Coupling Reactions of α-Bromocarboxylate with Non-Aromatic N-Heterocycles

Abstract
:1. Introduction
2. Results and Discussion


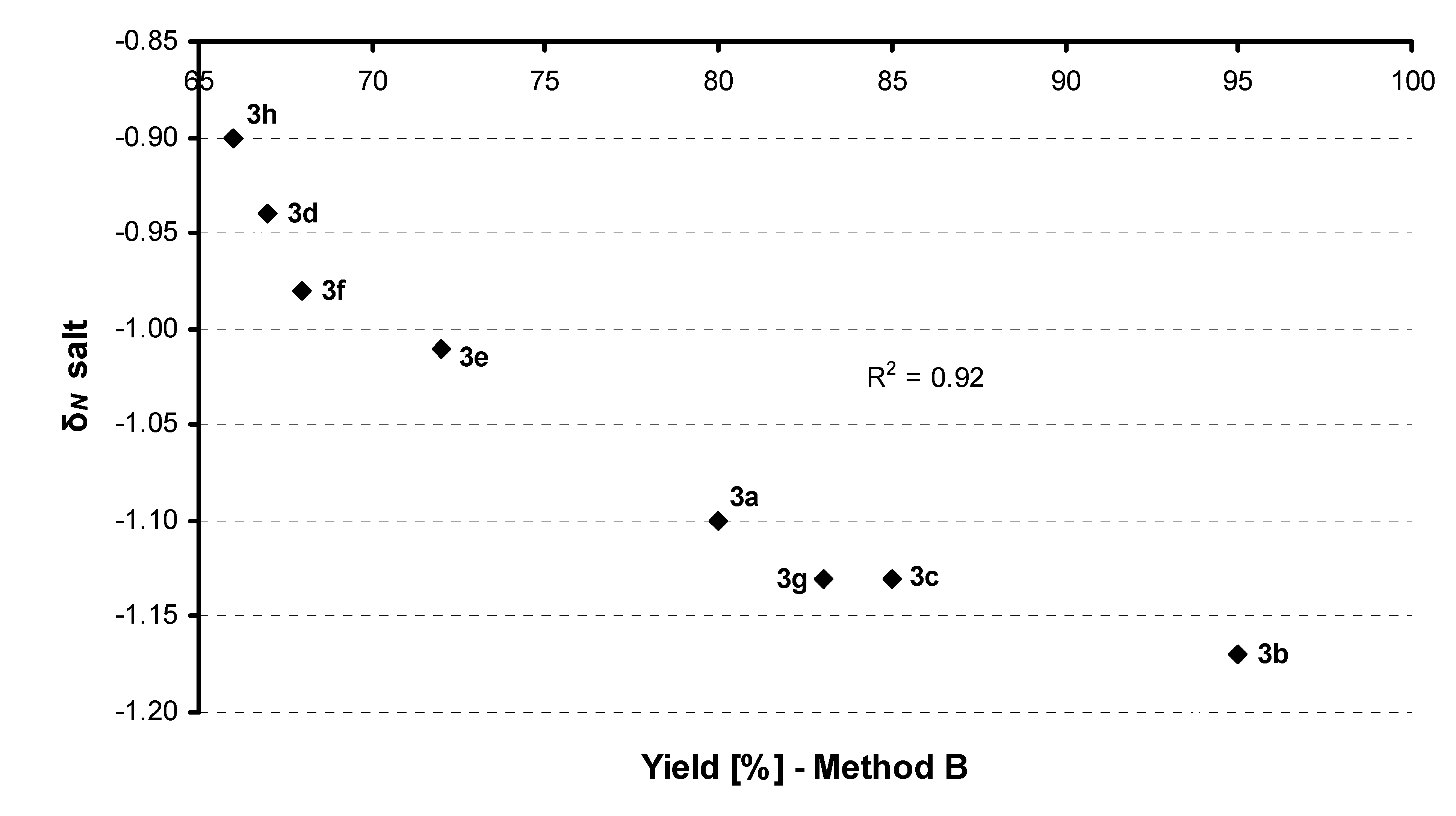
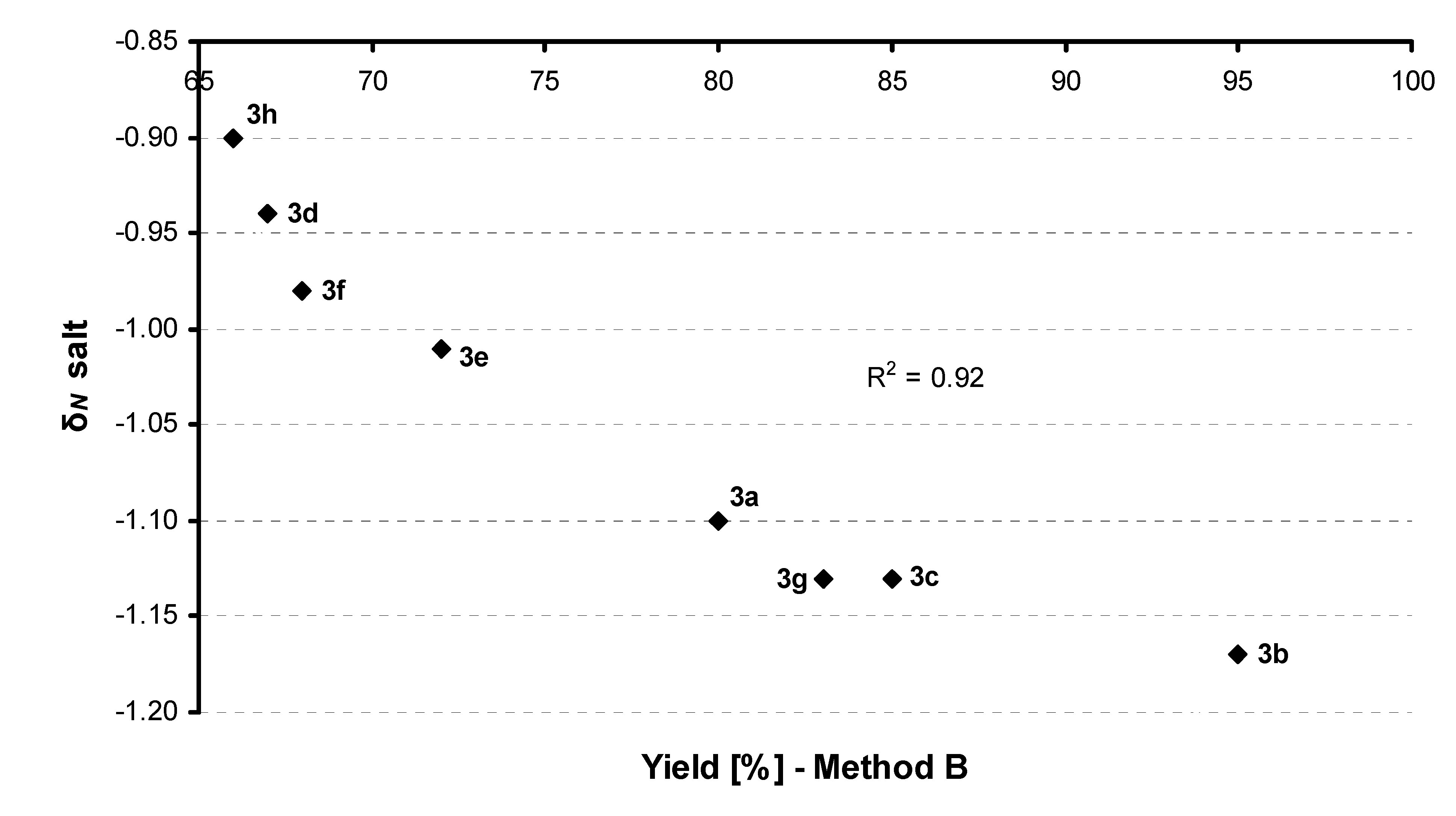{kind=link}
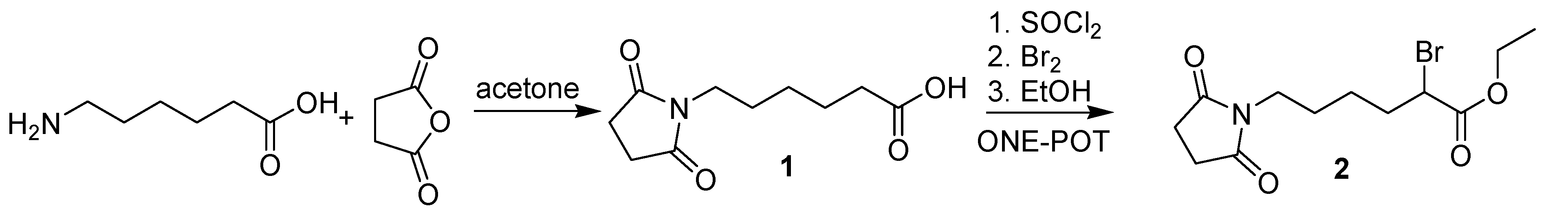{kind=link}
 | |||||
| Comp. | R | Conditionsa | Yield (%) | δN base | δN salt |
| 3a |  | Method A | 94 | –0.77 | –1.10 |
| Method B | 80 | ||||
| 3b |  | Method A | 77 | –0.73 | –1.17 |
| Method B | 95 | ||||
| 3c |  | Method A | 80 | –0.73 | –1.13 |
| Method B | 85 | ||||
| 3d |  | Method A | 0 | –0.50 | –0.94 |
| Method B | 67 | ||||
| 3e |  | Method A | 0 | –0.55 | –1.01 |
| Method B | 72 | ||||
| 3f |  | Method A | 0 | –0.53 | –0.98 |
| Method B | 68 | ||||
| 3g |  | Method A | 81 | –0.76 | –1.13 |
| Method B | 83 | ||||
| 3h |  | Method A | 0 | –0.48 | –0.90 |
| Method B | 66 | ||||
3. Conclusions
4. Experimental
4.1. General
4.2. Synthesis
4.3. Ab initio/DFT calculations
Acknowledgements
References
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv. Drug Deliv. Rev. 2004, 56, 603–618. [Google Scholar] [CrossRef]
- Hrabalek, A.; Dolezal, P.; Farsa, O.; Sklubalova, Z.; Kunes, J. Esters of 6-dimethylaminohexanoic acid as skin penetration enhancers. Pharmazie 2000, 55, 759–761. [Google Scholar]
- Pugh, W.J.; Wong, R.; Falson, F.; Michniak, B.B.; Moss, G.P. Discriminant analysis as a tool to identify compounds with potential as transdermal enhancers. J. Pharm. Pharmacol. 2005, 57, 1389–1396. [Google Scholar] [CrossRef]
- Sinha, V.R.; Kaur, M.P. Permeation enhancers for transdermal drug delivery. Drug. Dev. Ind. Pharm. 2000, 26, 1131–1140. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rees, C.W.; Scriven, E.F.V. (Eds.) Comprehensive Heterocyclic Chemistry II; Pergamon Press: Oxford, UK, 1996.
- Taylor, J.B.; Triggle, D.J. (Eds.) Comprehensive Medicinal Chemistry II; Elsevier: Oxford, UK, 2007.
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef]
- Xi, Z.; Liu, F.; Zhou, Y.; Chen, W. CuI/L(L=pyridine-functionalized 1,3-diketones) catalyzed C–N coupling reactions of aryl halides with NH-containing heterocycles. Tetrahedron 2008, 64, 4254–4259. [Google Scholar] [CrossRef]
- Halfen, J.A. Recent advances in metal-mediated carbon-nitrogen bond formation reactions. Aziridination and amidation. Curr. Org. Chem. 2005, 9, 657–669. [Google Scholar] [CrossRef]
- Shen, G.; Lv, X.; Qian, W.; Bao, W. Cu2O-catalyzed Ullmann-type reaction of vinyl bromides with imidazole and benzimidazole. Tetrahedron Lett. 2008, 49, 4556–4559. [Google Scholar] [CrossRef]
- Huang, Y.-Z.; Miao, H.; Zhang, Q.-H.; Chen, C.; Xu, J. Cu2O: a simple and efficient reusable catalyst for N-arylation of nitrogen-containing heterocycles with aryl halides. J. Catal. Lett. 2008, 122, 344–348. [Google Scholar] [CrossRef]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Raich, I.; Liska, F. 4-Substituted aryl bromides coupling with 4-methoxybenzene-1-thiol by means of copper catalysts. Chem. Pap. 2005, 59, 182–186. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Satinsky, D.; Raich, I. Novel regioselective preparation of 5-chloropyrazine-2-carbonitrile from pyrazine-2-carboxamide and coupling study of substituted phenylsulfanylpyrazine-2-carboxylic acid derivatives. Curr. Org. Chem. 2005, 9, 49–60. [Google Scholar] [CrossRef]
- Phillips, D.P.; Hudson, A.R.; Nguyen, B.; Lau, T.L.; McNeill, M.H.; Dalgard, J.E.; Chen, J.-H.; Penuliar, R.J.; Miller, T.A.; Zhi, L. Copper-catalyzed N-arylation of oxindoles. Tetrahedron Lett. 2006, 47, 7137–7138. [Google Scholar] [CrossRef]
- Filipski, K.J.; Kohrt, J.T.; Casimiro-Garcia, A.; Van Huis, C.A.; Dudley, D.A.; Cody, W.L.; Bigge, C.F.; Desiraju, S.; Sun, S.; Maiti, S.N.; Jaber, M.R.; Edmunds, J.J. A versatile copper-catalyzed coupling reaction of pyridin-2(1H)-ones with aryl halides. Tetrahedron Lett. 2006, 47, 7677–7680. [Google Scholar] [CrossRef]
- Kuil, M.; Bekedam, E.K.; Visser, G.M.; van den Hoogenband, A.; Terpstra, J.W.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; van Strijdonck, G.P.F. Mild copper-catalyzed N-arylation of azaheterocycles with aryl halides. Tetrahedron Lett. 2005, 46, 2405–2409. [Google Scholar] [CrossRef]
- Schwenk, E.; Papa, D. Bromination of dicarboxylic acids. J. Am. Chem. Soc. 1948, 70, 3626–3627. [Google Scholar] [CrossRef]
- Berry, J.P.; Isbell, A.F.; Hunt, G.E. Amino phosphonic acids. II. Aminoalkylphosphonic acids. J. Org. Chem. 1972, 37, 4396–4399. [Google Scholar] [CrossRef]
- Brychtova, K.; Farsa, O.; Csollei, J. Copper catalyzed coupling of α-bromocarboxylate with ω-lactam. Lett. Org. Chem. 2009, 6, 25–28. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comp. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comp. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Brychtova, K.; Farsa, O.; Csollei, J. Synthesis of esters of substituted 6-aminohexanoic acid as potential transdermal penetration enhancers. ECSOC-12. 1-30 November 2008, p. c0005. Available online: http://www.usc.es/congresos/ecsoc/12/ECSOC12.htm.
- Sample Availability: Samples of the compounds are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brychtova, K.; Slaba, B.; Placek, L.; Jampilek, J.; Raich, I.; Csollei, J. Coupling Reactions of α-Bromocarboxylate with Non-Aromatic N-Heterocycles. Molecules 2009, 14, 3019-3029. https://doi.org/10.3390/molecules14083019
Brychtova K, Slaba B, Placek L, Jampilek J, Raich I, Csollei J. Coupling Reactions of α-Bromocarboxylate with Non-Aromatic N-Heterocycles. Molecules. 2009; 14(8):3019-3029. https://doi.org/10.3390/molecules14083019
Chicago/Turabian StyleBrychtova, Katerina, Barbora Slaba, Lukas Placek, Josef Jampilek, Ivan Raich, and Jozef Csollei. 2009. "Coupling Reactions of α-Bromocarboxylate with Non-Aromatic N-Heterocycles" Molecules 14, no. 8: 3019-3029. https://doi.org/10.3390/molecules14083019