
Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and Virus DNA
2.2. Sequence Accession Numbers from Reference Exogenous Virus
2.3. Illumina Sequencing the CiHHV-6A Genomes and Sequence Assembly
2.4. Chromosomal Integration Site PCR Amplification
2.5. Multiple Alignments and Phylogenetic Analyses
3. Results
3.1. Genomic Analyses of CiHHV-6A
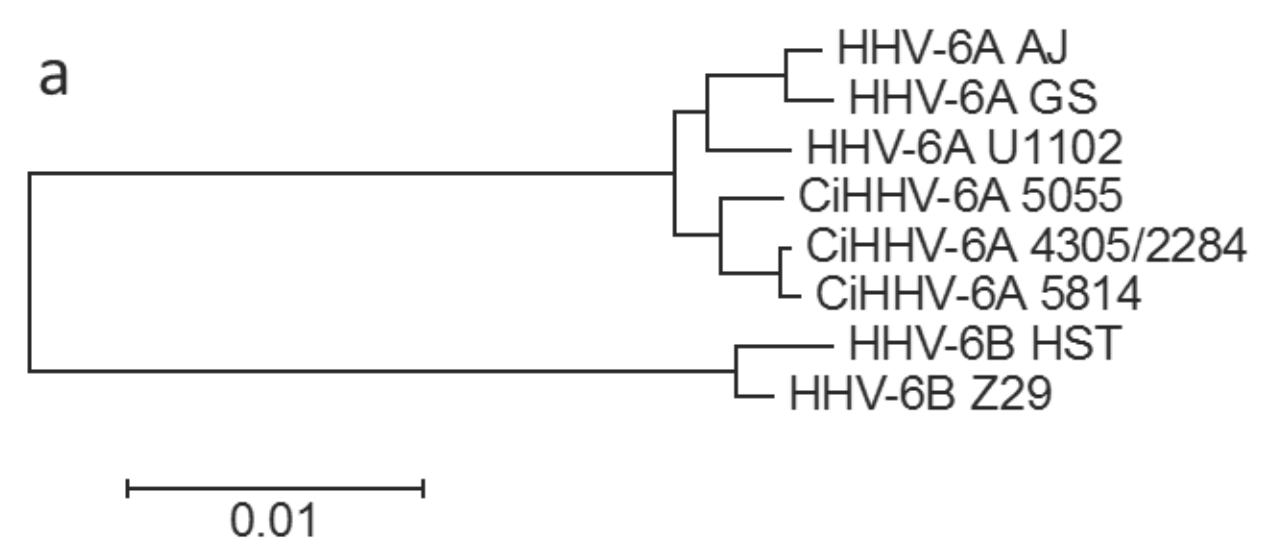
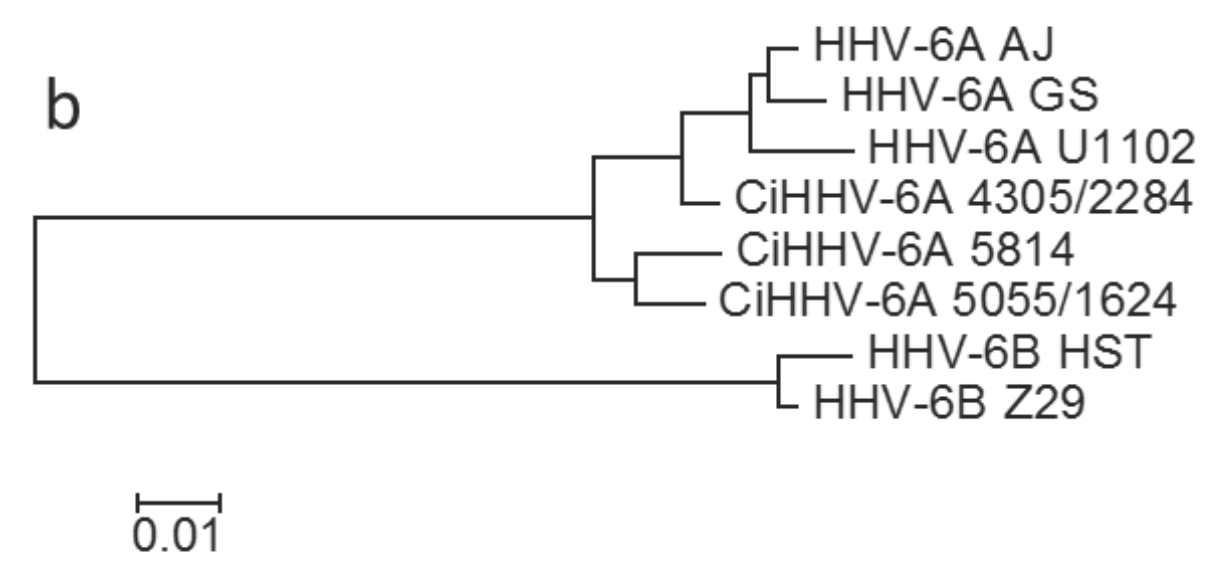
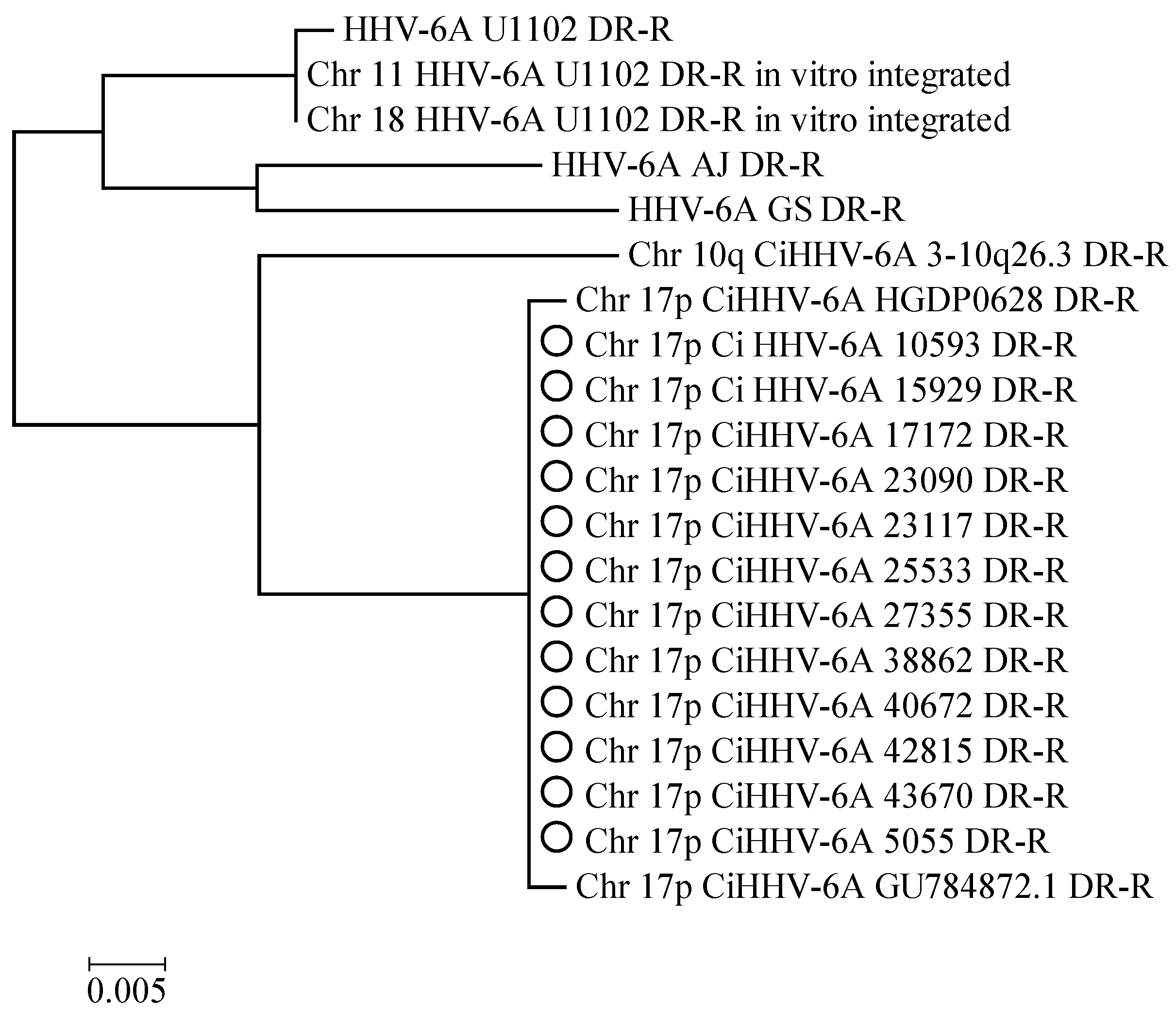
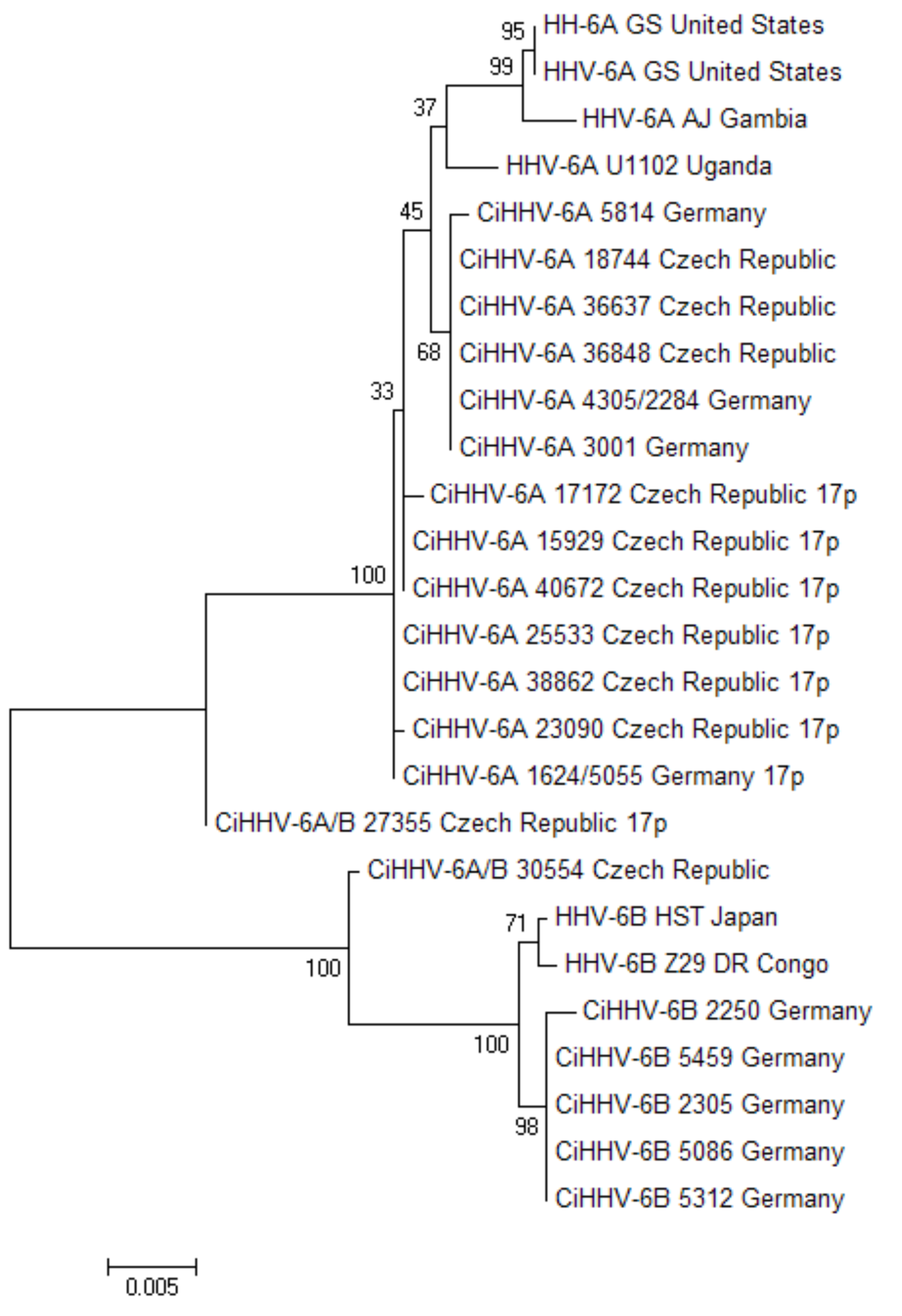
3.1.1. Phylogenetic Relationships between CiHHV-6A and HHV-6A


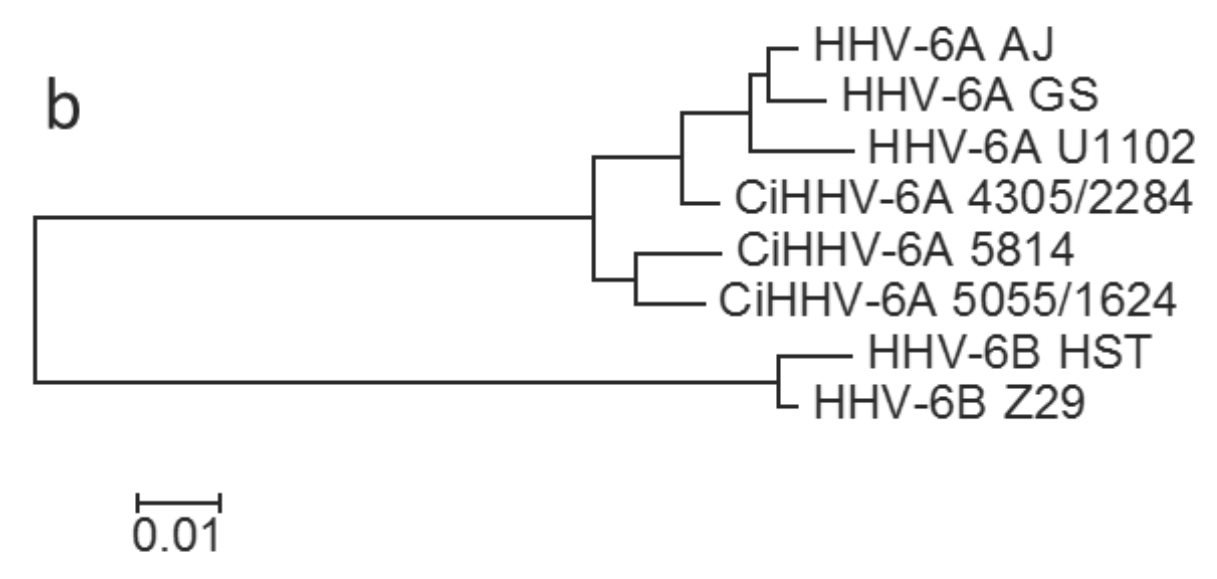

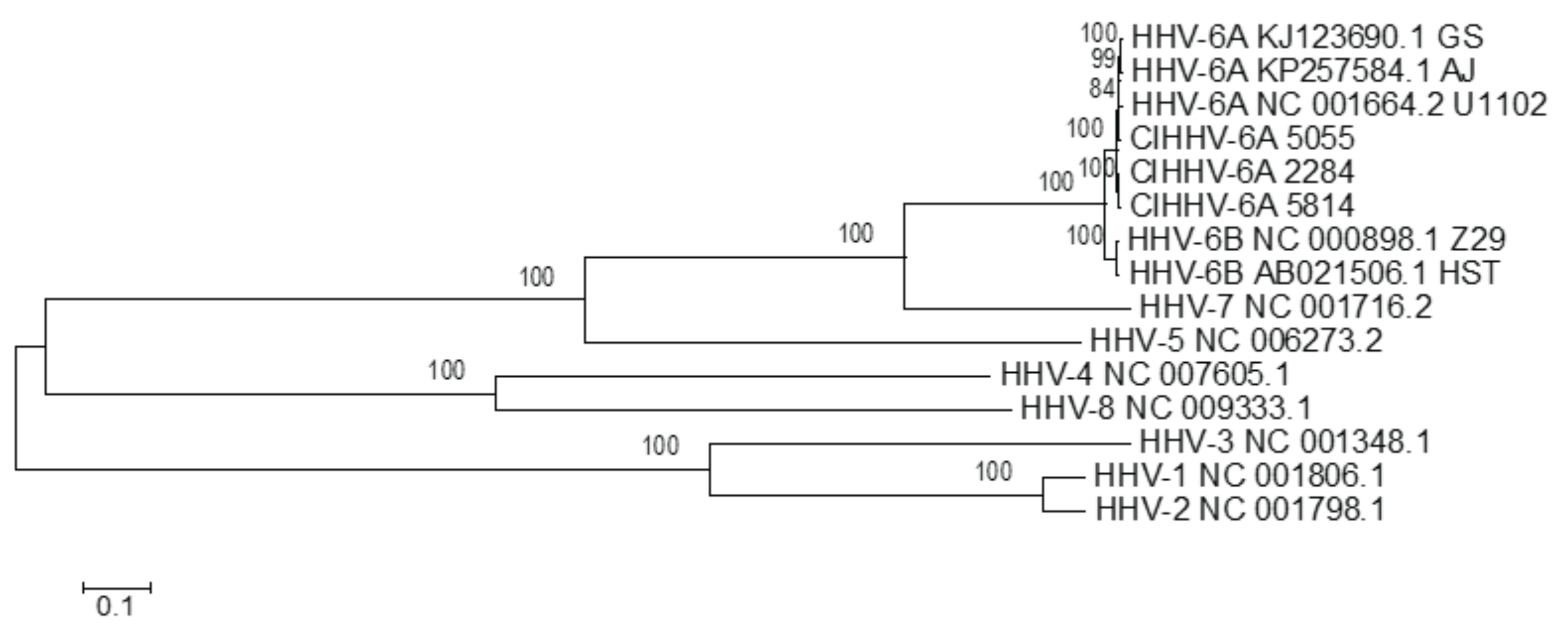{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | HHV-6A | CiHHV-6A | HHV-6B | ||||
|---|---|---|---|---|---|---|---|
| AJ | GS | 2284 | 5055 | 5814 | Z29 | ||
| % | % | % | % | % | % | Gene Function/Homologue, References | |
| DR1 * | 96.7 | 96.9 | 95.9 | 96.8 | 96.7i | 88.9 | Putative DNA directed RNA polymerase Z29 |
| DR6 * | 95.5 | 96.1 | 96.6 | 96.3 | 95.9 | 87.8 | DR6B binds p41 DNA processivity factor U27, inhibits replication, G2/M arrest [50,51] |
| U11 | 97.9 | 97.8 | 98.0 | 98.0 | 97.8 | 89.2 | Tegument phosphoprotein, pp100 major antigen (HCMV UL32) |
| U13 | 99.7 | 99.7 | 99.7 | 97.5 | 97.5 | 96.2 | |
| U14 | 99.6 | 99.7 | 99.6 | 96.5 | 96.0 | 90.5 | Virion tegument protein (HCMV UL25/35), P53 interaction cell cycle [52] |
| U15 * | 97.1 | 97.4 | 97.2 | 97.2 | 99.3 | 94.0 | |
| U19 | 97.9 | 97.7 | 97.9 | 97.7 | 98 | 94.2 | IE-B protein (HCMV US22 gene family, UL38) |
| U47 | 98.0 | 98.4 | 97.5 | 97.5 | 97.5 | 92.9 | Membrane glycoprotein gO complexes with gH/gL (HCMV gO U74) |
| U54 | 99.3 | 98.8 | 97.2 | 97.2 | 97.2 | 87.5 | Virion transactivator, (pp65, HCMV UL82/83) U54A activates NFAT [53] |
| U65 | 99.5 | 99.6 | 97.4 | 97.5 | 97.5 | 92.8 | Tegument protein (provisional HCMV UL94, HSV UL16) |
| U71 | 99.2 | 99.2 | 99.2 | 95.3 | 95.3 | 92.6 | Myristylated tegument protein; position HCMV pp28K (HSV UL11) |
| U79 * | 98.5 | 96.6 | 96.5 | 96.8 | 96.3 | 89.8 | DNA replication provisional, includes U80, (HCMV UL112/113,P34) |
| U86 * | 98.3 | 97.6 | 97.9 | 95.1 | 94.3 | 85.5 | IE2, IE-A protein; includes U87; (HCMV IE2 UL122), R1 repeats |
| U90 * | 97.8 | 97.6 | 97.4 | 97.5 | 97.3 | 83.8 | IE1, IE-A transactivator; includes U89; (position HCMV IE1) |
| U95 | 97.8 | 97.6 | 97.2 | 96.5 | 95.7 | 84.0 | (HCMV US22 gene family, position MCMV IE2), GRIM-19 interaction, mitochondria [54] |
| U100 * | 98.0 | 97.5 | 97.0 | 97.1 | 97.0 | 90.3 | Membrane glycoprotein gQ complexes with gH/gL binds CD46 |
| Mean | 98.2 | 98 | 97.6 | 97.0 | 96.8 | 90.5 | |
3.1.2. Divergent Genes in CiHHV-6A
| Taxa Comparisons | HHV-6A | CiHHV-6A | HHV-6B |
|---|---|---|---|
| Mean distance | |||
| HHV-6A | 0.015 | ||
| CiHHV-6A | 0.029 | 0.023 | |
| HHV-6B | 0.131 | 0.128 | 0.011 |
| Mean diversity | |||
| HHV-6A | 0.017 | ||
| CiHHV-6A | 0.025 (0.246) | 0.019 | |
| HHV-6B | 0.071 (0.844) | 0.085 (0.801) | 0.013 |
3.1.3. Identification of Exogenous HHV-6A “Superinfection” in CiHHV-6A Patients by Deep Sequencing
3.2. Characterization of a Common Integration Site at Chromosome 17p



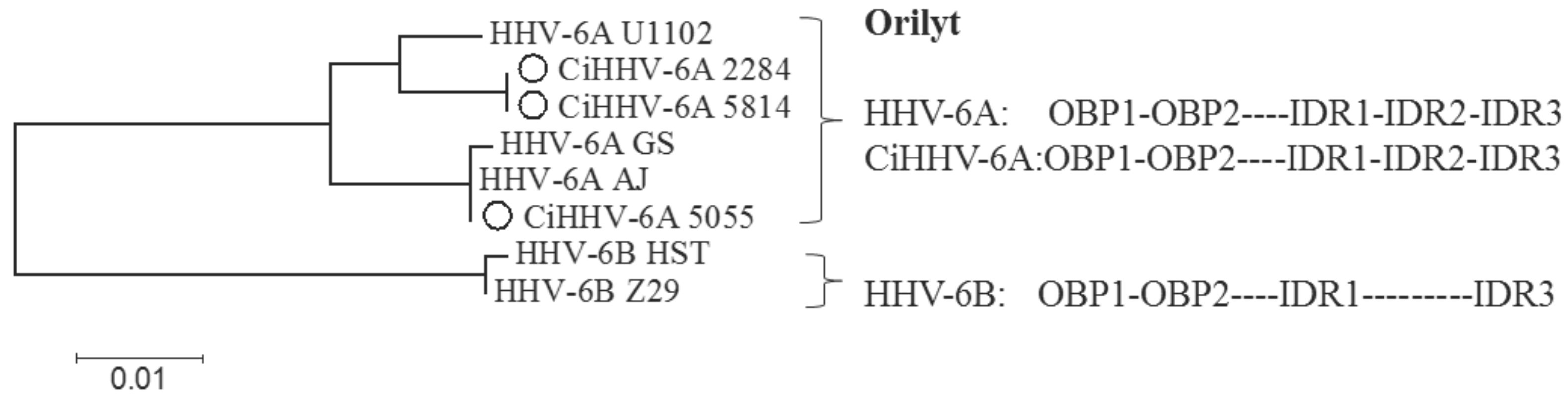
3.3. Conserved cis-Acting Signals for DNA Packaging, Replication and Gene Regulation

4. Discussion
4.1. Gene Divergence
4.2. Integration Site
4.3. Prevalence and Emergence
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, C.B.; Caserta, M.T.; Schnabel, K.; Shelley, L.M.; Marino, A.S.; Carnahan, J.A.; Yoo, C.; Lofthus, G.K.; McDermott, M.P. Chromosomal integration of human herpesvirus 6 is the major mode of congenital human herpesvirus 6 infection. Pediatrics 2008, 122, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Morissette, G.; Flamand, L. Herpesviruses and chromosomal integration. J. Virol. 2010, 84, 12100–12109. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.N.; Tuke, P.W.; Tedder, R.S.; Khanom, A.B.; Eglin, R.P.; Atkinson, C.E.; Ward, K.N.; Griffiths, P.D.; Clark, D.A. The prevalence of chromosomally integrated human herpesvirus 6 genomes in the blood of UK blood donors. J. Med. Virol. 2007, 79, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Taya, K.; Sashihara, J.; Kurahashi, H.; Amo, K.; Miyagawa, H.; Kondo, K.; Okada, S.; Yamanishi, K. Human herpesvirus 6 (HHV-6) is transmitted from parent to child in an integrated form and characterization of cases with chromosomally integrated HHV-6 DNA. J. Med. Virol. 2004, 73, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Caserta, M.T.; Hall, C.B.; Schnabel, K.; Lofthus, G.; Marino, A.; Shelley, L.; Yoo, C.; Carnahan, J.; Anderson, L.; Wang, H. Diagnostic assays for active infection with human herpesvirus 6 (HHV-6). J. Clin. Virol. 2010, 48, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; de Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, J.H.; Medveczky, P.G. The molecular biology of human herpesvirus-6 latency and telomere integration. Microbes Infect. 2011, 13, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA sequence of human herpesvirus-6: Structure, coding content, and genome evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Macaulay, H.A. Characterization of human telomeric repeat sequences from human herpesvirus 6 and relationship to replication. J. Gen. Virol. 1995, 76 Pt 2, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hidalgo-Bravo, A.; Zhang, E.; Cotton, V.E.; Mendez-Bermudez, A.; Wig, G.; Medina-Calzada, Z.; Neumann, R.; Jeffreys, A.J.; Winney, B.; et al. Human telomeres that carry an integrated copy of human herpesvirus 6 are often short and unstable, facilitating release of the viral genome from the chromosome. Nucleic Acids Res. 2014, 42, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ohye, T.; Inagaki, H.; Ihira, M.; Higashimoto, Y.; Kato, K.; Oikawa, J.; Yagasaki, H.; Niizuma, T.; Takahashi, Y.; Kojima, S.; et al. Dual roles for the telomeric repeats in chromosomally integrated human herpesvirus-6. Sci. Rep. 2014, 4, 4559. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, A.; Paul, S.; Hu, S.; Riethman, H. Human subtelomeric duplicon structure and organization. Genome Biol. 2007, 8, R151. [Google Scholar] [CrossRef] [PubMed]
- Linardopoulou, E.V.; Williams, E.M.; Fan, Y.; Friedman, C.; Young, J.M.; Trask, B.J. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 2005, 437, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Linardopoulou, E.V.; Parghi, S.S.; Friedman, C.; Osborn, G.E.; Parkhurst, S.M.; Trask, B.J. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet. 2007, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- Caserta, M.T.; Hall, C.B.; Canfield, R.L.; Davidson, P.; Lofthus, G.; Schnabel, K.; Carnahan, J.; Shelley, L.; Wang, H. Early Developmental Outcomes of Children with Congenital HHV-6 Infection. Pediatrics 2014, 134. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Caserta, M.T.; Schnabel, K.C.; Shelley, L.M.; Carnahan, J.A.; Marino, A.S.; Yoo, C.; Lofthus, G.K. Transplacental congenital human herpesvirus 6 infection caused by maternal chromosomally integrated virus. J. Infect. Dis. 2010, 201, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Watanabe, K.; Ohye, T.; Suzuki, K.; Matsubara, T.; Shimizu, N.; Kurahashi, H.; Yoshikawa, T.; Katano, H.; Inoue, N.; et al. Molecular and virological evidence of viral activation from chromosomally integrated HHV-6A in a patient with X-SCID. Clin. Infect. Dis. 2014, 59, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, U.; Lassner, D.; Wallaschek, N.; Gross, U.M.; Krueger, G.R.; Seeberg, B.; Kaufer, B.B.; Escher, F.; Poller, W.; Schultheiss, H.P. Chromosomally integrated human herpesvirus 6 in heart failure: Prevalence and treatment. Eur. J. Heart Fail. 2015, 17, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B. A Neonate with Acute Heart Failure: Chromosomally Integrated Human Herpesvirus 6-Associated Dilated Cardiomyopathy. J. Pediatr. 2015, 167, 188.e1–192.e1. [Google Scholar] [CrossRef] [PubMed]
- Gravel, A.; Dubuc, I.; Morissette, G.; Sedlak, R.H.; Jerome, K.R.; Flamand, L. Inherited chromosomally integrated human herpesvirus 6 as a predisposing risk factor for the development of angina pectoris. Proc. Natl. Acad. Sci. USA 2015, 112, 8058–8063. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Hubacek, P.; Kuhl, U.; Lassner, D.; Gompels, U.A. Analyses of germline, chromosomally integrated human herpesvirus 6A and B genomes indicate emergent infection and new inflammatory mediators. J. Gen. Virol. 2015, 96 Pt 2, 370–389. [Google Scholar] [CrossRef] [PubMed]
- Pantry, S.N.; Medveczky, M.M.; Arbuckle, J.H.; Luka, J.; Montoya, J.G.; Hu, J.; Renne, R.; Peterson, D.; Pritchett, J.C.; Ablashi, D.V.; et al. Persistent human herpesvirus-6 infection in patients with an inherited form of the virus. J. Med. Virol. 2013, 85, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Donaldson, C.D.; Depledge, D.; Breuer, J.; Gompels, U.A. Complete Genome Sequence of the Human Herpesvirus 6A Strain AJ from Africa Resembles Strain GS from North America. Genome Announc. 2015, 3, e01498-14. [Google Scholar] [CrossRef] [PubMed]
- Downing, R.G.; Sewankambo, N.; Serwadda, D.; Honess, R.; Crawford, D.; Jarrett, R.; Griffin, B.E. Isolation of human lymphotropic herpesviruses from Uganda. Lancet 1987, 2, 390. [Google Scholar] [CrossRef]
- Tedder, R.S.; Briggs, M.; Cameron, C.H.; Honess, R.; Robertson, D.; Whittle, H. A novel lymphotropic herpesvirus. Lancet 1987, 2, 390–392. [Google Scholar] [CrossRef]
- Gravel, A.; Ablashi, D.; Flamand, L. Complete Genome Sequence of Early Passaged Human Herpesvirus 6A (GS Strain) Isolated from North America. Genome Announc. 2013, 1, e00012–e00013. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, G.; Dambaugh, T.R.; Stamey, F.R.; Dewhurst, S.; Inoue, N.; Pellett, P.E. Human herpesvirus 6B genome sequence: Coding content and comparison with human herpesvirus 6A. J. Virol. 1999, 73, 8040–8052. [Google Scholar] [PubMed]
- Isegawa, Y.; Mukai, T.; Nakano, K.; Kagawa, M.; Chen, J.; Mori, Y.; Sunagawa, T.; Kawanishi, K.; Sashihara, J.; Hata, A.; et al. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J. Virol. 1999, 73, 8053–8063. [Google Scholar] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Gotz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Assefa, S.; Keane, T.M.; Otto, T.D.; Newbold, C.; Berriman, M. ABACAS: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics 2009, 25, 1968–1969. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis Comparison Tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011, 39, e57. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Megaw, A.G.; Rapaport, D.; Avidor, B.; Frenkel, N.; Davison, A.J. The DNA sequence of the RK strain of human herpesvirus 7. Virology 1998, 244, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Britt-Compton, B.; Rowson, J.; Locke, M.; Mackenzie, I.; Kipling, D.; Baird, D.M. Structural stability and chromosome-specific telomere length is governed by cis-acting determinants in humans. Hum. Mol. Genet. 2006, 15, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Scheffler, K.; Gravenor, M.B.; Poon, A.F.; Frost, S.D. Evolutionary fingerprinting of genes. Mol. Biol. Evol. 2010, 27, 520–536. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Lambert, D.M. Selective constraints determine the time dependency of molecular rates for human nuclear genomes. Genome Biol. Evolut. 2012, 4, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.; Monze, M.; Bima, H.; Kapambwe, M.; Clark, D.; Kasolo, F.C.; Gompels, U.A. Predominant human herpesvirus 6 variant A infant infections in an HIV-1 endemic region of Sub-Saharan Africa. J. Med. Virol. 2009, 81, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Schleimann, M.H.; Hoberg, S.; Solhoj Hansen, A.; Bundgaard, B.; Witt, C.T.; Kofod-Olsen, E.; Hollsberg, P. The DR6 protein from human herpesvirus-6B induces p53-independent cell cycle arrest in G2/M. Virology 2014, 452–453, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Schleimann, M.H.; Moller, J.M.; Kofod-Olsen, E.; Hollsberg, P. Direct Repeat 6 from human herpesvirus-6B encodes a nuclear protein that forms a complex with the viral DNA processivity factor p41. PLoS ONE 2009, 4, e7457. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Koike, M.; Mori, Y.; Yonemoto, S.; Sasamoto, Y.; Kondo, K.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J. Virol. 2005, 79, 13037–13046. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Morissette, G.; Gravel, A.; Flamand, L. Inhibition of Interleukin-2 Gene Expression by Human Herpesvirus 6B U54 Tegument Protein. J. Virol. 2014, 88, 12452–12463. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.M.; Isegawa, Y.; Chow, V.T. The U95 protein of human herpesvirus 6B interacts with human GRIM-19: Silencing of U95 expression reduces viral load and abrogates loss of mitochondrial membrane potential. J. Virol. 2008, 82, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Kasolo, F.C. HHV-6 Genome: Similar and Different. In Human Herpesvirus-6, 2nd ed.; Krueger, G., Ablashi, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 12, pp. 23–46. [Google Scholar]
- Ablashi, D.; Agut, H.; Alvarez-Lafuente, R.; Clark, D.A.; Dewhurst, S.; DiLuca, D.; Flamand, L.; Frenkel, N.; Gallo, R.; Gompels, U.A.; et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch. Virol. 2014, 159, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Gravel, A.; Fink, K.; Grandvaux, N.; Flamand, L. Inhibition of transcription of the beta interferon gene by the human herpesvirus 6 immediate-early 1 protein. J. Virol. 2007, 81, 5737–5748. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; DiLuca, D.; Gompels, U. Characterisation of a human herpesvirus 6 variant A “amplicon” and replication modulation by U94-Rep “latency gene”. J. Virol. Methods 2002, 105, 331–341. [Google Scholar] [CrossRef]
- Tuddenham, L.; Jung, J.S.; Chane-Woon-Ming, B.; Dolken, L.; Pfeffer, S. Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J. Virol. 2012, 86, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Nukui, M.; Mori, Y.; Murphy, E.A. A human herpesvirus 6A-encoded microRNA: Role in viral lytic replication. J. Virol. 2015, 89, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Gravel, A.; Flamand, L. Divergent susceptibilities of human herpesvirus 6 variants to type I interferons. Proc. Natl. Acad. Sci. USA 2010, 107, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Glosson, N.L.; Hudson, A.W. Human herpesvirus-6A and -6B encode viral immunoevasins that downregulate class I MHC molecules. Virology 2007, 365, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, A.; Oyaizu, H.; Maeki, T.; Tang, H.; Yamanishi, K.; Mori, Y. Analysis of a neutralizing antibody for human herpesvirus 6B reveals a role for glycoprotein Q1 in viral entry. J. Virol. 2011, 85, 12962–12971. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Kennedy, P.E.; Locatelli, G.; Malnati, M.S.; Berger, E.A.; Lusso, P. CD46 is a cellular receptor for human herpesvirus 6. Cell 1999, 99, 817–827. [Google Scholar] [CrossRef]
- Tang, H.; Hayashi, M.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor binding. J. Virol. 2011, 85, 11121–11130. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.; Chaubey, G.; Tamang, R.; Pathak, A.K.; Singh, V.K.; Karmin, M.; Singh, M.; Rani, D.S.; Anugula, S.; Yadav, B.K.; et al. The phylogeography of Y-chromosome haplogroup h1a1a-m82 reveals the likely Indian origin of the European Romani populations. PLoS ONE 2012, 7, e48477. [Google Scholar] [CrossRef] [PubMed]
- Achour, A.; Malet, I.; Deback, C.; Bonnafous, P.; Boutolleau, D.; Gautheret-Dejean, A.; Agut, H. Length variability of telomeric repeat sequences of human herpesvirus 6 DNA. J. Virol. Methods 2009, 159, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Leendertz, F.H.; Deckers, M.; Schempp, W.; Lankester, F.; Boesch, C.; Mugisha, L.; Dolan, A.; Gatherer, D.; McGeoch, D.J.; Ehlers, B. Novel cytomegaloviruses in free-ranging and captive great apes: Phylogenetic evidence for bidirectional horizontal transmission. J. Gen. Virol. 2009, 90 Pt 10, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, A.; Donato, D.; Gessain, A.; Niphuis, H.; Nerrienet, E.; Verschoor, E.J.; Lacoste, V. African great apes are naturally infected with roseoloviruses closely related to human herpesvirus 7. J. Virol. 2014, 88, 13212–13220. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, V.; Verschoor, E.J.; Nerrienet, E.; Gessain, A. A novel homologue of Human herpesvirus 6 in chimpanzees. J. Gen. Virol. 2005, 86 Pt 8, 2135–21340. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Davison, A.J.; Dolan, A.; Gatherer, D.; Sevilla-Reyes, E.E. Molecular Evolution of the Herpesvirales. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press, Elsevier Ltd.: Amsterdam, The Netherlands; London, UK, 2008; pp. 447–475. [Google Scholar]
- Aswad, A.; Katzourakis, A. The first endogenous herpesvirus, identified in the tarsier genome, and novel sequences from primate rhadinoviruses and lymphocryptoviruses. PLoS Genet. 2014, 10, e1004332. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.J.; Catusse, J.; Stacey, A.; Borrow, P.; Gompels, U. Activation of CCR2+ human proinflammatory monocytes by human herpesvirus-6B chemokine N-terminal peptide. J. Gen. Virol. 2013, 94, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Kasolo, F.C.; Mpabalwani, E.; Gompels, U.A. Infection with AIDS-related herpesviruses in human immunodeficiency virus-negative infants and endemic childhood Kaposi’s sarcoma in Africa. J. Gen. Virol. 1997, 78 Pt 4, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, B.; Klingel, K.; May, A.E.; Kandolf, R.; Gawaz, M.G. Human herpesvirus 6 subtype A-associated myocarditis with “apical ballooning”. Can. J. Cardiol. 2007, 23, 393–395. [Google Scholar] [CrossRef]
- Kuhl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Prusty, B.K.; Krohne, G.; Rudel, T. Reactivation of chromosomally integrated human herpesvirus-6 by telomeric circle formation. PLoS Genet. 2013, 9, e1004033. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tweedy, J.; Spyrou, M.A.; Pearson, M.; Lassner, D.; Kuhl, U.; Gompels, U.A. Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection. Viruses 2016, 8, 19. https://doi.org/10.3390/v8010019
Tweedy J, Spyrou MA, Pearson M, Lassner D, Kuhl U, Gompels UA. Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection. Viruses. 2016; 8(1):19. https://doi.org/10.3390/v8010019
Chicago/Turabian StyleTweedy, Joshua, Maria Alexandra Spyrou, Max Pearson, Dirk Lassner, Uwe Kuhl, and Ursula A. Gompels. 2016. "Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection" Viruses 8, no. 1: 19. https://doi.org/10.3390/v8010019