2010. "Recent Advances in the Synthesis of Ammonium-Based Rotaxanes" Molecules 15, no. 5: 3709-3730. https://doi.org/10.3390/molecules15053709
APA StyleThibeault, D., & Morin, J.-F. (2010). Recent Advances in the Synthesis of Ammonium-Based Rotaxanes. Molecules, 15(5), 3709-3730. https://doi.org/10.3390/molecules15053709