
A Synthetic Virus-Like Particle Streptococcal Vaccine Candidate Using B-Cell Epitopes from the Proline-Rich Region of Pneumococcal Surface Protein A

Abstract
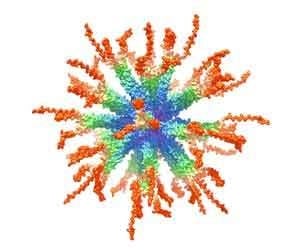
:
1. Introduction
2. Methods and Materials
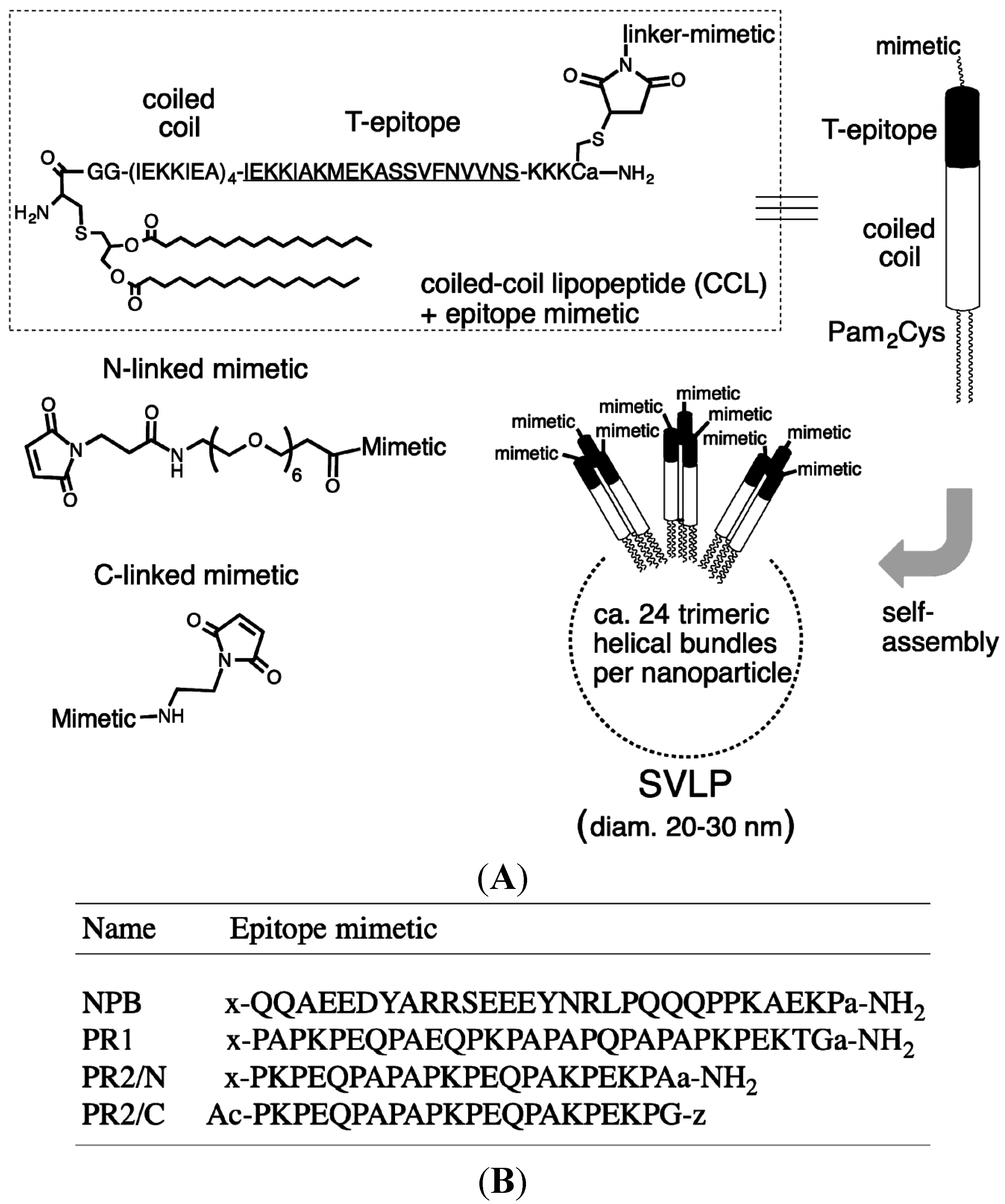
2.1. Synthesis of Peptides and Coiled-Coil Lipopeptides
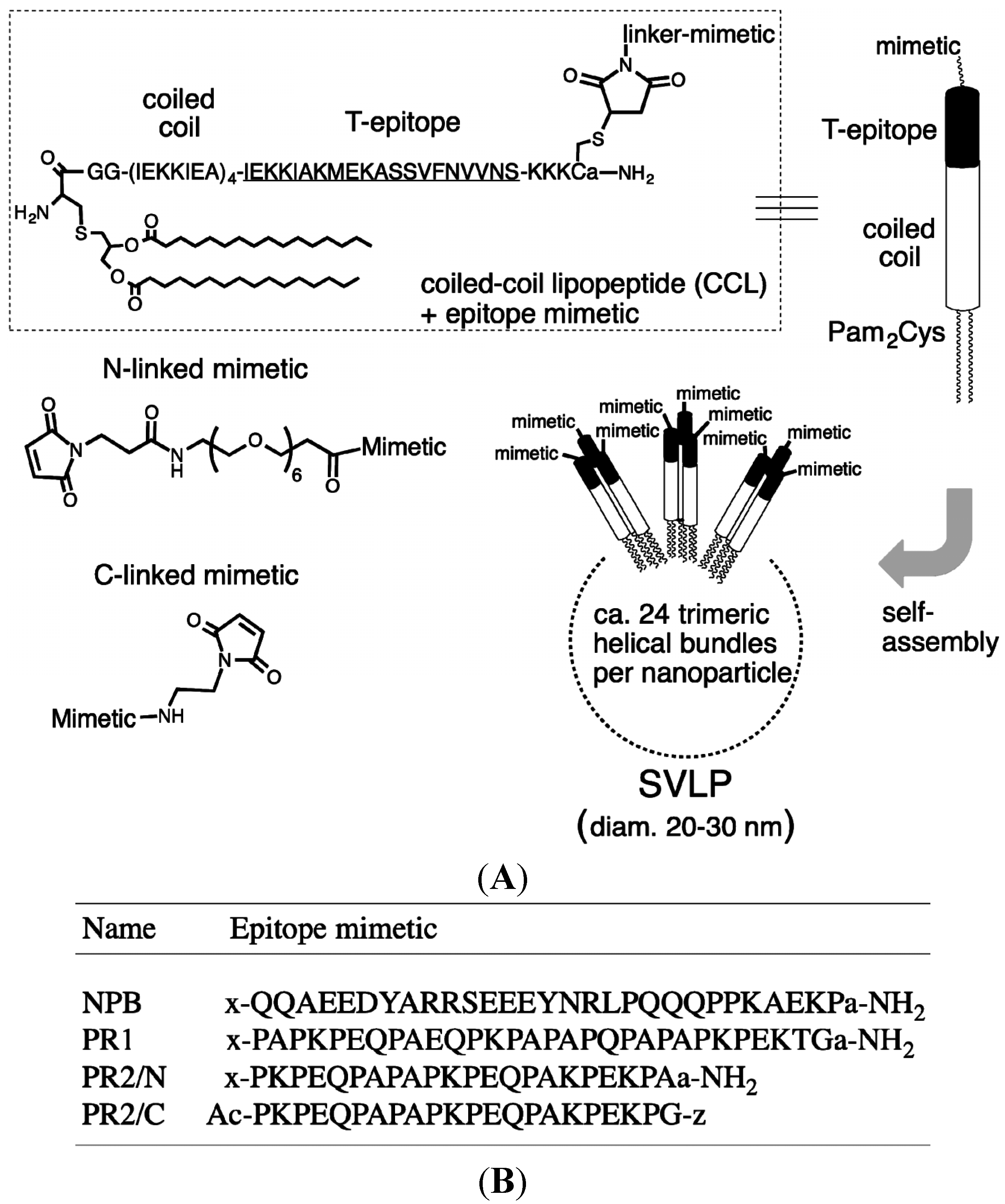
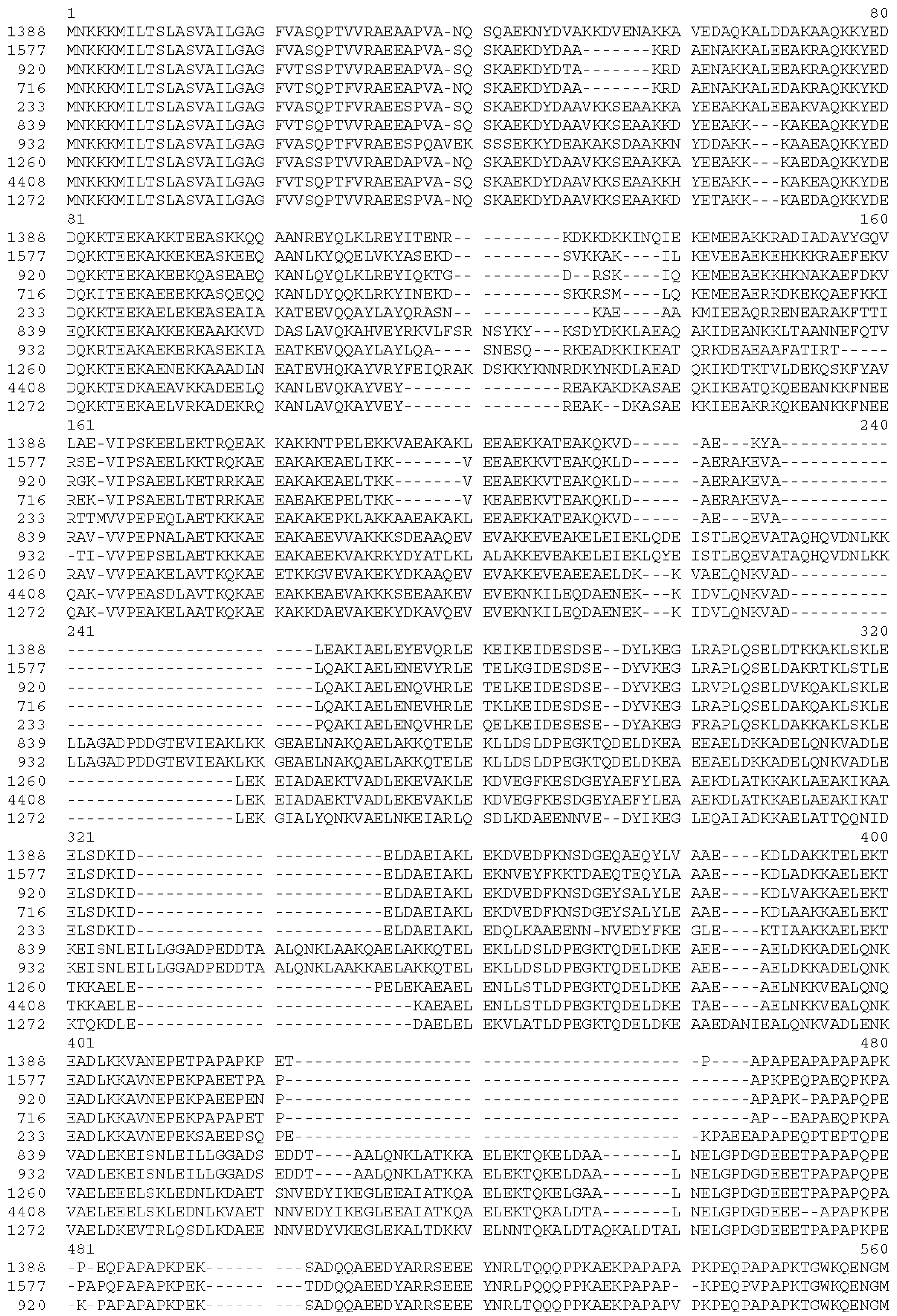
2.2. Cloning and Expression of the PRR from PspA Strain 1577 (rPspA)
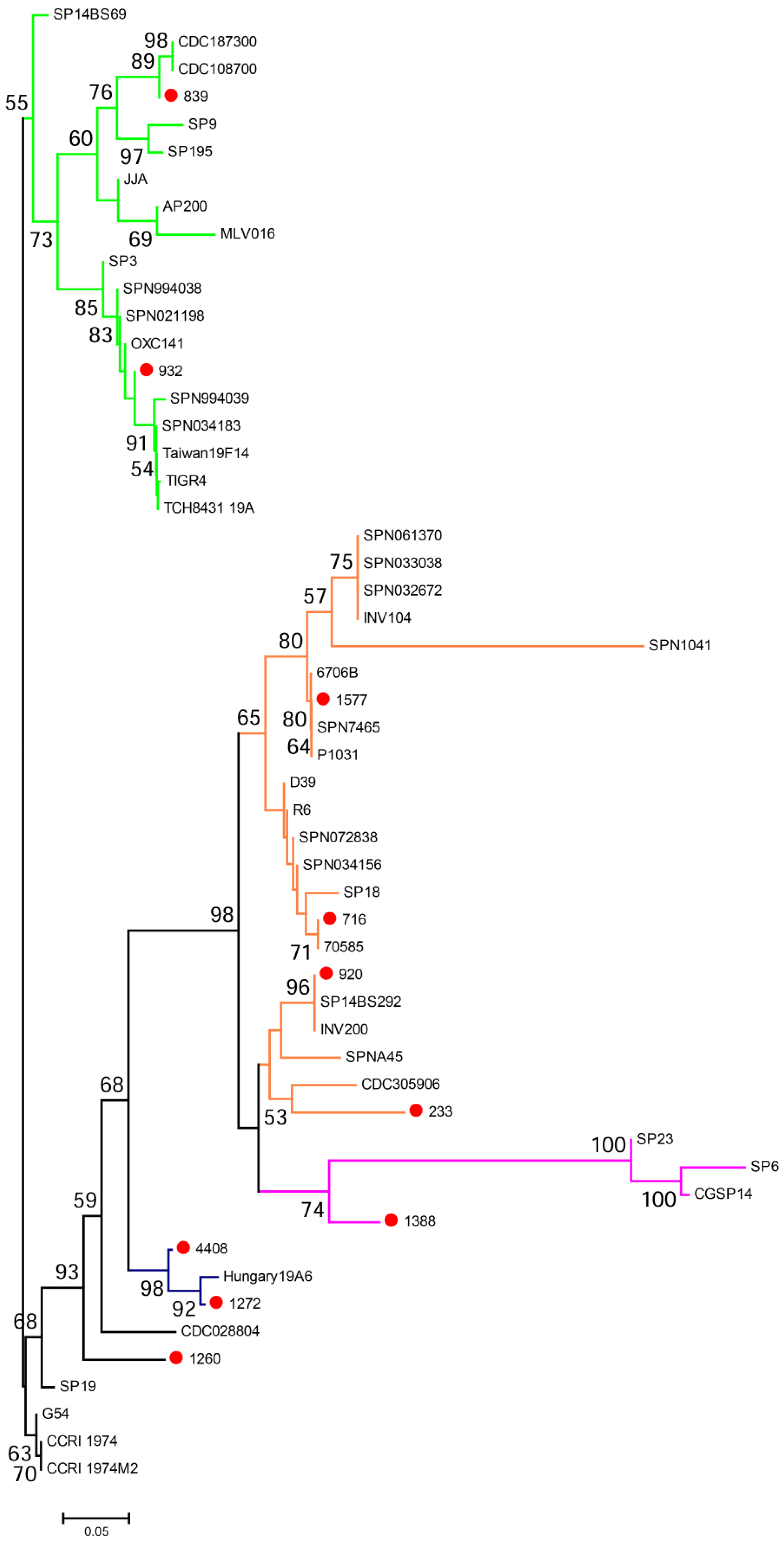
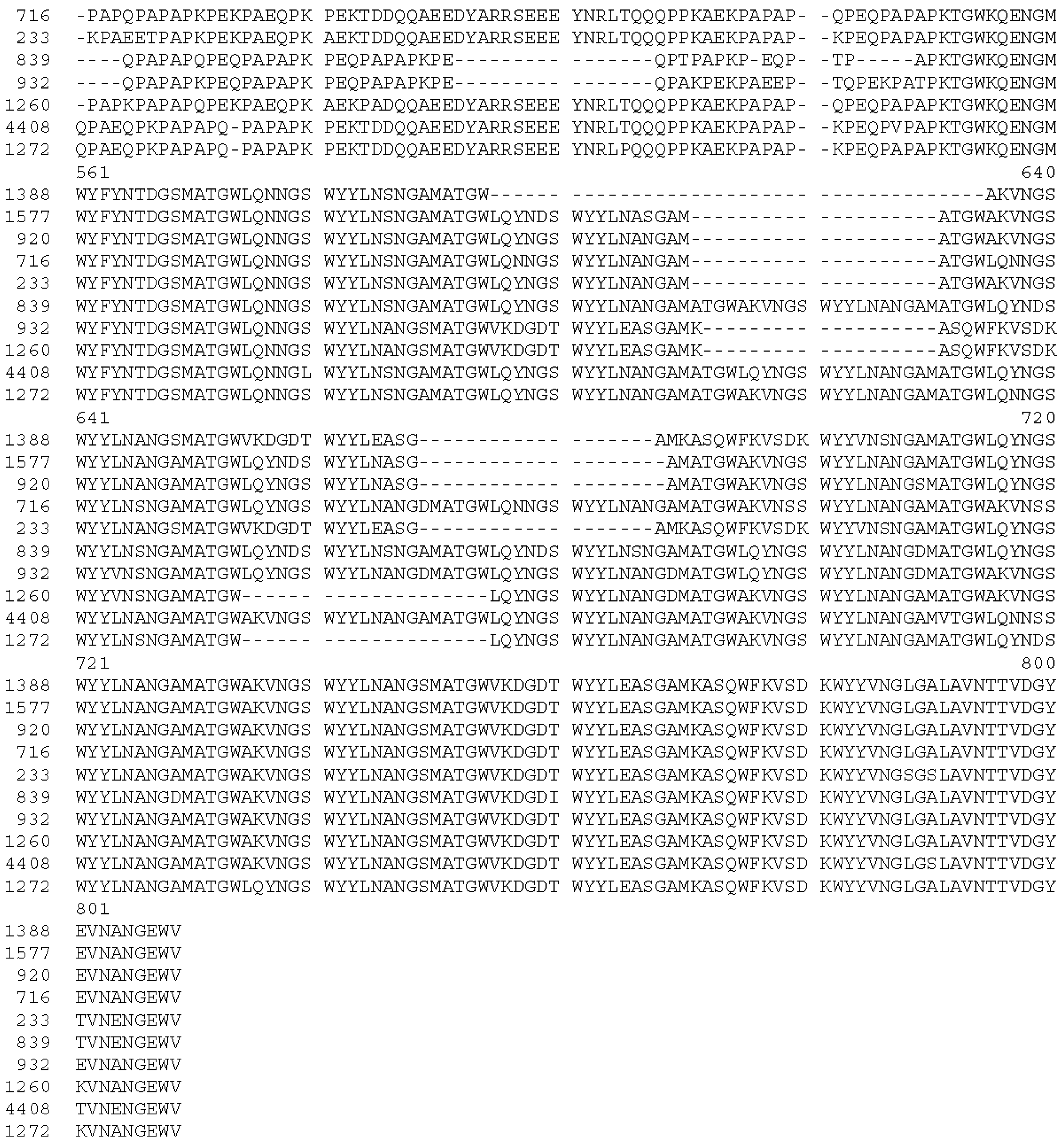
2.3. Pneumococcal Strains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Diagnosis of Patient | Site of Isolation | Origin | Year Isolated | pspA Clade | Capsule Type | Non-Proline Block | Cross-Reactivity of Sera Induced by Immunogen | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CCL-NPB | CCL-PR1 | CCL-PR2/N | CCL-PR2/C | ||||||||
| 1577 | case | CSF | Ghana | 2009 | 1 | 1 | present | + | + | + | + |
| 233 | case | CSF | PNG, Chimbu | 1997 | 1 | 4 | present | + | + | + | + |
| 716 | case | CSF | PNG, Lufa | 1999 | 1 | 5 | present | + | + | − | − |
| 920 | carriage | Naso-pharynx | PNG, Eastern Highlands | 2006 | 1 | 8 | present | + | + | + | + |
| 1388 | case | CSF | PNG, Lufa | 2002 | 2 | 4 | present | + | + | + | + |
| 932 | case | CSF | PNG, Goroka | 1999 | 3 | 6A/B | absent | − | + | + | + |
| 839 | case | CSF | PNG, Ung/Bena | 1999 | 3 | 7F | absent | − | − | + | + |
| 1260 | case | CSF | PNG, Goroka | 2001 | 4 | 4 | present | + | + | − | − |
| 1272 | case | CSF | PNG, Daulo | 2002 | 5 | 2 | present | + | + | + | + |
| 4408 | carriage | Naso-pharynx | PNG, Eastern Highlands | 2010 | 5 | 19A | present | + | + | + | + |
2.4. Animal Studies
2.5. Generation of Hybridoma Cell Lines Producing Epitope Mimetic-Specific mAbs
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. SDS-PAGE and Immunoblotting
2.8. Surface Staining of Bacterial Cells
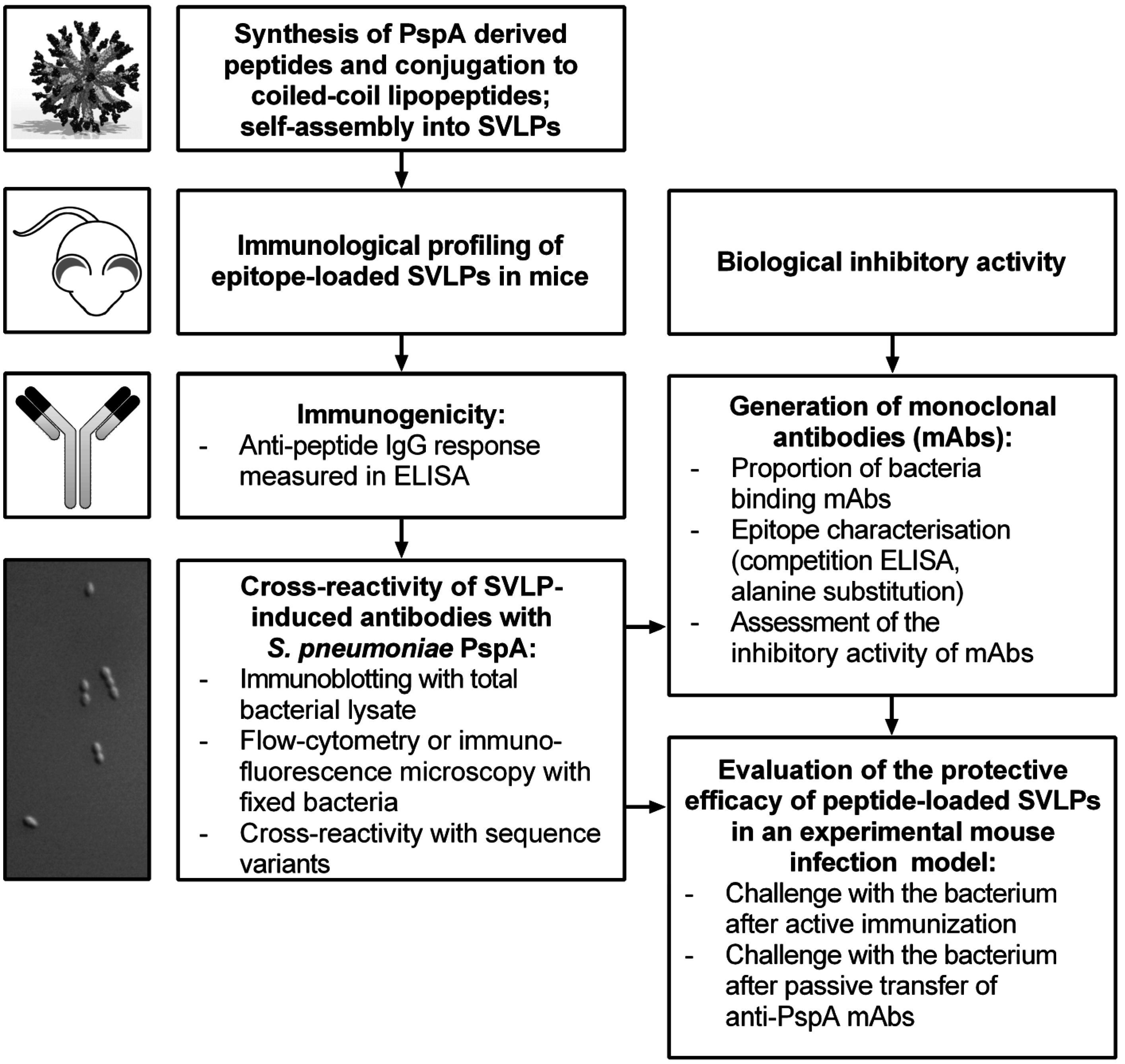
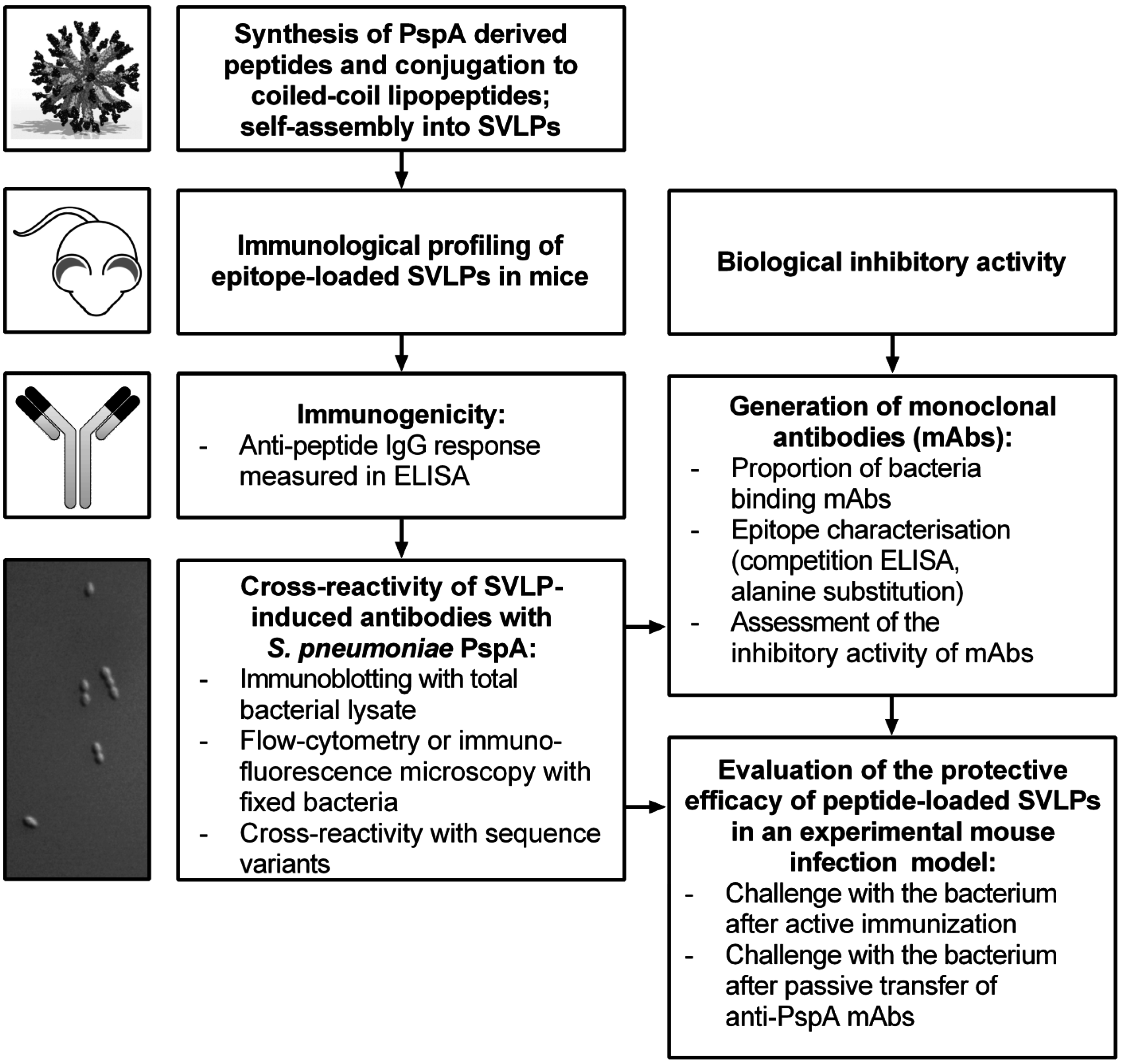
3. Results
3.1. Design and Synthesis of Epitope-Loaded SVLPs
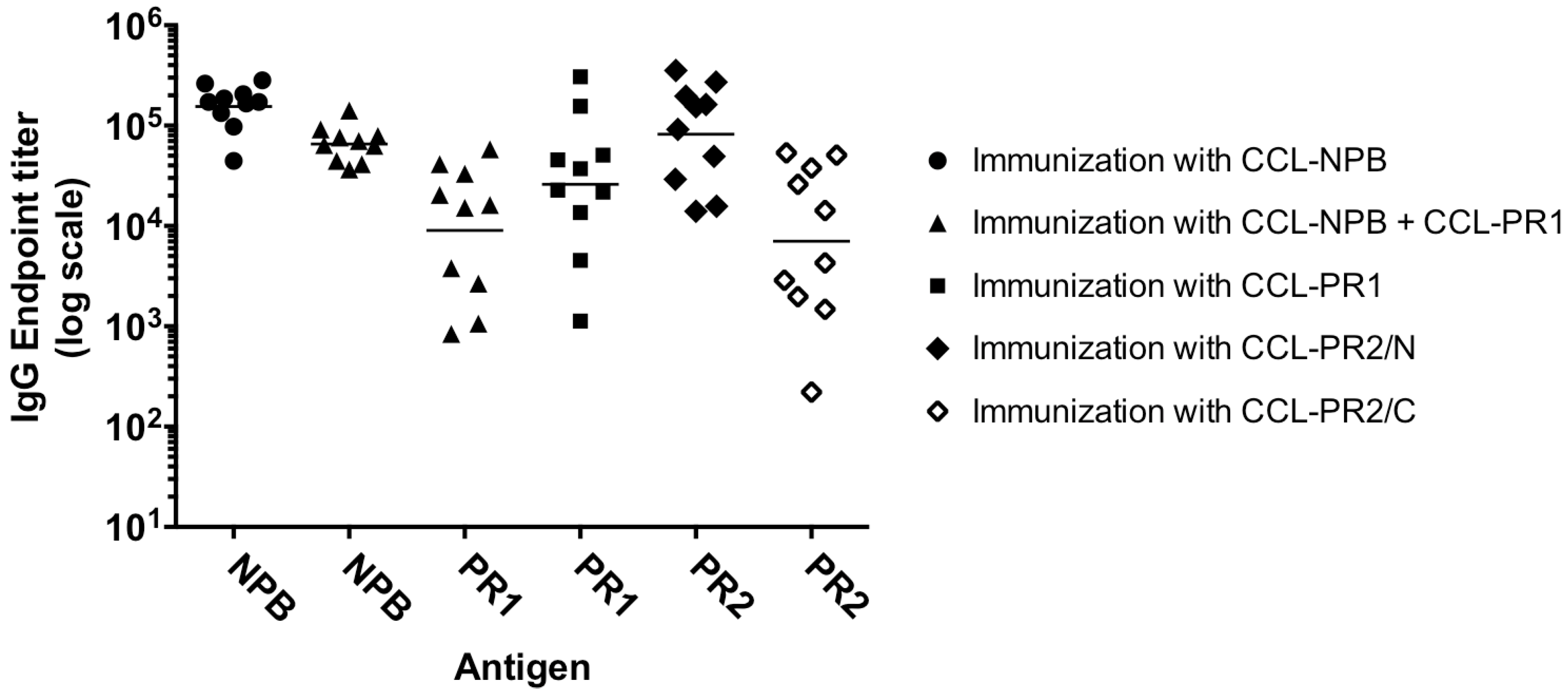
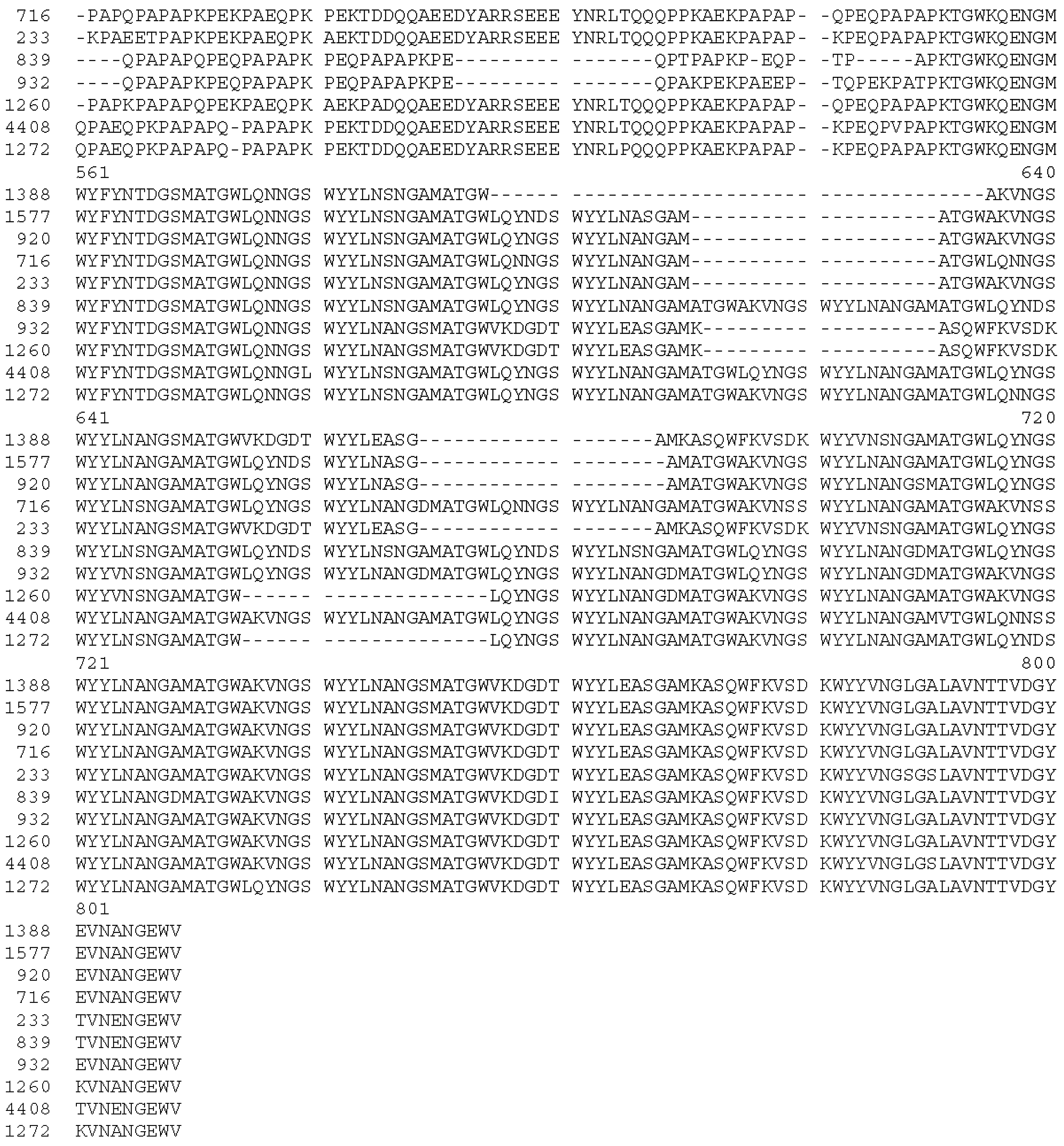
3.2. Immunogenicity of Epitope-Loaded SVLPs in Mice
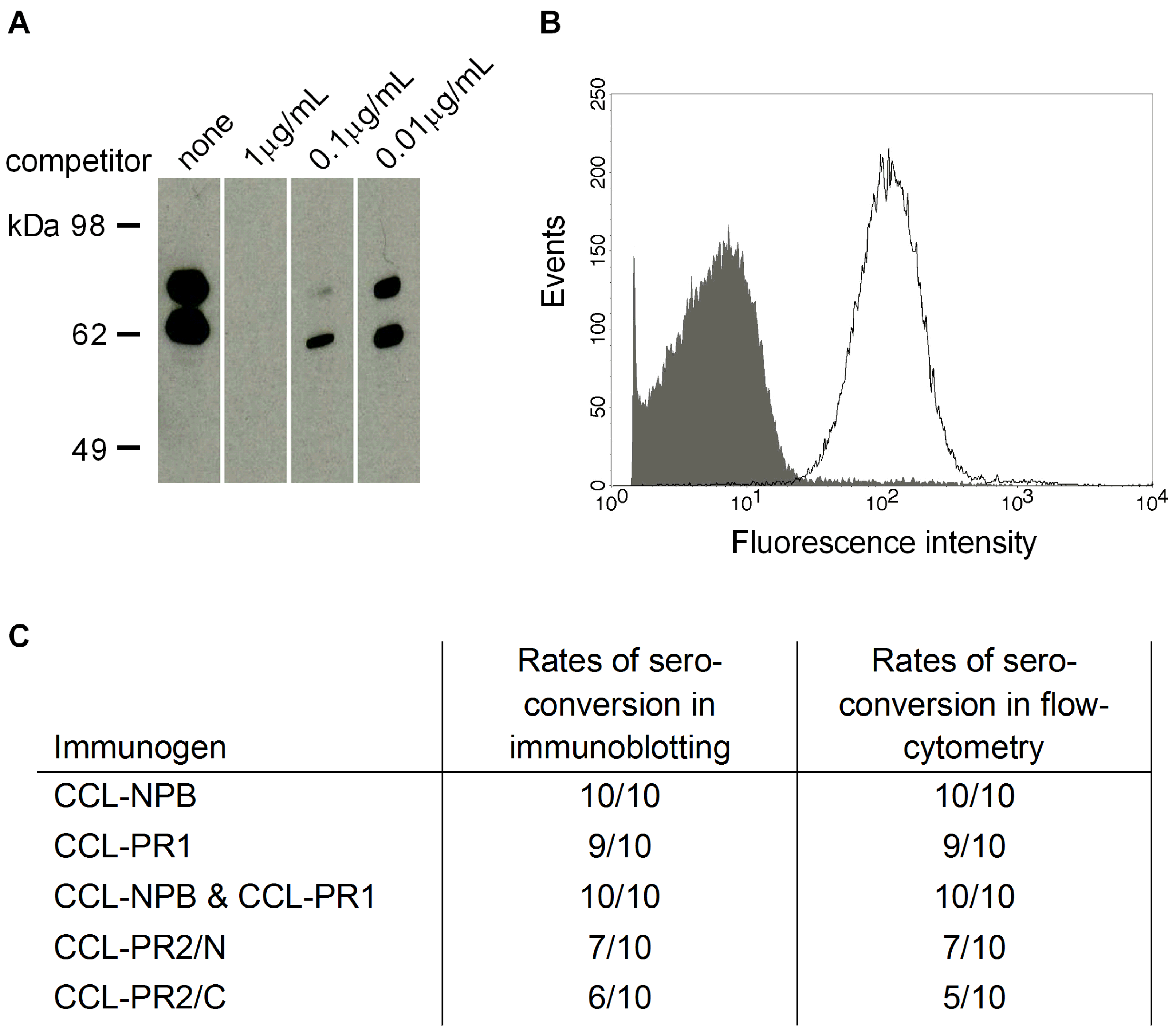
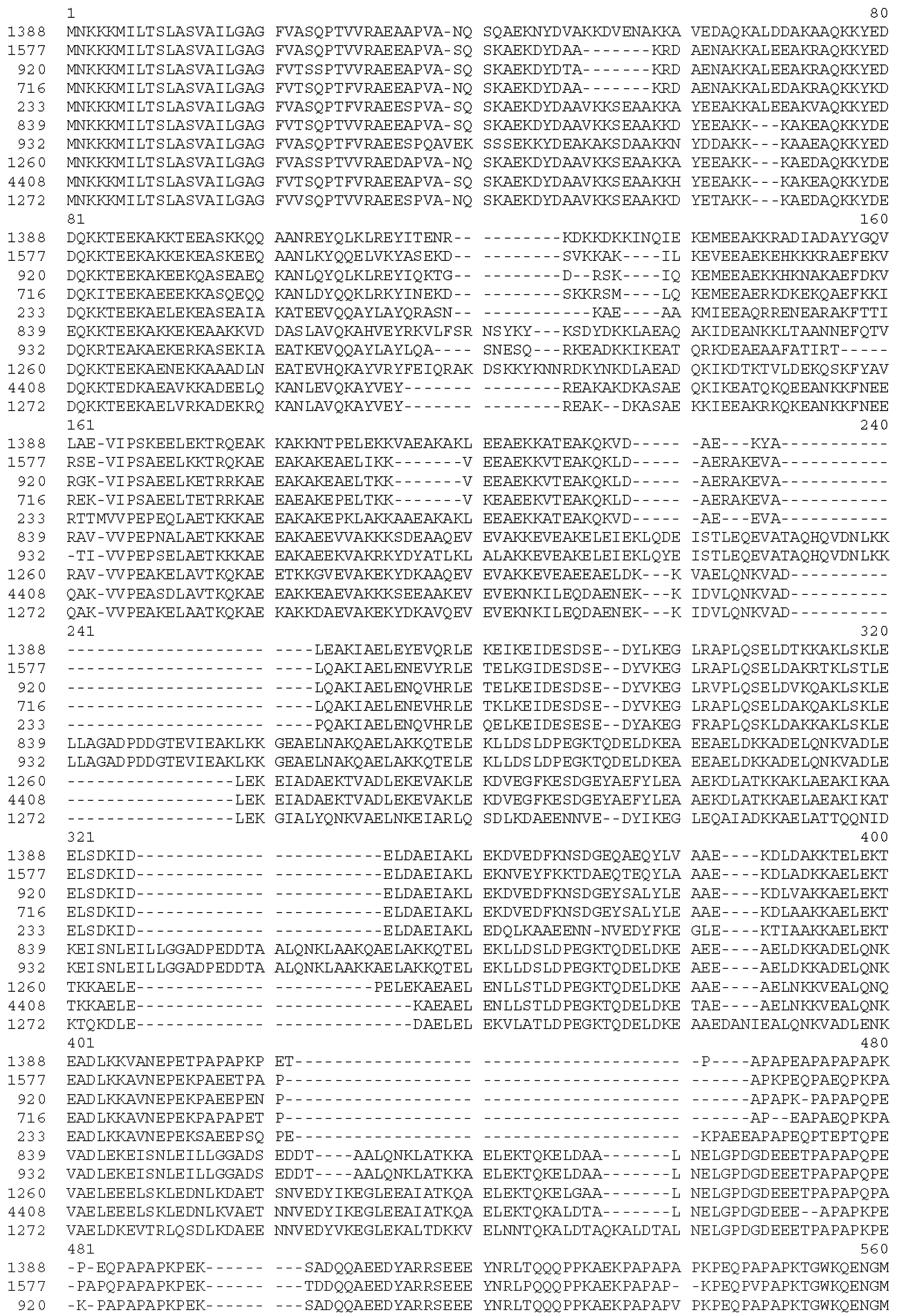
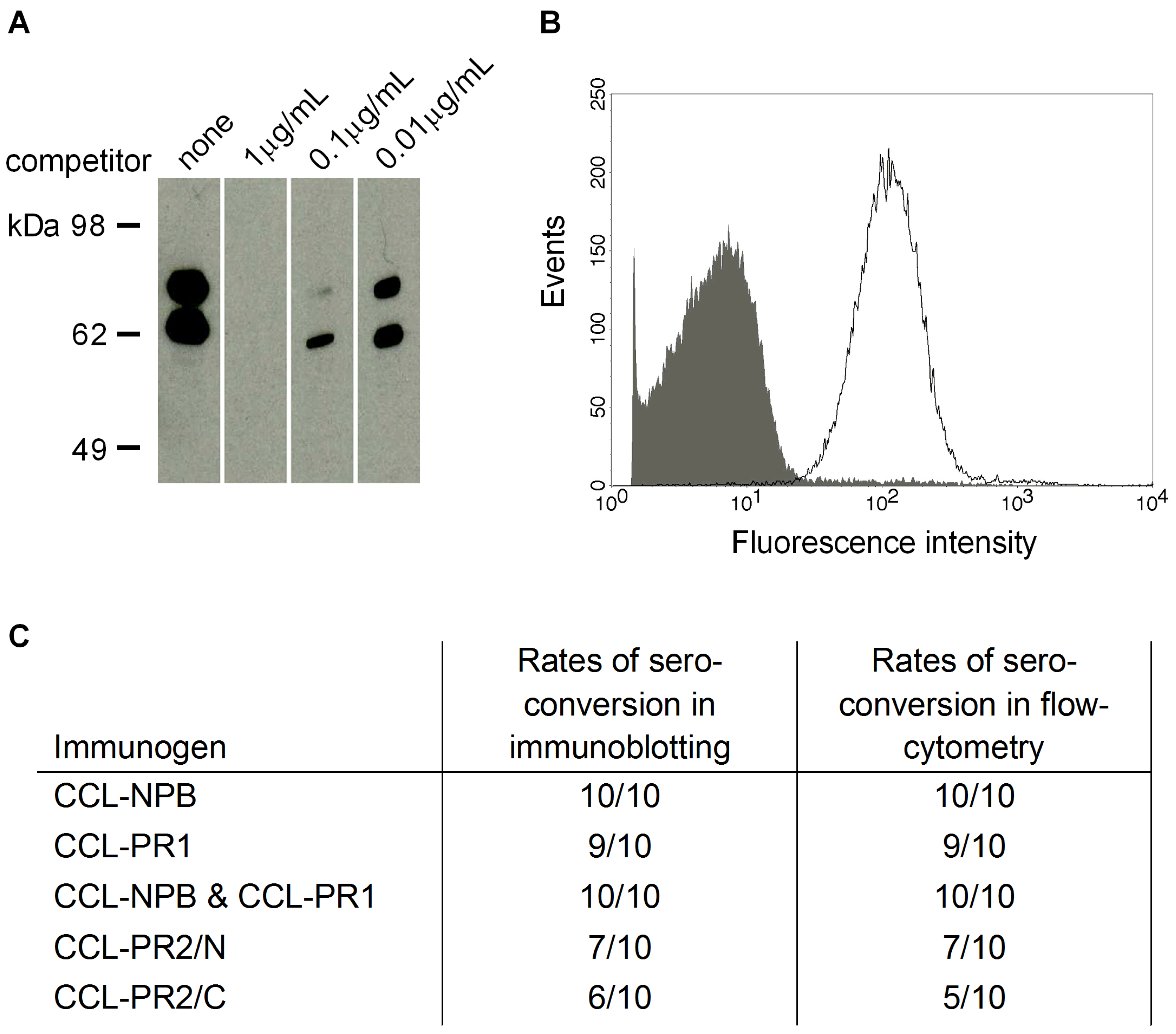
3.3. Cross-Reactivity of Induced Antibodies with S. Pneumoniae PspA

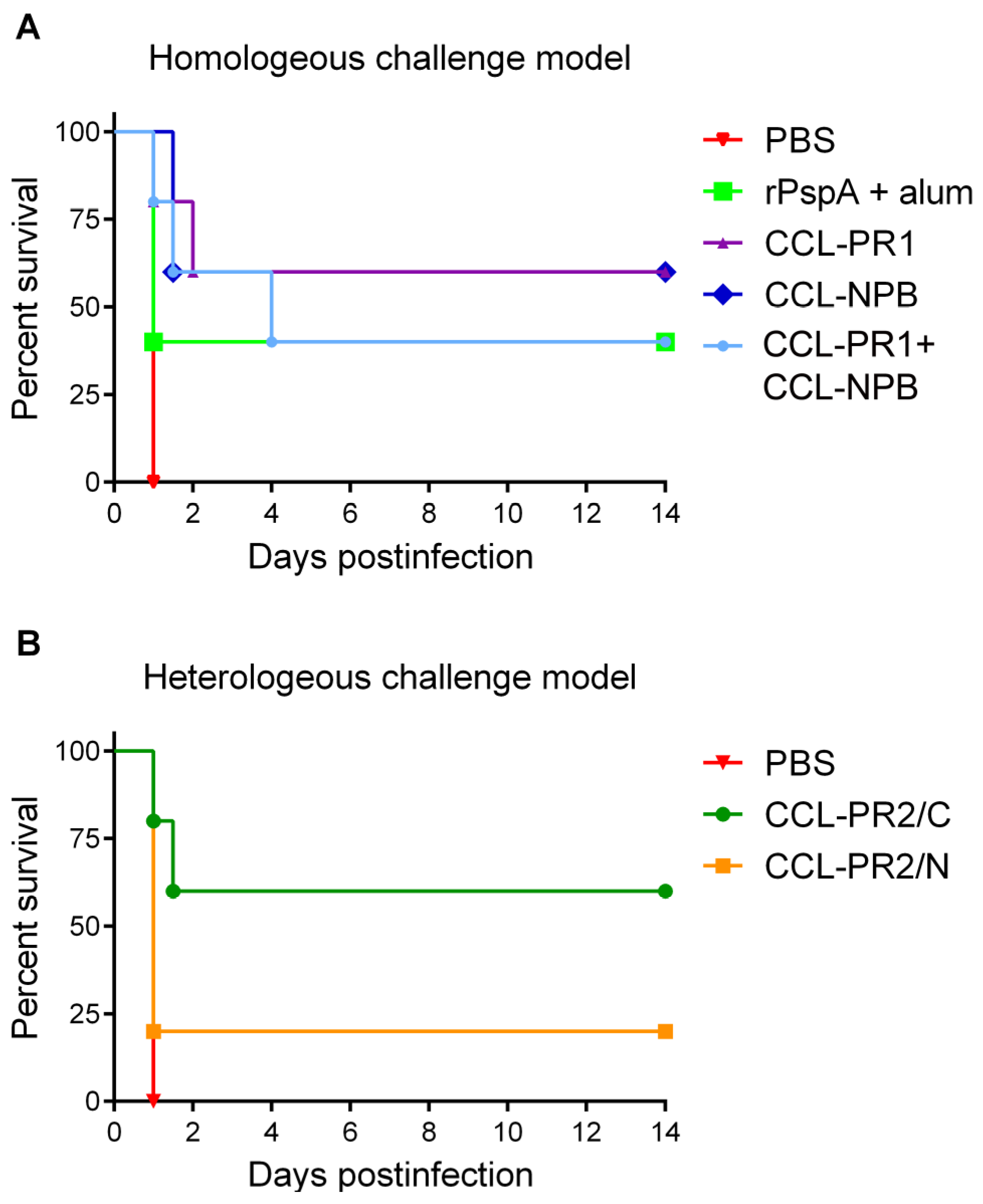
3.4. Cross-Reactivity with Strains Expressing Heterologous PspA
3.5. Protective Capacity of Epitope-Loaded SVLPs
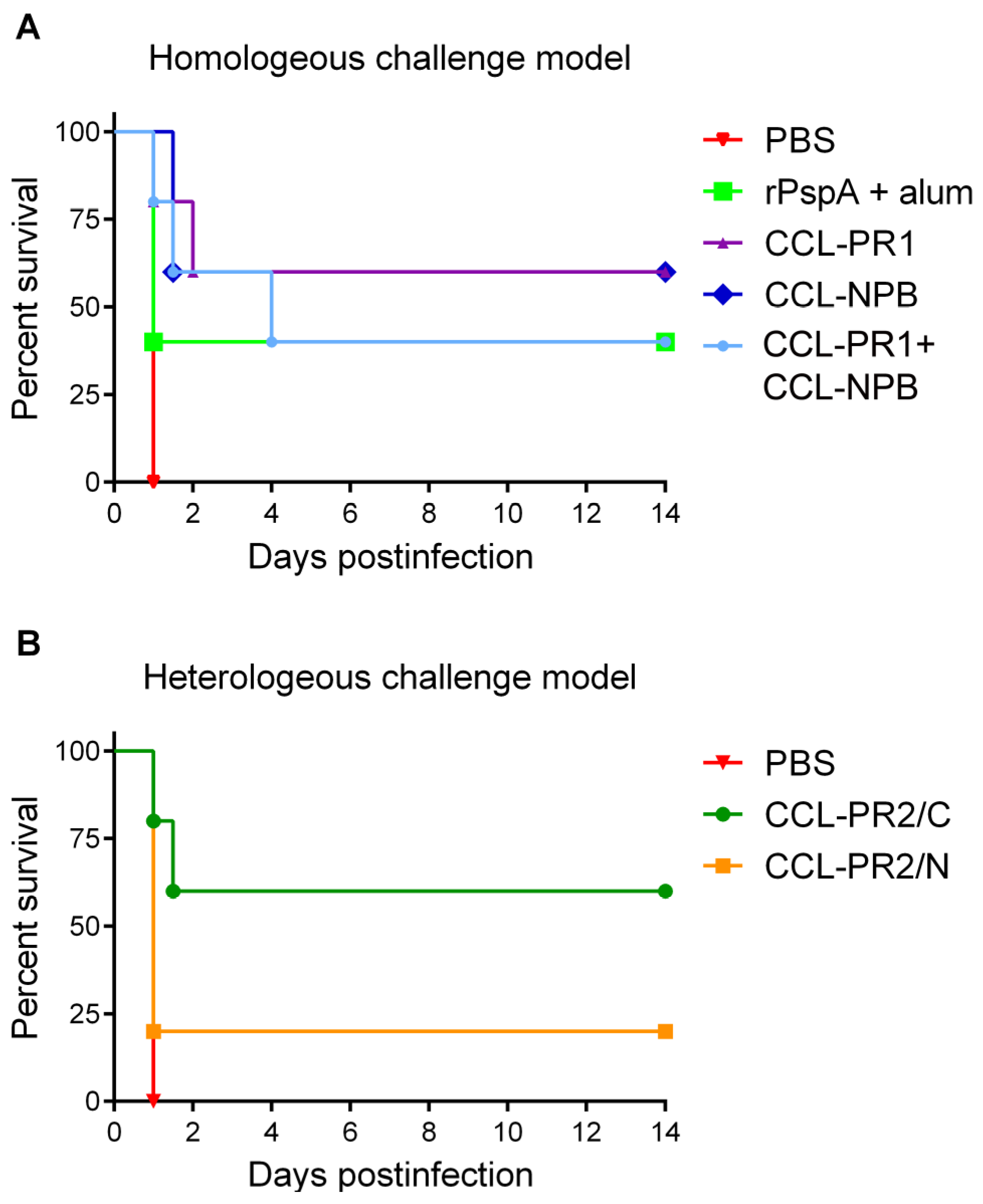
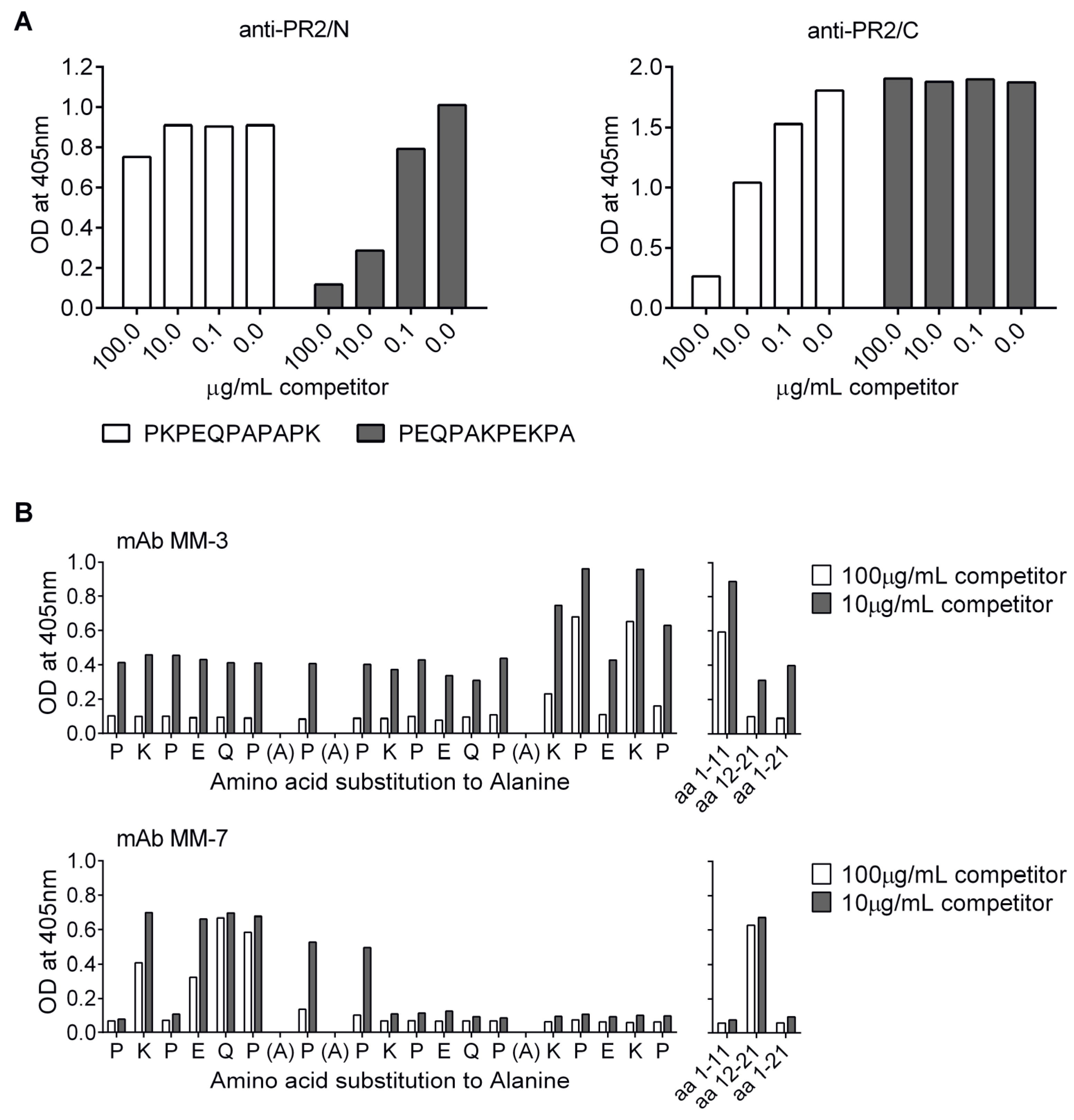
3.6. Localization of Cross-Protective Epitopes in PR2
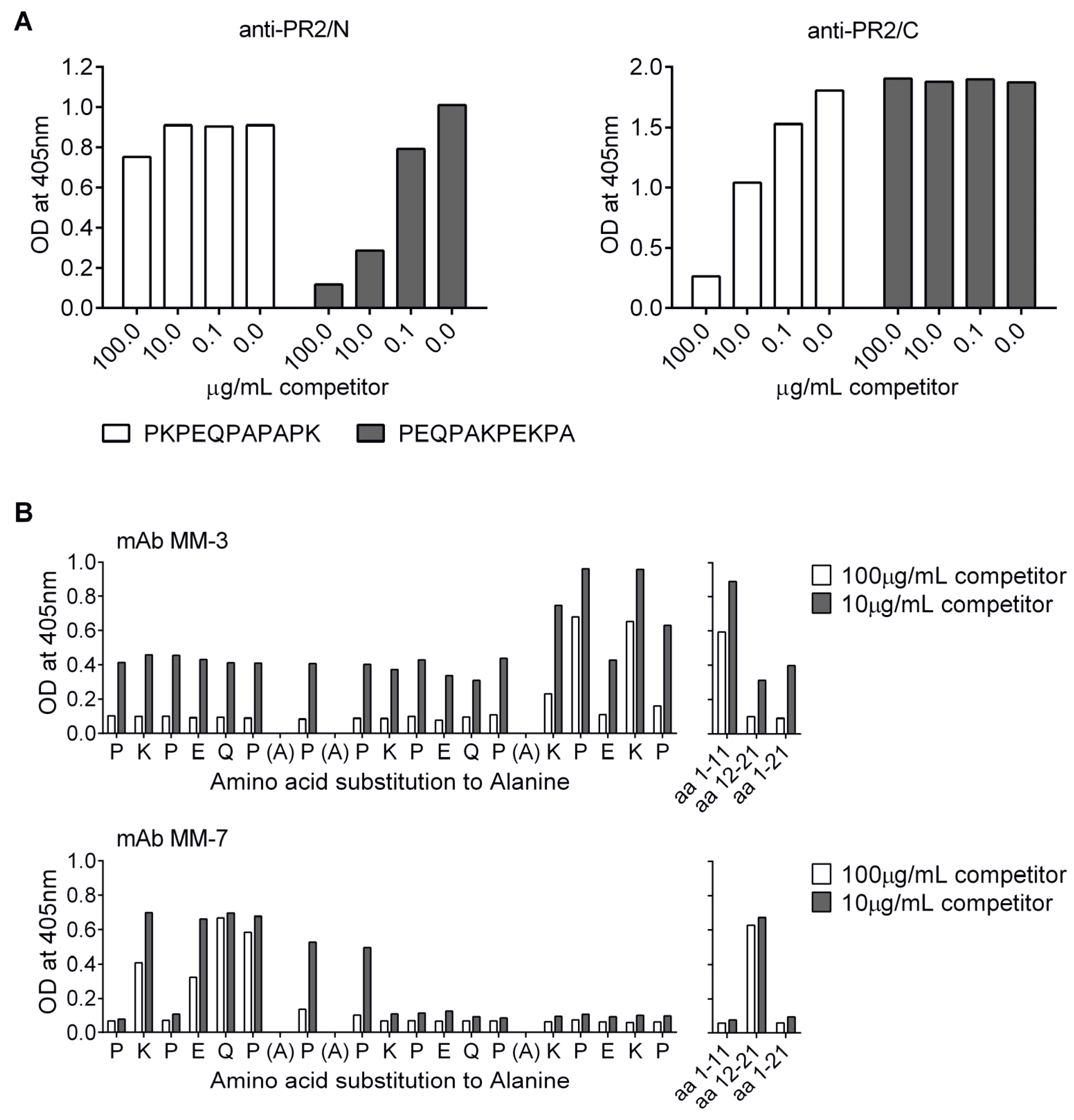
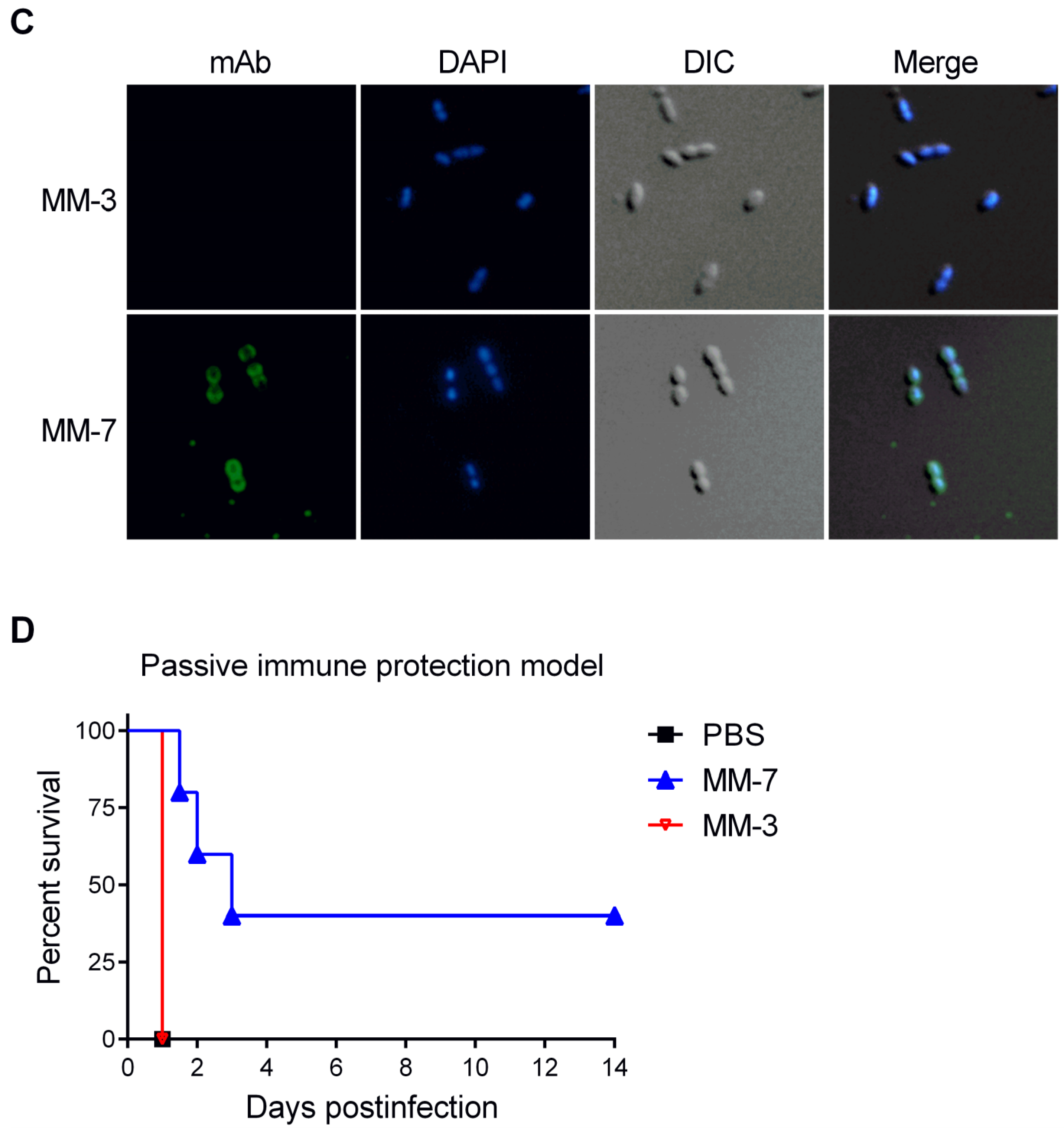
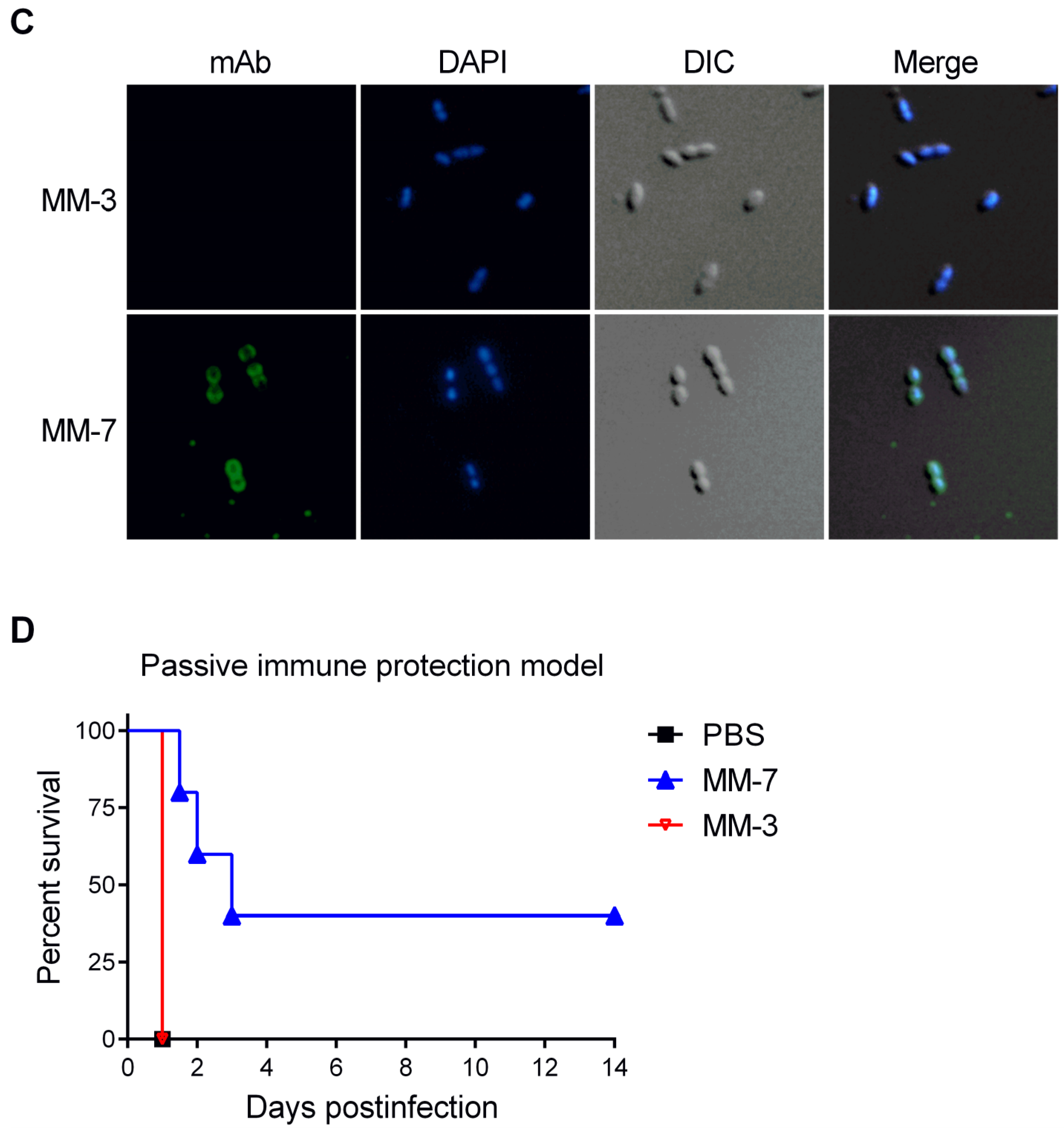
3.7. Protection by Passive Transfer of Anti-PspA mAbs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

| Primer | Sequence |
|---|---|
| PspA1 | GCAAAAGTAGTGTCTTTATCTTTTTGG |
| PspA2 | CCAGCGTCGCTATCTTAGGGGCTGGTT |
| PspA3 | CGAACATAAATCTATTCAATCGAACC |
| PspA4 | AATCAACACCACCACTCATCC |
| PspA5 | ACCACATACCGTTTTCTTGTTTCC |
| PspA6 | TGGAAACAAGAAAACGGTATGTGG |
| PspA7 | CGAACATAAATCTATTCAATCGAACC |
| PspA8 | AGGTCGTGTGTGCTTCACG |
| PspA9 | GGAAACAAGAAAACGGTATGTGGT |
| PspA10 | TCAACAAGCTGAAGAAGACTATGC |
| PspA11 | AATGAGTTAGGCCCTGATGG |
| PspA12 | CGGCTTAAACCCATTCACC |
| PspA13 | TCTCCATCTTTCACCCAACC |
| PspA14 | AATGAGTTAGGCCCTGATGG |

References
- Blasi, F.; Mantero, M.; Santus, P.; Tarsia, P. Understanding the burden of pneumococcal disease in adults. Clin. Microbiol. Infect. 2012, 18, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Drijkoningen, J.J.C.; Rohde, G.G.U. Pneumococcal infection in adults: Burden of disease. Clin. Microbiol. Infect. 2014, 20, 45–51. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- Feldman, C.; Anderson, R. Review: Current and new generation pneumococcal vaccines. J. Infect. 2014, 69, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Tarahomjoo, S. Recent approaches in vaccine development against Streptococcus pneumoniae. J. Mol. Microbiol. Biotechnol. 2014, 24, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, P.R. Pneumococcal conjugate vaccine for adults: A new paradigm. Clin. Infect. Dis. 2012, 55, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Poolman, J.T.; Peeters, C.; van den Dobbelsteen, G. The history of pneumococcal conjugate vaccine development: Dose selection. Exp. Rev. Vaccines 2013, 12, 1379–1394. [Google Scholar] [CrossRef] [PubMed]
- Pilishvili, T.; Lexau, C.; Farley, M.M.; Hadler, J.; Harrison, L.H.; Bennett, N.M.; Reingold, A.; Thomas, A.; Schaffner, W.; Craig, A.S.; et al. Sustained Reductions in Invasive Pneumococcal Disease in the Era of Conjugate Vaccine. J. Infect. Dis. 2010, 201, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, R.A.; Jefferies, J.M.; Faust, S.N.; Clarke, S.C. Pneumococcal 13-valent conjugate vaccine for the prevention of invasive pneumococcal disease in children and adults. Exp. Rev. Vaccines 2012, 11, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, A.S.; Alderson, M.R. New conjugate vaccines for the prevention of pneumococcal disease in developing countries. Drugs Today 2011, 47, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Brandau, D.T.; Jones, L.S.; Wiethoff, C.M.; Rexroad, J.; Middaugh, C.R. Thermal stability of vaccines. J. Pharm. Sci. 2003, 92, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Dagan, R.; Goldblatt, D.; Maleckar, J.R.; Yaïch, M.; Eskola, J. Reduction of Antibody Response to an 11-Valent Pneumococcal Vaccine Coadministered with a Vaccine Containing Acellular Pertussis Components. Infect. Immun. 2004, 72, 5383–5391. [Google Scholar] [CrossRef] [PubMed]
- Singleton, R.J.; Hennessy, T.W.; Bulkow, L.R.; Hammitt, L.L.; Zulz, T.; Hurlburt, D.A.; Butler, J.C.; Rudolph, K.; Parkinson, A. INvasive pneumococcal disease caused by nonvaccine serotypes among alaska native children with high levels of 7-valent pneumococcal conjugate vaccine coverage. J. Am. Med. Assoc. 2007, 297, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.M.; Malley, R.; Lipsitch, M. Serotype replacement in disease after pneumococcal vaccination. Lancet 2011, 378, 1962–1973. [Google Scholar] [CrossRef]
- Wyres, K.L.; Lambertsen, L.M.; Croucher, N.J.; McGee, L.; von Gottberg, A.; Linares, J.; Jacobs, M.R.; Kristinsson, K.G.; Beall, B.W.; Klugman, K.P.; et al. Pneumococcal Capsular Switching: A Historical Perspective. J. Infect. Dis. 2013, 207, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, E.N.; Oliveira, M.L.S.; Carvalho, E.; Ho, P.L. Serotype-independent pneumococcal vaccines. Cell. Mol. Life Sci. 2013, 70, 3303–3326. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, A.S.; Nahm, M.H.; Khambaty, F.M.; Alderson, M.R. Issues and challenges in the development of pneumococcal protein vaccines. Expert Rev. Vaccines 2012, 11, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Tobin, G.J.; Trujillo, J.D.; Bushnell, R.V.; Lin, G.; Chaudhuri, A.R.; Long, J.; Barrera, J.; Pena, L.; Grubman, M.J.; Nara, P.L. Deceptive imprinting and immune refocusing in vaccine design. Vaccine 2008, 26, 6189–6199. [Google Scholar] [CrossRef] [PubMed]
- Boato, F.; Thomas, R.M.; Ghasparian, A.; Freund-Renard, A.; Moehle, K.; Robinson, J.A. Synthetic virus-like particles from self-assembling coiled-coil lipopeptides and their use in antigen display to the immune system. Angew. Chem. Int. Ed. 2007, 46, 9015–9018. [Google Scholar] [CrossRef] [PubMed]
- Ghasparian, A.; Riedel, T.; Koomullil, J.; Moehle, K.; Gorba, C.; Svergun, D.I.; Perriman, A.W.; Mann, S.; Tamborrini, M.; Pluschke, G.; et al. Engineered Synthetic Virus-Like Particles and Their Use in Vaccine Delivery. Chembiochem 2011, 12, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Perriman, A.W.; Williams, D.S.; Jackson, A.J.; Grillo, I.; Koomullil, J.M.; Ghasparian, A.; Robinson, J.A.; Mann, S. Synthetic viruslike particles and hybrid constructs based on lipopeptide self-assembly. Small 2010, 6, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Ghasparian, A.; Moehle, K.; Rusert, P.; Trkola, A.; Robinson, J.A. Synthetic Virus-Like Particles and Conformationally Constrained Peptidomimetics in Vaccine Design. Chembiochem 2011, 12, 2829–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effio, C.L.; Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015, 10, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Limas, W.A.; Sekar, K.; Tyo, K.E.J. Virus-like particles: The future of microbial factories and cell-free systems as platforms for vaccine development. Curr. Opin. Biotechnol. 2013, 24, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xin, L.; Li, S.W.; Fang, M.J.; Zhang, J.; Xia, N.S.; Zhao, Q.J. Lessons learned from successful human vaccines: Delineating key epitopes by dissecting the capsid proteins. Hum. Vaccines Immunother. 2015, 11, 1277–1292. [Google Scholar] [CrossRef] [PubMed]
- Mukerji, R.; Mirza, S.; Roche, A.M.; Widener, R.W.; Croney, C.M.; Rhee, D.-K.; Weiser, J.N.; Szalai, A.J.; Briles, D.E. Pneumococcal Surface Protein A Inhibits Complement Deposition on the Pneumococcal Surface by Competing with the Binding of C-Reactive Protein to Cell-Surface Phosphocholine. J. Immunol. 2012, 189, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Li, J.; Genschmer, K.; Hollingshead, S.K.; Briles, D.E. The Absence of PspA or Presence of Antibody to PspA Facilitates the Complement-Dependent Phagocytosis of Pneumococci In Vitro. Clin. Vaccines Immunol. 2012, 19, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Shaper, M.; Wilson, L.; Benjamin, W.H.; Novak, J.; Barnes, S.; Hollingshead, S.K.; Briles, D.E. Serine protease PrtA from Streptococcus pneumoniae plays a role in the killing of S. pneumoniae by apolactoferrin. Infect. Immun. 2011, 79, 2440–2450. [Google Scholar]
- Shaper, M.; Hollingshead, S.K.; Benjamin, W.H.; Briles, D.E. PspA Protects Streptococcus pneumoniae from Killing by Apolactoferrin, and Antibody to PspA Enhances Killing of Pneumococci by Apolactoferrin. Infect. Immun. 2004, 72, 5031–5040. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, S.K.; Becker, R.; Briles, D.E. Diversity of PspA: Mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 2000, 68, 5889–5900. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.C.; Coan, P.; King, J.; Hale, J.; Benton, K.A.; Briles, D.E.; Hollingshead, S.K. The proline-rich region of pneumococcal surface proteins A and C contains surface-accessible epitopes common to All pneumococci and elicits antibody-mediated protection against sepsis. Infect. Immun. 2010, 78, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Brooks-Walter, A.; Briles, D.E.; Hollingshead, S.K. The pspC Gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive Antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect. Immun. 1999, 67, 6533–6542. [Google Scholar] [PubMed]
- Melin, M.; Coan, P.; Hollingshead, S. Development of cross-reactive antibodies to the proline-rich region of pneumococcal surface protein A in children. Vaccine 2012, 30, 7157–7160. [Google Scholar] [CrossRef] [PubMed]
- Atherton, E.; Sheppard, R.C. Solid Phase Peptide Synthesis A Practical Approach; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Pai, R.; Gertz, R.E.; Beall, B. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J. Clin. Microbiol. 2006, 44, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Goulart, C.; Darrieux, M.; Rodriguez, D.; Pimenta, F.C.; Brandileone, M.C.C.; de Andrade, A.L.S.S.; Leite, L.C.C. Selection of family 1 PspA molecules capable of inducing broad-ranging cross-reactivity by complement deposition and opsonophagocytosis by murine peritoneal cells. Vaccine 2011, 29, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hiroaki, H.; Kohda, D.; Tanaka, T. An isoleucine zipper peptide forms a native-like triple stranded coiled coil in solution. Protein Eng. 1998, 11, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Kilgus, J.; Jardetzky, T.; Gorga, J.C.; Trzeciak, A.; Gillessen, D.; Sinigaglia, F. Analysis of the permissive association of a malaria T cell epitope with DR molecules. J. Immunol. 1991, 146, 307–315. [Google Scholar] [PubMed]
- Sinigaglia, F.; Guttinger, M.; Kilgus, J.; Doran, D.M.; Matile, H.; Etlinger, H.; Trzeciak, A.; Gillessen, D.; Pink, J.R.L. A malaria T-cell epitope recognized in association with most mouse and human MHC class II molecules. Nature 1988, 336, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Senkovich, O.; Cook, W.J.; Mirza, S.; Hollingshead, S.K.; Protasevich, I.I.; Briles, D.E.; Chattopadhyay, D. Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein A provides insight into microbial defense mechanism. J. Mol. Biol. 2007, 370, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Oggioni, M.R.; Pozzi, G. Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene 2002, 284, 63–71. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.G.; Grundling, A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Sternberg, M.J.E.; Makarov, A.A. Polyproline-II Helix in Proteins: Structure and Function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Areschoug, T.; Linse, S.; Stålhammar-Carlemalm, M.; Hedén, L.-O.; Lindahl, G. A Proline-Rich Region with a Highly Periodic Sequence in Streptococcal β Protein Adopts the Polyproline II Structure and Is Exposed on the Bacterial Surface. J. Bacteriol. 2002, 184, 6376–6383. [Google Scholar] [CrossRef] [PubMed]
- Brückner, R.; Nuhn, M.; Reichmann, P.; Weber, B.; Hakenbeck, R. Mosaic genes and mosaic chromosomes–genomic variation in Streptococcus pneumoniae. Int. J. Med. Microbiol. 2004, 294, 157–168. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamborrini, M.; Geib, N.; Marrero-Nodarse, A.; Jud, M.; Hauser, J.; Aho, C.; Lamelas, A.; Zuniga, A.; Pluschke, G.; Ghasparian, A.; et al. A Synthetic Virus-Like Particle Streptococcal Vaccine Candidate Using B-Cell Epitopes from the Proline-Rich Region of Pneumococcal Surface Protein A. Vaccines 2015, 3, 850-874. https://doi.org/10.3390/vaccines3040850
Tamborrini M, Geib N, Marrero-Nodarse A, Jud M, Hauser J, Aho C, Lamelas A, Zuniga A, Pluschke G, Ghasparian A, et al. A Synthetic Virus-Like Particle Streptococcal Vaccine Candidate Using B-Cell Epitopes from the Proline-Rich Region of Pneumococcal Surface Protein A. Vaccines. 2015; 3(4):850-874. https://doi.org/10.3390/vaccines3040850
Chicago/Turabian StyleTamborrini, Marco, Nina Geib, Aniebrys Marrero-Nodarse, Maja Jud, Julia Hauser, Celestine Aho, Araceli Lamelas, Armando Zuniga, Gerd Pluschke, Arin Ghasparian, and et al. 2015. "A Synthetic Virus-Like Particle Streptococcal Vaccine Candidate Using B-Cell Epitopes from the Proline-Rich Region of Pneumococcal Surface Protein A" Vaccines 3, no. 4: 850-874. https://doi.org/10.3390/vaccines3040850