Is Endothelial Nitric Oxide Synthase a Moonlighting Protein Whose Day Job is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease †


Abstract
:1. Introduction
2. The Central Inference
3. Cholesterol Sulfate and eNOS
3.1. Biological Roles for Cholesterol Sulfate
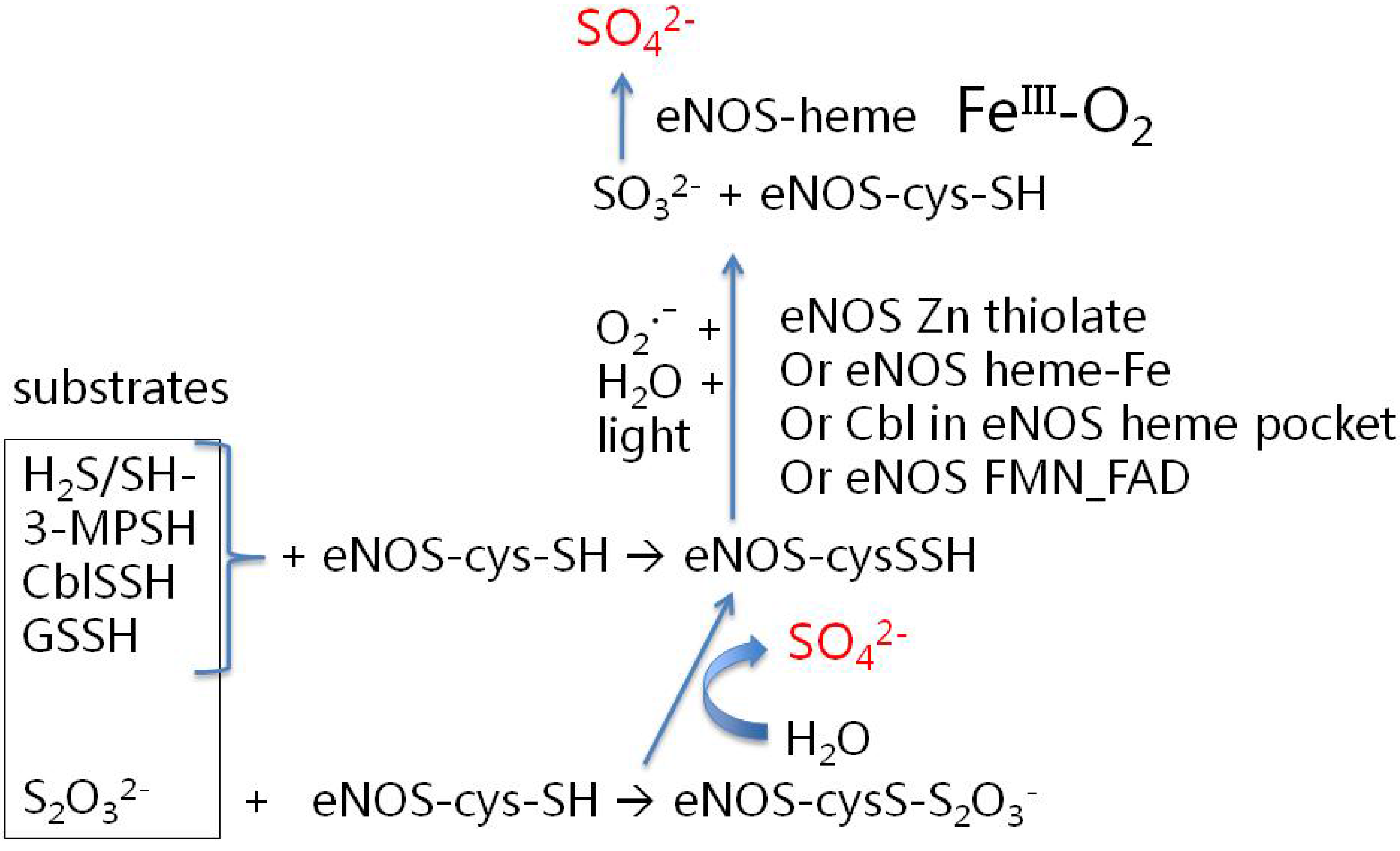
3.2. How Might eNOS Synthesize Sulfate? Some Possibilities
- (a)
- use of caveolin-bound, calmodulin-free eNOS;
- (b)
- use of various candidate reduced sulfur substrate compounds (see discussion below), preferably at physiologically-realistic concentrations;
- (c)
- activity assays aimed at detecting sulfate (not just NO); and
- (d)
- appropriate controls to suppress the facile non-enzymatic oxidation of reduced sulfur compounds in the presence of oxygen and trace metals [63].
4. Red Blood Cells and Cardiovascular Disease
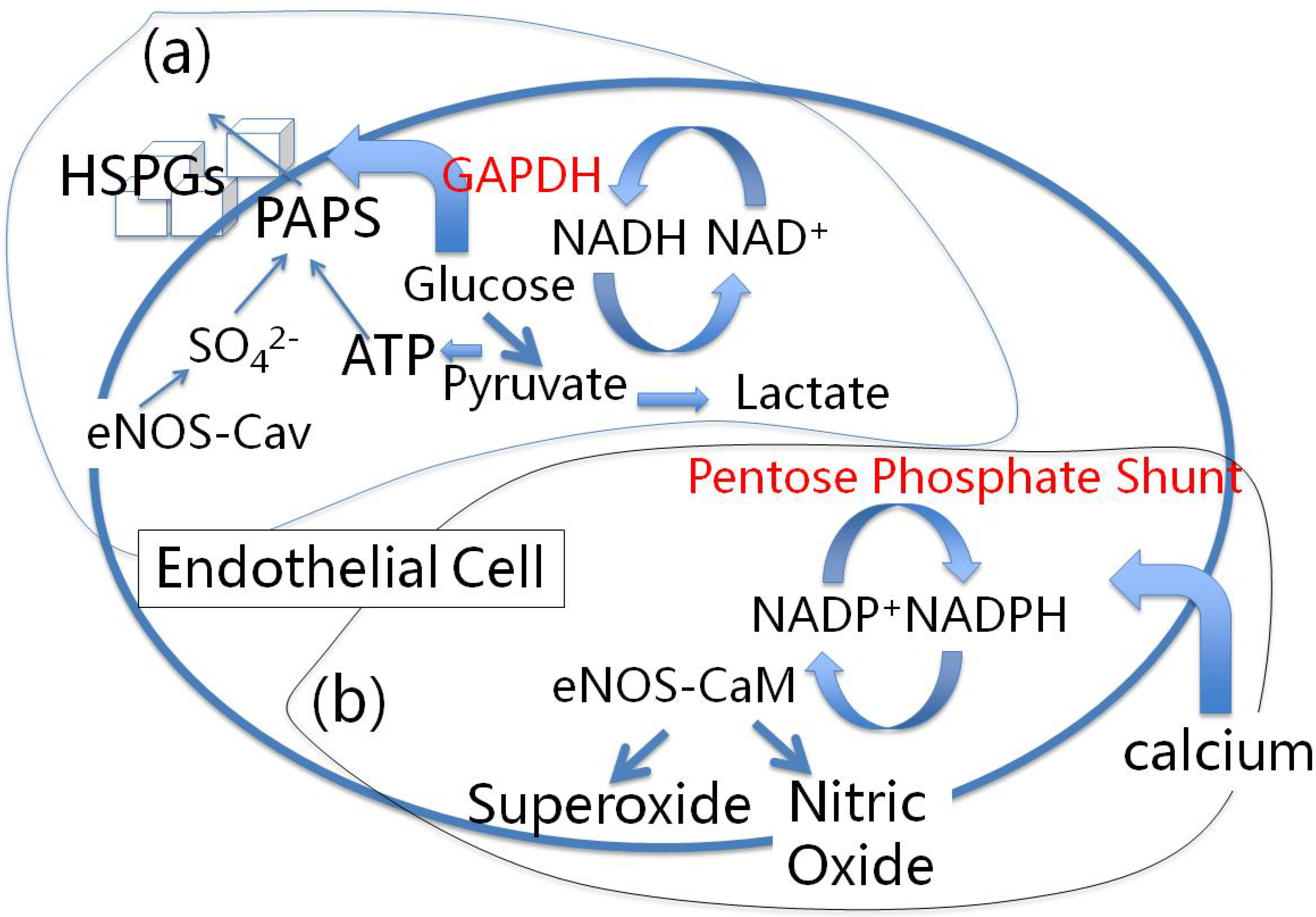
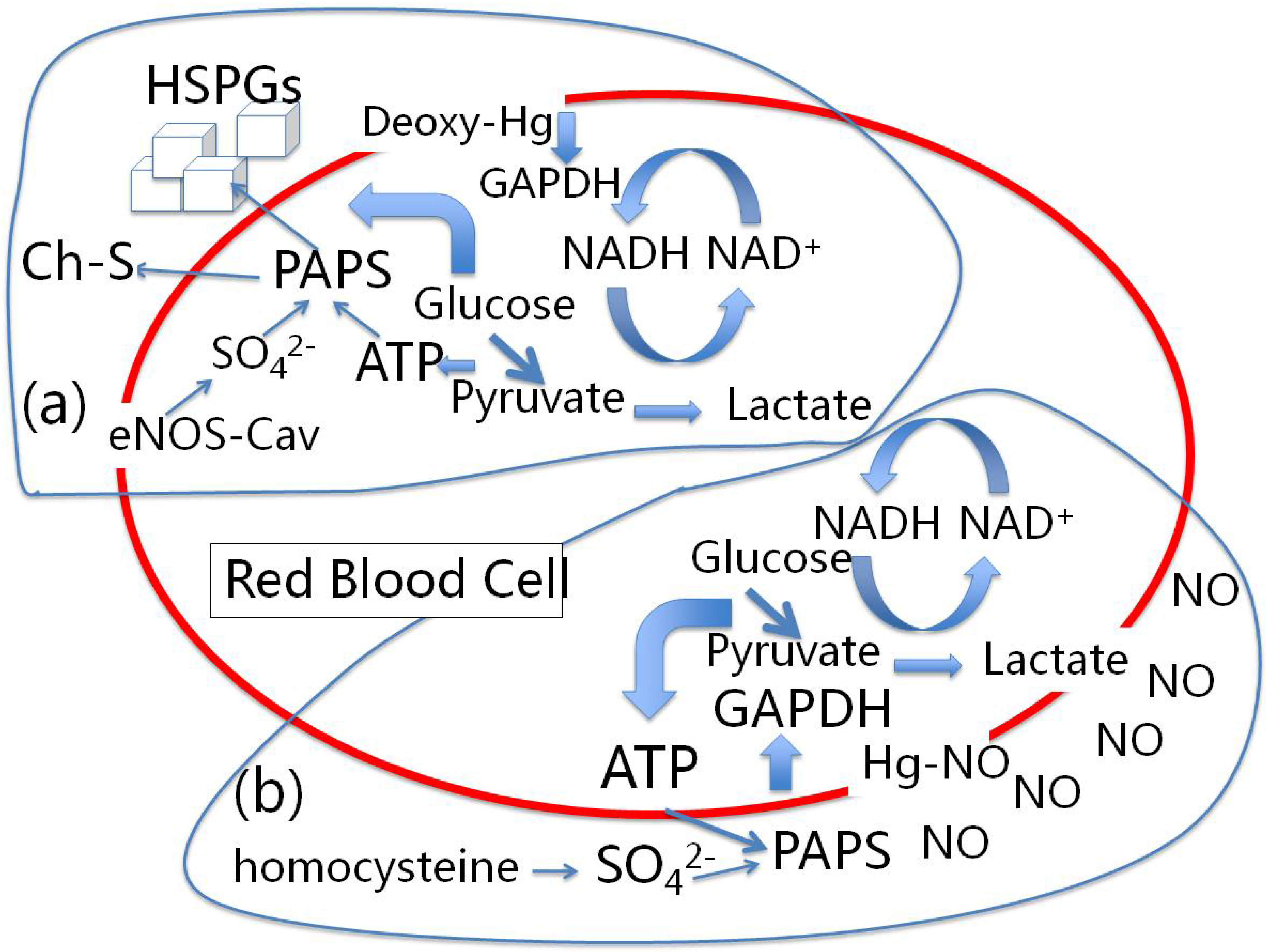
4.1. Red Blood Cells, eNOS and Cholesterol Sulfate
4.2. Nitrite-Induced Vasodilation by RBCs
4.3. A Proposed Role for the Atheroma
5. The Hofmeister Series, the Glycocalyx and the Effect of Sulfate on Water Structure
6. Heparan Sulfate Proteoglycans
6.1. Heparan Sulfate Proteoglycans and Glucose Metabolism
6.2. eNOS and Sulfated Glycosaminoglycan Synthesis
6.3. The Lysosomal Connection
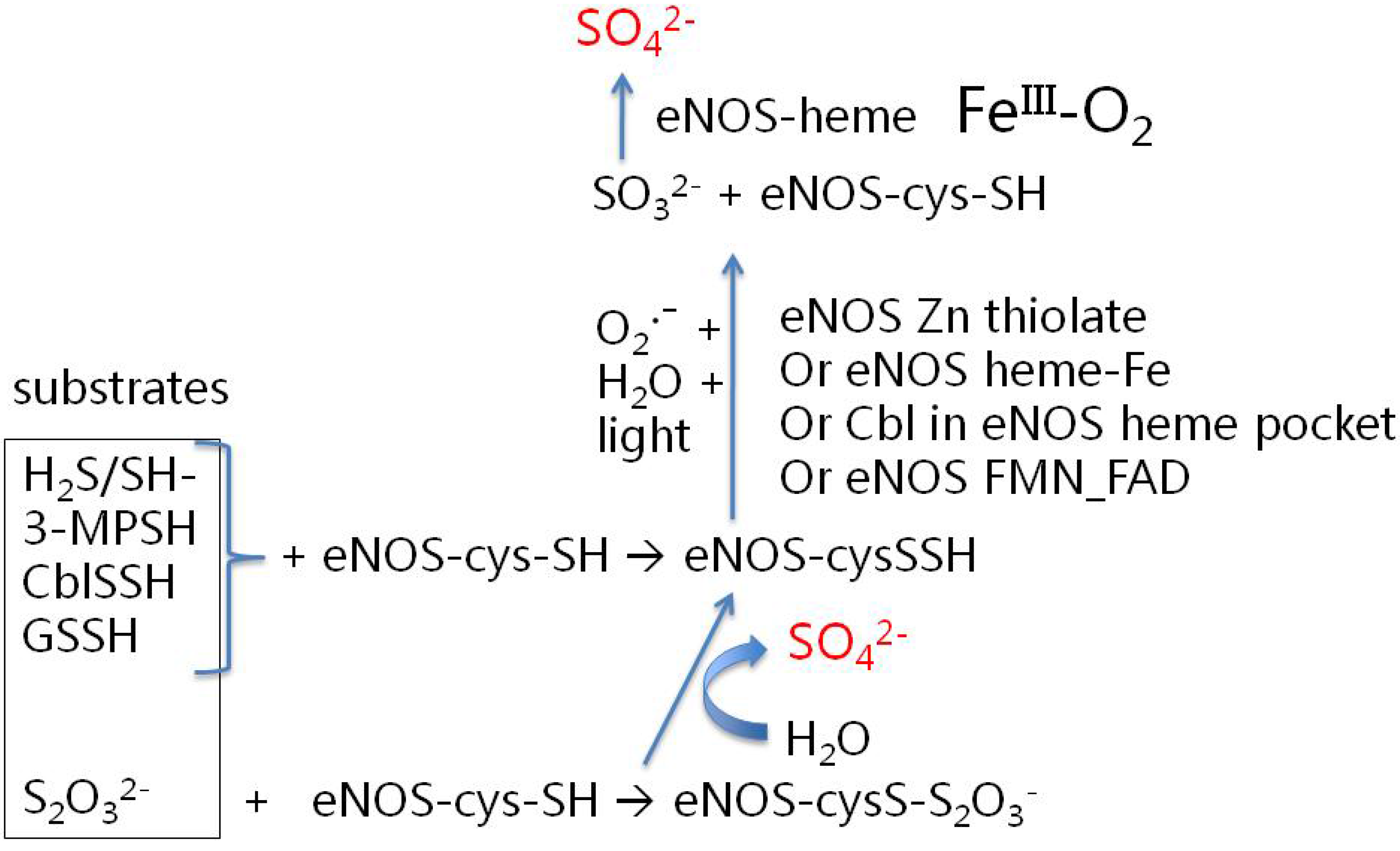
7. Signaling Gases: H2S, NO and CO

{kind=link}
{kind=link}
{kind=link}
| 2H2S | ➜ | S2O32− | ➜ | 2SO42− |
| NO | ➜ | NO32− | ||
| CO | ➜ | CO32− |
8. NO and Autophagy Suppression
9. Discussion
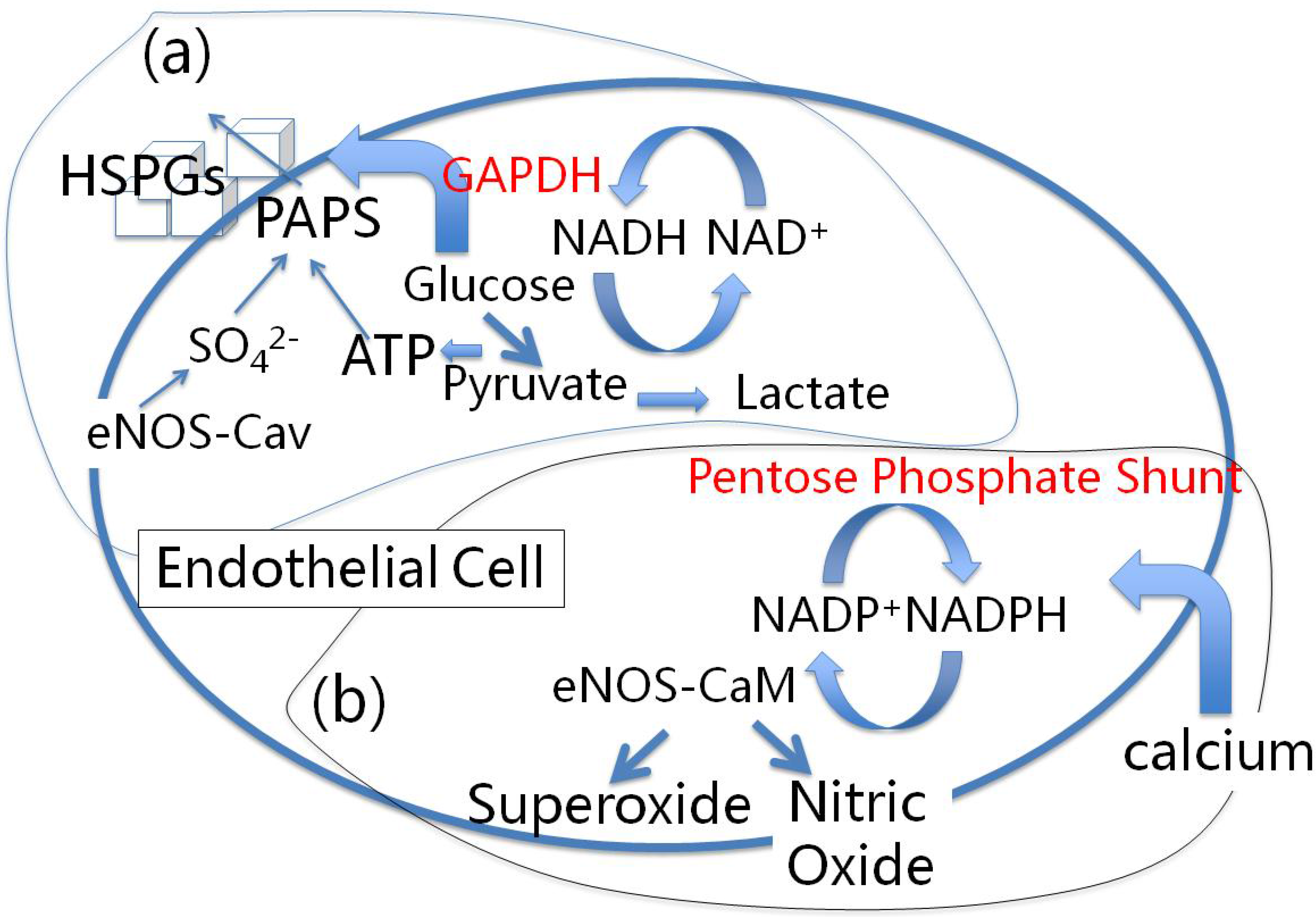
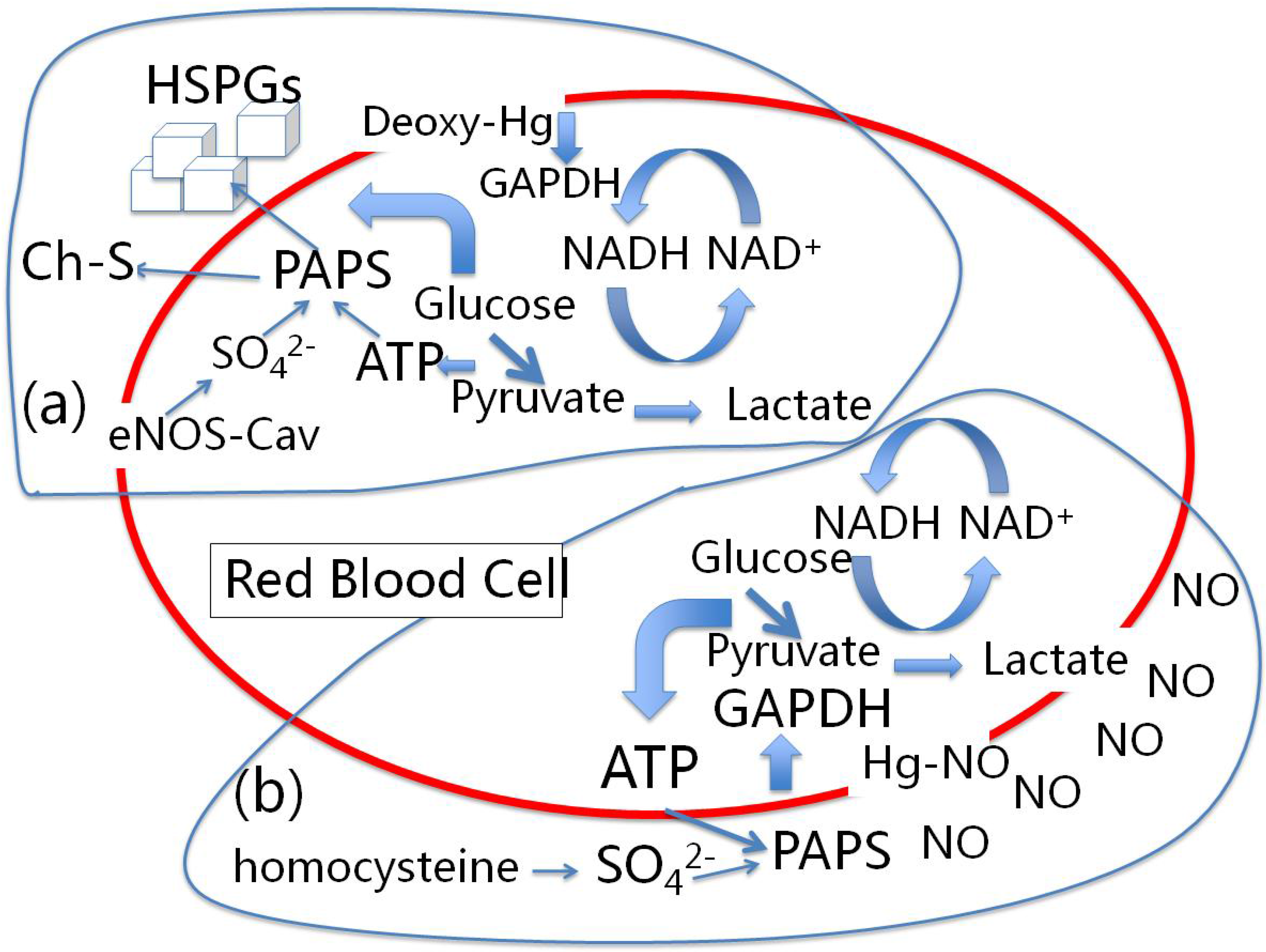
10. Conclusions
Acknowledgments
Abbreviations
| ADMA | asymmetrical dimethylarginine |
| Akt | protein kinase B (PKB) |
| ATP | adenosine triphosphate |
| BH4 | tetrahydrobiopterin |
| Cbl | cobalamin |
| CblSSH | cobalamin persulfide |
| Ch-S | cholesterol sulfate |
| deoxyHb | deoxyhemoglobin |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| EZ | exclusion zone |
| FAD | flavin adenine dinucleotide |
| FMN | flavin mononucleotide |
| GAG | glycosaminoglycan |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| Glc | glucose |
| GLUT | glucose transporter |
| GSH | glutathione |
| GSNO | S-nitrosylglutathione |
| GSSH | glutathione persulfide |
| Hb | hemoglobin |
| HDL | high-density lipoprotein |
| HMG-CoA reductase | 3-hydroxy-3-methyl-glutaryl-Coenzyme A reductase |
| HS | heparan sulfate |
| HSPG | heparan sulfate proteoglycan |
| iNOS | inducible nitric oxide synthase |
| IR | infrared |
| LDH | lactate dehydrogenase |
| LDL | low-density lipoprotein |
| LRP | LDL receptor-related protein |
| LPS | lipopolysaccharide |
| metHb | methemoglobin |
| mTOR | mammalian target ofrapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NAc | N-acetyl |
| NAD(P) | nicotinamide adenine dinucleotide (phosphate) |
| NAD(P)H | nicotinamide adenine dinucleotide (phosphate), reduced form |
| NF-κB | nuclear factor κB |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| oxyHb | oxyhemoglobin |
| PAPS | 3’-phosphadenosine-5’-phosphosulfate |
| PFK | phosphofructokinase |
| PI3K | phosphatidylinositol-3-kinase |
| PKB | protein kinase B, also known as Akt |
| PKC | protein kinase C |
| PON1 | paraoxonase 1 |
| RBC | red blood cell |
| ROS | reactiveoxygenspecies |
| SR | scavenger receptor |
| SULT | sulfotransferase |
References
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Xie, Q. Nitric oxide synthases: Roles, tolls and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Florio, T.; Arena, S.; Pattarozzi, A.; Thellung, S.; Corsaro, A.; Villa, V.; Massa, A.; Diana, F.; Spoto, G.; Forcella, S.; et al. Basic fibroblast growth factor activates endothelial nitric-oxide synthase in CHO-K1 cells via the activation of ceramide synthesis. Mol. Pharmacol. 2003, 63, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Saldana, F.; Michel, J.B.; Michel, T. The endothelial nitric-oxide synthase-caveolin regulatory cycle. J. Biol. Chem. 1998, 273, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Randriamboavonjy, V.; Fleming, I. Endothelial nitric oxide synthase (eNOS) in platelets: How is it regulated and what is it doing there? Pharmacol. Rep. 2005, 57, 59–65. [Google Scholar] [PubMed]
- Kleinbongard, P.; Schulz, R.; Rassaf, T.; Lauer, T.; Dejam, A.; Jax, T.; Kumara, I.; Gharini, P.; Kabanova, S.; Özüyaman, B.; et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006, 107, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Zou, R.; Venema, V.J.; Venema, R.C. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 1997, 272, 18522–18525. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Sampaio, J.L. Membrane Organization and Lipid Rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Lipid rafts: Bringing order to chaos. J. Lipid Res. 2003, 44, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Jones, C.; Hattori, Y.; Raman, C. Tetrahydrobiopterin: An essential co-factor of nitric oxide synthase with an elusive role. In Nitric Oxide Biology and Pathobiology; Jignarro, L., Ed.; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Rafikov, R.; Fonseca, F.V.; Kumar, S.; Pardo, D.; Darragh, C.; Elms, S.; Fulton, D.; Black, S.M. eNOS activation and NO function: Structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011, 210, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Smith, R.S., Jr.; Hsieh, C.-M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Invest. 2002, 109, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Lottspeich, F.; Brüne, B. Nitric oxide causes ADP-ribosylationand inhibition of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1992, 267, 16771–16774. [Google Scholar] [PubMed]
- OBrien, M.L.; Tew, K.D. Glutathione and related enzymes in multidrug resistance. Eur. J. Cancer 1996, 32A, 967–978. [Google Scholar] [CrossRef]
- Mayer, B.; Pfeiffer, S.; Schrammel, A.; Koesling, D.; Schmidt, K.; Brunner, F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J. Biol. Chem. 1998, 273, 3264–3270. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex Inhibitory Effects of Nitric Oxide on Autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic Degeneration and Death of Cardiomyocytes in Heart Failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Moonlighting proteins: Old proteins learning new tricks. Trends Genet. 2003, 19, 415–417. [Google Scholar] [CrossRef]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Teien, A.N.; Abildgaard, U.; Höök, M. The anticoagulant effect of heparansulfate and dermatan sulfate. Thromb. Res. 1976, 8, 859–867. [Google Scholar] [CrossRef]
- Shriver, Z.; Liu, D.; Sasisekharan, R. Emerging views of heparan sulfate glycosaminoglycan structure/activity relationships modulating dynamic biological functions. Trends Cardiovas. Med. 2002, 12, 71–77. [Google Scholar] [CrossRef]
- Deng, D.; Jiang, N.; Hao, S.J.; Sun, H.; Zhang, G.J. Loss of membrane cholesterol influences lysosomal permeability to potassium ions and protons. Biochim. Biophys. Acta 2009, 1788, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Jadot, M.; Andrianaivo, F.; Dubois, F.; Wattiaux, R. Effects of methylcyclodextrin on lysosomes. Eur. J. Biochem. 2001, 268, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, M.; Kjeken, R.; Kolset, S.O.; Berg, T.; Prydz, K. Internalization and stepwise degradation of heparan sulfate proteoglycans in rat hepatocytes. Biochim. Biophys. Acta 2001, 1541, 135–149. [Google Scholar] [CrossRef]
- Harper, G.S.; Rozaklis, T.; Bielicki, J.; Hopwood, J. Lysosomal sulfate efflux following glycosaminoglycan degradation: Measurements in enzyme-supplemented Maroteaux-Lamy syndrome fibroblasts and isolated lysosomes. Glycoconjugate J. 1993, 10, 407–415. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Dwek, R.A.; Butters, T.D.; Platt, F.M. Storage solutions: Treating lysosomal disorders of the brain. Nature Rev. Neurosci. 2005, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, J.J.; Ballabio, A. Multiple sulfatase deficiency and the nature of the sulfatase family. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: Columbus, OH, USA, 2001; pp. 3725–3732. [Google Scholar]
- Strott, C.A. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.V.; Wheeler, J.J.; Klimuk, S.K.; Kitson, C.N.; Hope, M.J. Transbi-layer movement and net flux of cholesterol and cholesterol sulfate between liposomal membranes. Biochemistry 1995, 34, 6208–6217. [Google Scholar] [CrossRef]
- Haines, T.H. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog. Lipid Res. 2001, 40, 299–324. [Google Scholar] [CrossRef]
- Astrup, J.; Sørensen, P.M.; Sørensen, H.R. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke 1981, 12, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Ye, Q.; Tian, J.; Jing, R.; Xie, Z. Regulation of α1 Na/K-ATPase expression by cholesterol. J. Biol. Chem. 2011, 286, 15517–15524. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Na+, K+ ATPase: Functions in the nervous system and involvement in neurologic disease. Neurology 2011, 76, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.J.; Hou, J.F.; Jiang, N.; Zhang, G.J. Loss of membrane cholesterol affects lysosomal osmotic stability. Gen. Physiol. Biophys. 2008, 27, 278–283. [Google Scholar] [PubMed]
- Yang, A.J.; Chandswangbhuvana, D.; Margol, L.; Glabe, C.G. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Aβ1–42 pathogenesis. J. Neurosci. Res. 1998, 52, 691–698. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Haskins, M.E.; Schuchman, E.H. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: A possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab. Invest. 2001, 81, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, M.; Faber, M.; Zaborowski, A.; Tosawski, J.; Bryszewska, M. Morphological changes of human erythrocytes induced by cholesterol sulphate. Clin. Biochem. 1998, 31, 73–79. [Google Scholar] [CrossRef]
- Cignarelli, M.; Damato, A.; Cospite, M.R.; Guastamacchia, E.; Nardelli, G.M.; Giorgino, R. Erythrocyte cholesterol and red blood cells: Deformability in diabetes mellitus. Boll. Soc. Ital. Biol. Sper. 1982, 58, 1115–1118. [Google Scholar] [PubMed]
- Nakae, H.; Hanyu, O.; Fuda, H.; Strott, C.A. Novel role of cholesterol sulfate in gene regulation during skin development. FASEB J. 2008, 22, 782. [Google Scholar]
- McGrath, J.A.; Uitto, J. The filaggrin story: Novel insights into skin-barrier function and disease. Trends Molec. Med. 2007, 14, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, D.A.; Sud’ina, G.F. Suppression of 5-Lipoxygenase Activity by Anionic Cholesterol Derivatives. (in Russian). Biochemistry (Mosc.) Supplement Series B: Biomed. Khim. 2007, 1, 168–171. [Google Scholar] [CrossRef]
- Langlais, J.; Zollinger, M.; Plante, L.; Chapdelaine, A.; Bleau, G.; Roberts, K.D. Localization of cholesteryl sulfate in human spermatozoa in support of a hypothesis for the mechanism of capacitation. Proc. Natl. Acad. Sci. USA 1981, 78, 7266–7270. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kubushiro, K.; Akiba, Y.; Cui, Y.; Tsukazaki, K.; Nozawa, S.; Iwamori, M. Alteration of acidic lipids in human sera during the course of pregnancy: Characteristic increase in the concentration of cholesterol sulfate. J. Chromatog. B 1997, 704, 99–104. [Google Scholar] [CrossRef]
- Seneff, S.; Davidson, R.; Mascitelli, L. Might cholesterol sulfate deficiency contribute to the development of autistic spectrum disorder? Med. Hypotheses 2012, 78, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Tierney, E.; Bukelis, I.; Thompson, R.; Ahmed, K.; Aneja, A.; Kratz, L.; Kelley, R. Abnormalities of cholesterol metabolism in autism spectrum disorders. Amer. J. Med. Genet. B 2006, 141B, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Waring, R.H.; Klovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Tai, S.C.; Robb, G.B.; Marsden, P.A. Endothelial Nitric oxide Synthase: A new paradigm for gene regulation in the injured blood vessel. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Marletta, M.A. Another activation switch for endothelial nitric oxide synthase: why does it have to be so complicated? Trends Biochem. Sci. 2001, 26, 519–521. [Google Scholar] [CrossRef]
- Marzinzig, M.; Nussler, A.K.; Stadler, J.; Marzinzig, E.; Barthlen, W.; Nussler, N.C.; Beger, H.G.; Morris, S.M., Jr.; Brückner, U.B. Improved methods to measure end products of nitric oxide in biological fluids: nitrite, nitrate, and S-nitrosothiols. Nitric Oxide 1997, 1, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, N.; Biebel, H. The dissimilatory sulfate-reducing bacteria. In The prokaryotes: A Handbook on Habitats, Isolation and Identification of Bacteria; Springer-Verlag: New York, NY, USA, 1986. [Google Scholar]
- Hockin, S.L.; Gadd, G.M. Linked redox precipitation of sulfur and selenium under anaerobic conditions by sulfate-reducing bacterial biofilms. Appl. Environ. Microbiol. 2003, 69, 7063–7072. [Google Scholar] [CrossRef] [PubMed]
- Teske, A.; Ramsing, N.B.; Habicht, K.; Fukui, M.; Kver, J.; Jrgensen, B.B.; Cohen, Y. Sulfate-reducing bacteria and their activities in cyanobacterial mats of Solar Lake (Sinai, Egypt). Appl. Environ. Microbiol. 1998, 64, 2943–2951. [Google Scholar] [PubMed]
- Wahlund, T.M.; Woese, C.R.; Castenholz, R.W.; Madigan, M.T. A thermophilic green sulfur bacterium from New Zealand Hot Springs, Chlorobium tepidum sp. Arch. Microbiol. 1991, 159, 81–90. [Google Scholar] [CrossRef]
- Olson, J.M.; Blankenship, R.E. Thinking about the evolution of photosynthesis. Photosynth. Res. 2004, 80, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Brune, D. Sulfur compounds as photosynthetic electron donors. Photosynth. Res. 2004, 2, 847–870. [Google Scholar]
- Moss, M.; Waring, R.H. The plasma cysteine/sulphate ratio: A possible clinical biomarker. J. Nutr. Environ. Med. 2003, 13, 215–229. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, B. Mechanism of oxidation of inorganic thiosulfate and thiosulfate esters in mammals. Acta Chem. Scand. 1964, 18, 821–823. [Google Scholar] [CrossRef]
- Friedrich, C.G.; Rother, D.; Bardischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Bamford, V.A.; Bruno, S.; Rasmussen, T.; Appia-Ayme, C.; Cheesman, M.R.; Berks, B.C.; Hemmings, A.M. Structural basis for the oxidation of thiosulfate by a sulfur cycle enzyme. EMBO J. 2002, 21, 5599–5610. [Google Scholar] [CrossRef] [PubMed]
- Tapley, D.W.; Buettner, G.R.; Shick, J.M. Free radicals and chemiluminescence as products of the spontaneous oxidation of sulfide in seawater, and their biological implications. Biol. Bull. 1999, 196, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Derivative of cobalamin containing persulfide sulfur and glutathione. U.S. Patent 4,751,285, 1988. [Google Scholar]
- Weinberg, J.B.; Chen, Y.; Jiang, N.; Beasley, B.E.; Salerno, J.C.; Ghosh, D.K. Inhibition of nitric oxide synthase by cobalamins and cobinamides. Free Radic. Biol. Med. 2009, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.P.; Bahnemann, D.W.; Hoffman, M.R. Cobalt(II) tetrasulfophthalocyanine on titanium dioxide. 2. Kinetics and mechanisms of the photocatalytic oxidation of aqueous sulfur dioxide. J. Phys. Chem. 1987, 91, 6245–6251. [Google Scholar] [CrossRef]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hermann, C.; Hassan Talukder, M.A.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Chen, C.-A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Sign. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Marcus, R.A. On the Theory of Organic Catalysis “on Water”. J. Am. Chem. Soc. 2007, 129, 5492–5502. [Google Scholar] [CrossRef] [PubMed]
- Markovitch, O.; Chen, H.; Izvekov, S.; Paesani, F.; Voth, G.A.; Agmon, N. Special Pair Dance and Partner Selection: Elementary Steps in Proton Transport in Liquid Water. J. Phys. Chem. B 2008, 112, 9456–9466. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Born, B.; Heyden, M.; Tworowski, D.; Fields, G.B.; Sagi, I.; Havenith, M. Correlated structural kinetics and retarded solvent dynamics at the metalloprotease active site. Nat. Struct. Mol. Biol. 2011, 18, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Born, B.; Sagi, I.; Havenith, M. Water's contribution and enzyme's work: a KITA study. In Proceeding of Imaging, Manipulation, and Analysis of Biomolecules, Cells, and Tissues X, San Francisco, CA, USA, 21 January 2012.
- O’Brien, J.T.; Prell, J.S.; Bush, M.F.; Williams, E.R. Sulfate Ion Patterns Water at Long Distance. J. Am .Chem. Soc. 2010, 132, 8248–8249. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, A.; Everson, W.V.; Matveev, S.V.; Smart, E.J. Cholesteryl ester is transported from caveolae to internal membranes as part of a caveolin-annexin II lipid- protein complex. J. Biol. Chem. 2002, 277, 4925–4931. [Google Scholar] [CrossRef] [PubMed]
- Fuda, H.; Javitt, N.B.; Mitamura, K.; Ikegawa, S.; Strott, C.A. Oxysterols are substrates for cholesterol sulfotransferase J. Lipid Res. 2007, 48, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. J. Am. Med. Assoc. 1986, 256, 2835–2838. [Google Scholar] [CrossRef]
- Foster, M.; Samman, S. Zinc and redox signaling: Perturbations associated with cardiovascular disease and diabetes mellitus. Antioxid. Redox Sign. 2010, 13, 1549–1573. [Google Scholar] [CrossRef] [PubMed]
- Stehouwer, C.D.A.; Weijenberg, M.P.; van den Berg, M.; Jakobs, C.; Feskens, E.J.M.; Kromhout, D. Serum Homocysteine and Risk of Coronary Heart Disease and Cerebrovascular Disease in Elderly Men. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Invest. 2001, 108, 793–797. [Google Scholar] [CrossRef] [PubMed]
- DeVries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. JCB 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Invest. 2002, 110, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M.; Seneff, S. The initial common pathway of inflammation, disease, and death. Entropy 2012, 14, 1399–1442. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Keymel, S.; Kelm, M. New functional aspects of the L-arginine-nitric oxide metabolism within the circulating blood. Thromb. Haemost. 2007, 98, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.S.; Ford, K.; Grokulsky, G.; Wang, Y.B.; Chiang, T.M.; Acchiardo, S.R. Normal circulating adult human red blood cells contain inactive NOS proteins. J. Lab. Clin. Med. 2000, 135, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.E.; Chu, H.; Low, P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.H.; Murthy, S.N.; Steck, T.L. Effect of red cell membrane binding on the catalytic activity of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1982, 257, 1438–1442. [Google Scholar] [PubMed]
- Messana, I.; Orlando, M.; Cassiano, L.; Pennacchietti, L.; Zuppi, C.; Castagnola, M.; Giardina, B. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett. 1996, 390, 25–28. [Google Scholar] [CrossRef]
- Bleau, G.; Bodley, F.H.; Longpré, J.; Chapdelaine, A.; Roberts, K.D. Cholesterol sulfate. I. Occurrence and possible biological functions as an amphipathic lipid in the membrane of the human erythrocyte. Biochim. Biophys. Acta 1974, 352, 1–9. [Google Scholar] [CrossRef]
- Jennings, M.L. Rapid electrogenic sulfate-chloride exchange mediated by chemically modified band 3 in human erythrocytes. J. Gen. Physiol. 1995, 105, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Jagger, J.E.; Bateman, R.M.; Ellsworth, M.L.; Ellis, C.G. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2833–H2839. [Google Scholar] [PubMed]
- Gladwin, M.T.; Grubina, R.; Doyle, M.P. The new chemical biology of nitrite reactions with hemoglobin: R-state catalysis, oxidative denitrosylation, and nitrite reductase/anhydrase. Acc. Chem. Res. 2009, 42, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.W.; Piantadosi, C.A. How do red blood cells cause hypoxic vasodilation? The SNO-hemoglobin paradigm. Am. Physiological Soc. 2006, 291, H1507–H1512. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.H.; Isbell, T.S.; Huang, Z.; Shiva, S.; Chacko, B.K.; Schechter, A.N.; Darley-Usmar, V.M.; Kerby, J.D.; Lang, J.D., Jr.; Kraus, D.; et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 2006, 107, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Bonaventura, C.; Bonaventura, J.; Stamler, J.S. S-nitrosohaemoglobin: A dynamic activity of blood involved in vascular control. Nature 1996, 380, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Ramasamy, S.; Rifkind, J.M. S-nitrosohemoglobin: A mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 2006, 15, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Ramasamy, S.; Rifkind, J.M. Intermediates detected by visible spectroscopy during the reaction of nitrite with deoxyhemoglobin: The effect of nitrite concentration and diphosphoglycerate. Biochemistry 2007, 46, 11650–11659. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Bell, J.B.; Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: A new mechanism for nitrite-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1494–H1503. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Javitt, N.B.; Higashi, Y.; Fuda, H.; Strott, C.A. Expression of Cholesterol Sulfotransferase (SULT2B1b) in Human Platelets. Circulation 2004, 109, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Steeber, D.A.; Chen, A.; Engel, P. The selectins: Vascular adhesion molecules. Selectins 1995, 9, 866–873. [Google Scholar]
- Girard, J.-P.; Baekkevold, E.S.; Amalric, F. Sulfation in high endothelial venules: cloning and expression of the human PAPS synthetase. FASEB J. 1998, 12, 603–612. [Google Scholar] [PubMed]
- Drayer, N.M.; Lieberman, S. Isolation of cholesterol sulfate from human aortas and adrenal tumors. J. Clin. Endocrinol. Metab. 1967, 27, 136–139. [Google Scholar] [PubMed]
- Lentz, S.R. Homocysteine and vascular dysfunction. Life Sci. 1997, 61, 1205–1215. [Google Scholar] [CrossRef]
- McCully, K.S. Chemical pathology of homocysteine V: Thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Ann. Clin. Lab. Sci. 2011, 41, 300–313. [Google Scholar]
- Schmitz, G.; Kaminski, W.E.; Porsch-Ozcürümez, M.; Klucken, J.; Orsó, E.; Bodzioch, M.; Büchler, C.; Drobnik, W. ATP-binding cassette transporter A1 (ABCA1) in macrophages: A dual function in inflammation and lipid metabolism? Pathobiology 1999, 67, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.K.; Brown, M.S.; Goldstein, J.L. Hydrolysis and excretion of cytoplasmic cholesteryl esters by macrophages: Stimulation by high density lipoprotein and other agents. J. Lipid Res. 1980, 21, 391–398. [Google Scholar] [PubMed]
- Stanger, O.; Weger, M. Interactions of homocysteine, nitric oxide, folate and radicals in the progressively damaged endothelium. Clin. Chem. Lab. Med. 2003, 41, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Tsujino, T.; Watari, Y.; Emoto, N.; Yokoyama, M. Taurine prevents the decrease in expression and secretion of extracellular superoxide dismutase induced by homocysteine. Circulation 2001, 104, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Hara, H.; Adachi, T. Effects of homocysteine on the binding of extracellular-superoxide dismutase to the endothelial cell surface. FEBS Lett. 2000, 486, 159–162. [Google Scholar] [CrossRef]
- Wang, G.; Siow, Y.L.O.K. Homocysteine stimulates nuclear factor κB activity and monocyte chemoattractant protein-1 expression in vascular smooth muscle cells: a possible role for protein kinase. Biochem. J. 2000, 352, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Edwards, J.G.; Kaminski, P.M.; Wolin, M.S.; Kaley, G.; Koller, A. Increased superoxide production in coronary arteries in hyperhomocysteinemia. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, R.N.; Lentz, S.R. The homocysteine paradox. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Vernier, R.L.; Klein, D.J.; Sisson, S.P.; Mahan, J.D.; Oegema, T.R.; Brown, D.M. Heparan sulfate--rich anionic sites in the human glomerular basement membrane. Decreased concentration in congenital nephrotic syndrome. N. Engl. J. Med. 1983, 309, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009, 30, 1142–1450. [Google Scholar] [CrossRef] [PubMed]
- Yui, Y.; Aoyama, T.; Monshita, H.; Takahashi, M.; Takatsu, Y.; Kawal, C. Serum prostacyclin stabilizing factor is identical to Apolipoprotein A-I (Apo A-I) a novel function of Apo A-I. J. Clin. Invest. 1988, 82, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.B.; Zhang, Q.; Lim, Y.K.; Fang, D.; Retnam, L.; Lim, S.K. Expression of major HDL-associated antioxidant PON-1 is gender dependent and regulated during inflammation. Free Radic. Biol. Med. 2003, 34, 824–829. [Google Scholar]
- Aviram, M.; Rosenblat, M.; Billecke, S.; Erogul, J.; Sorenson, R.; Bisgaier, C.L.; Newton, R.S.; Du, B.L. Human serum paraoxonase (PON1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic. Biol. Med. 1999, 26, 892–904. [Google Scholar] [CrossRef]
- Jakubowski, H. The role of paraoxonase-1 in the detoxification of homocysteine thiolactone. Adv. Exp. Med. Biol. 2010, 660, 113–127. [Google Scholar] [PubMed]
- Billecke, S.; Draganov, D.; Counsell, R.; Stetson, P.; Watson, C.; Hsu, C.; la Du, B.N. Human serum paraoxonase (PON1) isozymes Q and R hydrolyze lactones and cyclic carbonate esters. Drug Metab. Dispos. 2007, 28, 1336–1342. [Google Scholar]
- Gugliucci, A.; Hermo, R.; Tsuji, M.; Kimura, S. Lower serum paraoxonase-1 activity in type 2 diabetic patients correlates with nitrated apolipoprotein A-I levels. Clin. Chim. Acta 2006, 368, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Pollack, G. Cells, Gels and the Engines of Life; Ebner and Sons, publishers: New York, NY, USA, 2001. [Google Scholar]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflugers Arch. EJP 2000, 440, 653–666. [Google Scholar] [CrossRef]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Vlachy, N.; Jagoda-Cwiklik, B.; Vacha, R.; Touraud, D.; Jungwirth, P.; Kunz, W. Hofmeister series and specific interactions of charged headgroups with aqueous ions. Adv. Colloid Interface Sci. 2009, 146, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Huh, N.; Kim, J.H. Isolation of total RNA from Escherichia coli using kosmotropic Hofmeister salts. Anal. Biochem. 2008, 381, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Pollack, G.H.; Figueroa, X.; Zhao, Q. Molecules, water, and radiant energy: New clues for the origin of life. Int. J. Mol. Sci. 2009, 10, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Voeikov, V.L.; Del Giudice, E. Water respiration-the basis of the living state. Water 2009, 1, 52–75. [Google Scholar]
- Mohri, K.; Fukushima, M. Milligauss magnetic field triggering reliable self-organization of water with long-range ordered proton transport through cyclotron resonance. IEEE T. Magn. 2003, 39, 3328–3330. [Google Scholar] [CrossRef]
- Vossen, C.Y.; Conard, J.; Fontcuberta, J.; Makris, M.; van Der Meer, F.J.M.; Pabinger, I.; Palareti, G.; Preston, F.E.; Scharrer, I.; Souto, J.C.; et al. Familial thrombophilia and lifetime risk of venous thrombosis. J. Thromb. Haemost. 2004, 2, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Engström-Laurent, A.; Mörner, S.; Johansson, B. Hyaluronan and Collagen in Human Hypertrophic Cardiomyopathy: A Morphological Analysis. Cardiol. Res. and Pract. 2012, 545219. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P.; Wight, T.N.; Tammi, M.I. Hyaluronan-Cell Interactions in Cancer and Vascular Disease. J. Biol. Chem. 2002, 277, 4593–4596. [Google Scholar] [CrossRef] [PubMed]
- Sasisekharan, R.; Shriver, Z.; Venkataraman, G.; Narayanasami, U. Roles of Heparan-sulphate glycosaminoglycans in cancer. Nature Reviews 2002, 2, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, W.C.; Kalus, I.; Padva, M.; Baldwin, R.J.; Merry, C.L.R.; Dierks, T. The heparanome –The enigma of encoding and decoding heparan sulfate sulfation. J. Biotech. 2007, 129, 290–307. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Powell, A.; Guimond, S. Heparan sulfate: Decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001, 11, 75–82. [Google Scholar] [CrossRef]
- Gorsi, B.; Stringer, S.E. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol. 2007, 17, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R. Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.H.; Aquino, R.S.; Park, P.W. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Manon-Jensen, T.; Itoh, Y.; Couchman, J.R. Proteoglycansin health and disease. The multiple roles of syndecan shedding. FEBS J. 2010, 277, 3876–3889. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumor microenvironment, and angiogenesis. J. Cell Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Anttonen, A.; Eriksson, M.; Mákitaro, R.; Alfthan, H.; Kinnula, V.; Leppä, S. Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res. 2002, 62, 5210–5217. [Google Scholar] [PubMed]
- Seidel, C.; Sundan, A.; Hjorth, M.; Turesson, I.; Dahl, I.M.; Abildgaard, N.; Waage, A.; Borset, M. Serum syndecan-1: A new independent prognostic marker in multiple myeloma. Blood 2000, 95, 388–392. [Google Scholar] [PubMed]
- Ott, V.L.; Rapraeger, A.C. Tyrosine phosphorylation of syndecan-1 and -4 cytoplasmic domains in adherent B82 fibroblasts. J. Biol. Chem. 1998, 273, 35291–35298. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Iwamori, M.; Iwamori, Y.; Ito, N. Sulfated lipids as inhibitors of pancreatic trypsin and chymotrypsin in epithelium of the mammalian digestive tract. Biochem. Biophys. Res. Comm. 1997, 237, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Pruett, P.S.; Thompson, C.A.; DeLucas, L.D.; Sanderson, R.D. Heparan Sulfate Chains of Syndecan-1 Regulate Ectodomain Shedding. J. Biol. Chem. 2012, 287, 9952–9961. [Google Scholar] [CrossRef] [PubMed]
- Bode, L.; Salvestrini, C.; Park, P.-W.; Li, J.-P.; Esko, J.D.; Yamaguchi, Y.; Murch, S.; Freeze, H.H. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J. Clin. Invest. 2008, 118, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Levin, M.; Lionetti, P.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar] [CrossRef]
- Wang, J.Y.; Roehrl, M.H. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2002, 99, 14362–14367. [Google Scholar] [CrossRef] [PubMed]
- Parildar, Z.; Uslu, R.; Tanyalcin, T.; Doganavsargil, E.; Kutay, F. The urinary excretion of glycosaminoglycans and heparan sulphate in lupus nephritis. Clin. Rheumatol. 2002, 21, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowski, A.F.; Popp, S.K.; Freeman, C.; Parish, C.R.; Simeonovic, C.J. Heparan sulfate and heparanase play key roles in mouse cell survival and autoimmune diabetes. J. Clin. Invest. 2012, 122, 132–1421. [Google Scholar] [CrossRef] [PubMed]
- Gepts, W.; Lecompte, P.M. The pancreatic islets in diabetes. Am. J. Med. 1981, 70, 105–115. [Google Scholar] [CrossRef]
- Smeeth, L.; Juan, P.; Casas, J.P.; Hingorani, A.D. The role of infection in cardiovascular disease: more support but many questions remain. Eur. Heart J. 2007, 28, 1178–1179. [Google Scholar] [CrossRef] [PubMed]
- Kalayoglu, M.V.; Libby, P.; Byrne, G.I. Chlamydia pneumoniae as an Emerging Risk Factor in Cardiovascular Disease. JAMA 2002, 288, 2724–2731. [Google Scholar] [CrossRef] [PubMed]
- Saikku, P.; Leinonen, M.; Tenkanen, L.; Linnanmäki, E.; Ekman, M.R.; Manninen, V.; Mänttäri, M.; Frick, M.H.; Huttunen, J.K. Chronic Chlamydia pneumoniae infection as a risk factor for coronary heart disease in the Helsinki Heart Study. Ann. Intern. Med. 1992, 116, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen-Lathrop, S.J.; Koshiyama, K.; Phillips, N.; Stephens, R.S. Chlamydia-dependent biosynthesis of a heparan sulphate-like compound in eukaryotic cells. Cell Microbiol. 2000, 2, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, C.M.; Als-Nielsen, B.; Damgaard, M.; Hansen, J.F.; Hansen, S.; Helø, O.H.; Hildebrandt, P.; Hilden, J.; Jensen, G.B.; Kastrup, J.; et al. Randomised placebo controlled multicentre trial to assess short term clarithromycin for patients with stable coronary heart disease: CLARICOR trial. BMJ 2006, 332, 22. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Kinscherf, R. Aberrant insulin receptor signaling and amino acid homeostasis as a major cause of oxidative stress in aging. Antioxid. Redox Sign. 2008, 10, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Groffen, A.J.A.; Veerkamp, J.H.; Monnens, L.A.H.; van den Heuvel, L.P.W.J. Recent insights into the structure and functions of heparan sulfate proteoglycans in the human glomerular basement membrane. Nephrol. Dial. Transplant. 1999, 14, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Tamsma, J.T.; van den Born, J.; Bruijn, J.A.; Assmann, K.J.; Weening, J.J.; Berden, J.H.; Wieslander, J.; Schrama, E.; Hermans, J.; Veerkamp, J.H.; et al. Expression of glomerular extracellular matrix components in human diabetic nephropathy: Decrease of heparan sulphate in the glomerular basement membrane. Diabetologia 1994, 37, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Van den Born, J.; van Kraats, A.A.; Bakker, M.A.; Assmann, K.J.; van den Heuvel, L.P.; Veerkamp, J.H.; Berden, J.H. Selective proteinuria in diabetic nephropathy in the rat is associated with a relative decrease in glomerular basement membrane heparan sulphate. Diabetologia 1995, 38, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Fridén, V.; Dasgupta, I.; Foster, R.R.; Welsh, G.I.; Tooke, J.E.; Haraldsson, B.; Mathieson, P.W.; Satchell, S.C. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am. J. Physiol. Renal Physiol. 2011, 300, F40–F48. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D. Ions from the Hofmeister series and osmolytes: Effects on proteins in solution and in the crystallization process. Macromolecular Crystallization 2004, 34, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Akimoto, Y. The O-GlcNAc Modification. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 18. [Google Scholar]
- Buse, M.G.; Robinson, K.A.; Marshall, B.A.; Hresko, R.C.; Mueckler, M.M. Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1- overexpressing muscles. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E241–E250. [Google Scholar] [CrossRef] [PubMed]
- Dias, W.B.; Hart, G.W. O-GlcNAc modification in diabetes and Alzheimer's disease. Mol. BioSyst. 2007, 3, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Deniziak, M.A.; Barciszewski, J. Methionyl-tRNA synthetase. Acta Biochim. Pol. 2001, 48, 337–350. [Google Scholar] [PubMed]
- Jakubowski, H. Proofreading in vivo: Editing of homocysteine by methionyl-tRNA synthetase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 4504–4508. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Sarafanov, A.G.; Ananyeva, N.M.; Shima, M.; Saenko, E.L. Cell Surface heparan sulfate proteoglycans participate in factor VIII catabolism mediated by low density lipoprotein receptor-related protein. JBC 2001, 276, 11970–11979. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Strickland, D.K. LRP: A multifunctional scavenger and signaling receptor. J. Clin. Invest. 2001, 108, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Brickman, Y.G.; Ford, M.D.; Gallagher, J.T.; Nurcombe, V.; Bartlett, P.E.; E. Turnbull, J.E. Structural Modification of Fibroblast Growth Factor-binding Heparan Sulfate at a Determinative Stage of Neural Development. J. Biol. Chem. 1998, 273, 4350–4359. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.L.; Buczek-Thomas, J.A.; Nugent, M.A. Heparan sulphate proteoglycans modulate fibroblast growth factor-2 binding through a lipid raft-mediated mechanism. Biochem. J. 2004, 379, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. EJP 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Cordelier, P.; Esteve, J.-P.; Rivard, N.; Marletta, M.; Vaysse, N.; Susini, C.; and Buscail, L. The activation of neuronal NO synthase is mediated by G-protein βγ subunit and the tyrosine phosphatase SHP-2. FASEB J. 1999, 13, 2037–2050. [Google Scholar] [PubMed]
- Hopwood, J.J.; Dorfman, A. Glycosaminoglycan synthesis by cultured human skin fibroblasts after transformation with simian virus 40. J. Biol. Chem. 1977, 252, 4777–4785. [Google Scholar] [PubMed]
- Lajoix, A.-D.; Reggio, H.; Chardès, T.; Péraldi-Roux, S.; Tribillac, F.; Roye, M.; Dietz, S.; Broca, C.; Manteghetti, M.; Ribes, G.; et al. A Neuronal isoform of nitric oxide synthase expressed in pancreatic β-cells controls insulin secretion. Diabetes 2001, 50, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Kröncke, K.D.; Kolb-Bachofen, V.; Berschick, B.; Burkart, V.; Kolb, H. Activated macrophages kill pancreatic syngeneic islet cells via arginine-dependent nitric oxide generation. Biochem. Biophys. Res. Commun. 1991, 175, 752–758. [Google Scholar] [CrossRef]
- Gajkowska, B.; Gniadecki, R. Induction of endothelial nitric oxide synthase in perivascular mast cells in rat neurohypophysis after ischemia. Neuro. Endocrinol. Lett. 1999, 20, 189–193. [Google Scholar] [PubMed]
- Wu, K.; Aoki, C.; Elste, A.; Rogalski-Wilk, A.A.; Siekevitz, P. The synthesis of ATP by glycolytic enzymes in the postsynaptic density and the effect of endogenously generated nitric oxide. Proc. Natl. Acad. Sci. USA 1997, 94, 13273–13278. [Google Scholar] [CrossRef] [PubMed]
- Damon, D.H.; D’Amore, P.A.; Wagner, J.A. Sulfated glycosaminoglycans modify growth factor-induced neurite outgrowth in PC12 cells. J. Cell Physiol. 1988, 135, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lander, A.D.; Fujii, D.K.; Gospodarowicz, D.K.; Reichardt, L.F. Characterization of a factor that promotes neurite outgrowth: Evidence linking activity to a heparan sulfate proteoglycan. J. Cell Biol. 1982, 94, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Burbacha, B.J.; Friedl, A.; Mundhenke, C.; Rapraeger, A.C. Syndecan-1 accumulates in lysosomes of poorly differentiated breast carcinoma cells. Matrix Biol. 2003, 22, 163–177. [Google Scholar] [CrossRef]
- Ignarro, L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J. Physiol. Pharmacol. 2002, 53, 503–514. [Google Scholar] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Al-Magableh, M.R.; Hart, J.L. Mechanism of vasorelaxation and role of endogenous hydrogen sulfide production in mouse aorta. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Nagai, Y.; Umemura, K.; Kimura, Y. Physiological Roles of Hydrogen Sulfide: Synaptic Modulation, Neuroprotection, and Smooth Muscle Relaxation Antioxid. Redox Sign. 2005, 7, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen sulfide in biochemistry and medicine. Antioxid. Redox Sign. 2012, 17, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Pae, H.O.; Lee, B.S.; Kim, B.N.; Kim, J.M.; Kim, H.R.; Jeon, S.B.; Jeon, W.K.; Chae, H.J.; Chung, H.T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-κB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Bacher, S.; Schmitz, M.L. The NF-kB pathway as a potential target for autoimmune disease therapy. Curr. Pharm. Design 2004, 10, 2827–2837. [Google Scholar] [CrossRef]
- Hu, L.-F.; Lu, M.; Wu, Z.-Y.; Wong, P.T.-H.; Bian, J.S. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol. Pharmacol. 2009, 75, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.G.; Sass, N.L.; Hill, L.; Tarka, S.; Truex, R. The Synthesis of taurine from sulfate IV: An alternate pathway for taurine synthesis by the rat. Exp. Biol. Med. 1972, 141, 632–633. [Google Scholar] [CrossRef]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. Wiley Interdiscip. Rev. RNA 2011, 2, 376–386. [Google Scholar] [PubMed]
- Hansen, S.H.; Andersen, M.L.; Cornett, C.; Gradinaru, R.; Grunnet, N. A role for taurine in mitochondrial function. J. Biomed. Sci. 2010, 17, S23. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Sunami, O.; Nakajima, H.; Nishio, H.; Takeuchi, T.; Hata, F. Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochem. Pharmacol. 1999, 58, 133–143. [Google Scholar] [CrossRef]
- Song, S.; Finkel, T. GAPDH and the search for alternative energy. Nature Cell Biol. 2007, 9, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Ricci, J.-E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; et al. GAPDH and autophagy preserve survival after apoptotic cytochrome C release in the absence of caspase activation. Cell 2007, 129, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Ding, W.X.; Gao, W. Autophagy in the Liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic Degeneration and Death of Cardiomyocytes in Heart Failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence, of insulin resistance and hyperinsulinemia: Inhibition of Fox01-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Scherer, P.E. Adipose Tissue, Inflammation, and Cardiovascular Disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Yeagle, P.L. Cholesterol modulation of (Na+ + K+)-ATPase ATP hydrolyzing activity in the human erythrocyte. Biochim. Biophys. Acta 1983, 727, 39–44. [Google Scholar] [CrossRef]
- Nakazato, R.; Gransar, H.; Berman, D.S.; Cheng, V.Y.; Lin, F.Y.; Achenbach, S.; Al-Mallah, M.; Budoff, M.J.; Cademartiri, F.; Callister, T.Q.; et al. Statins use and coronary artery plaque composition: Results from the International Multicenter CONFIRM Registry. Atherosclerosis 2012, 225, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.S.; Hindle, E.; Dyer, T. Sunlight, cholesterol and coronary heart disease. QJM 1996, 89, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Michos, E.D.; Blumenthal, R.S. Vitamin D supplementation and cardiovascular disease risk. Circulation 2007, 115, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Siegel, N.; Haug, A. Aluminum interaction with calmodulin. Evidence for altered structure and function from optical and enzymatic studies. Biochim. Biophys. Acta 1983, 744, 36–45. [Google Scholar] [CrossRef]
- Rearick, J.I.; Jetten, A.M. Accumulation of cholesterol 3-sulfate during in vitro squamous differentiation of rabbit tracheal epithelial cells and its regulation by retinoids. J. Biol. Chem. 1986, 261, 13898–13904. [Google Scholar] [PubMed]
- Halliday, G.M.; Robertson, B.O.; Barnetson, R.C. Topical retinoic acid enhances, and a dark tan protects, from subedemal solar-simulated photocarcinogenesis. J. Invest. Dermatol. 2000, 114, 923–927. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Seneff, S.; Lauritzen, A.; Davidson, R.; Lentz-Marino, L. Is Endothelial Nitric Oxide Synthase a Moonlighting Protein Whose Day Job is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease. Entropy 2012, 14, 2492-2530. https://doi.org/10.3390/e14122492
Seneff S, Lauritzen A, Davidson R, Lentz-Marino L. Is Endothelial Nitric Oxide Synthase a Moonlighting Protein Whose Day Job is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease. Entropy. 2012; 14(12):2492-2530. https://doi.org/10.3390/e14122492
Chicago/Turabian StyleSeneff, Stephanie, Ann Lauritzen, Robert Davidson, and Laurie Lentz-Marino. 2012. "Is Endothelial Nitric Oxide Synthase a Moonlighting Protein Whose Day Job is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease" Entropy 14, no. 12: 2492-2530. https://doi.org/10.3390/e14122492
APA StyleSeneff, S., Lauritzen, A., Davidson, R., & Lentz-Marino, L. (2012). Is Endothelial Nitric Oxide Synthase a Moonlighting Protein Whose Day Job is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease. Entropy, 14(12), 2492-2530. https://doi.org/10.3390/e14122492