
Polymer–Zeolite Composites: Synthesis, Characterization and Application
by
Galymzhan Kulamkadyrovich Mamytbekov
*,
Dmitry Anatol’evich Zheltov
,
Olga Sergeevna Milts
and
Yernat Rashidovich Nurtazin
Institute of Nuclear Physics, Ibragimov Street 1, Almaty 050032, Kazakhstan
*
Author to whom correspondence should be addressed.
Colloids Interfaces 2024, 8(1), 8; https://doi.org/10.3390/colloids8010008
Submission received: 20 November 2023
/
Revised: 20 December 2023
/
Accepted: 26 December 2023
/
Published: 9 January 2024
Abstract
:Although the potential of natural minerals for purification of liquid radioactive wastes (LRW) from radionuclides has been widely studied, the use of hybrid polymer composites made of zeolite is still rather scarce. This article reports on the preparation of zeolite-based hybrid polymer composites using the in situ polymerization technique in the body of mineral matrix and its intercalated with copper ferrocyanide (CuFC) forms. This hybrid polymer composites have shown unique and enhanced properties for the removal of micropollutants from wasted water as compared to the individual mineral. The change in conventional properties of two mixed minerals, such as zeolite and bentonite, and their intercalated with CuFC forms were probed using techniques such as scanning electron microscopy (SEM), X-ray diffraction (XRD), Mössbauer spectroscopy (MS) and FT-IR analysis. The totality of analysis showed a coexistence of intercalated and percolated zeolite phases. The hybrid polymer composites exhibited both adsorption and ion-exchange properties in the removal of 134,137Cs+, 57,60Co2+ and 85Sr2+ radionuclides from LRW.
1. Introduction
It is well known [1,2,3] that the main factor that completely excludes the presence of radionuclides (RN) and their further migration in the environment is natural radioactive decay, the rate of which cannot be changed. Taking this into account, a number of researchers believe that the only safe way to prevent the entry of RN into geochemical and biological chains (cycles) is to slow down the migration of radionuclides in environmental objects to the rates comparable to the rate of natural decay.
The theoretical and experimental results obtained so far make a great contribution to the development of a topical scientific direction—the development of advanced methods for fixing and concentrating radionuclides based on the use of natural raw materials and chemical industry waste in combination with synthetic polymer matrices, which makes it possible to ensure the economic feasibility of measures for eliminating the consequences of nuclear power plants activities and radiation accidents and catastrophes [4,5,6,7]. The issue deals with the hybrid composite materials consisting of natural aluminosilicates of various structural organizations (clays, zeolites, shungites, carbonates) and polymers of various chemical composition and structure. In this case, it is necessary to pay special attention to the “programmed” functionalization of natural and synthetic sorbents in terms of their composition and structure, which is expressed in enhancing the effect of synergism of sorption parameters with respect to both natural and artificial radionuclides. It is necessary to develop directions for the synthesis of new classes of hybrid polymeric materials for selective sorption of certain types of radionuclides that pose the greatest hazard to the surrounding biosphere and the population life.
The major disadvantages of natural inorganic ion-exchangers are their unsuitable aggregative, granule and mechanical properties, which limit their use in practical applications. Inclusion of metal oxide particles in organic polymer matrixes via sol-gel processes is an effective approach to enhancing the tensile properties of the matrix [8,9,10]. Inorganic materials that can be broken down into their nanoscale building blocks, such as smectite clays, can be superior alternatives to the sol-gel process for the preparation of nanostructured hybrid organic–inorganic composites. Intercalated polymer–clay nanocomposites have been synthesized by direct polymer intercalation and in situ intercalative polymerization of monomers in the clay galleries. Owing to the spatial confinement of the polymer between the dense clay layers, intercalated polymer–clay nanocomposites exhibited impressive conductivity and barrier properties [8,9,10,11,12].
Thus, the progress in our understanding of the adsorption characteristics of hydrosoluble polymers and polyelectrolytes requires more theoretical, numerical and experimental studies insofar as real systems combine various interactions not only between adsorbent and polymers but also between the polymer molecules. Adsorption of a polymer at a solid–liquid interface can be a very efficient strategy in many industrial, environmental and agricultural spheres [10]. In the noted study, PAA was used as a binding material for preparation of composites materials of zeolite (ZT) and bentonite (BT) to enable their practical use. The adsorptive features of BT and ZT and their composites were investigated for Pb2+ removal from various solution environments with increasing ionic strength.
Based on the methodological approaches developed by the research group of the Institute of Nuclear Physics (Almaty), a wide range of effective organo-mineral hybrid compositions have been prepared, characterized by a low cost of production and ability to be used for fixing and concentrating and non-proliferation of radionuclides in the composition of liquid radioactive waste [13,14].
This study focuses on the design and evaluation of a mineral-filled hybrid polymer composition intended for providing protective barriers against the spread of radionuclides during transportation, interim storage and final disposal of radioactive wastes as well as to avoid leaching out and migration of radionuclides to surroundings.
2. Experimental Section
2.1. Materials
Monomers of acrylic acid (AAc), acrylamide (AAm) and crosslinking agent N,N′-methylene-bis-acrylamide (MBA) and initiators ammonium persulfate (NH4)2S2O8 (PSA) and sodium metabisulfite Na2S2O3 (SMB) from Sigma-Aldrich, Inc. (Missouri, St. Louis, MO 68178, USA) of chemically pure grade were used with no additional purification.
Bentonite of the JSC “Ilsky Zavod Utyazhelitel” (Russia) TU-2164-003-00136716-2015 was used after washing with distilled water to remove soluble components such as carbonates and chlorides of monovalent alkali metals and other impurities, then dried to a constant weight at 70 °C temperature.
Zeolite was received from Chankanai field (Almaty region, Kazakhstan, LTD “Taza Su”). The zeolite rocks were crushed, ground and sieved to 140 mesh (or 105 μm) in size. No pretreatment was applied to the zeolite. The certified chemical composition of natural zeolite is provided in Table 1.
2.2. Preparation of Composite Materials
Intercalated complexes (ICC) was synthesized by mixing aqueous solutions of CuSO4 (0.1 M) and K4[Fe(CN)6] (0.1 M) in a ratio of 2:1 in a volume of zeolite and bentonite swollen in water with vigorous stirring on a magnetic stirrer. After washing and filtering the suspension, the solid residue of ICC was dried at 60 °C to constant weight.
Synthesis of percolated complexes (PCC) consisted of the following stages: 5 g of ICC sample swollen in water, 5 g of AAm, 5 mL of AAc and the calculated amount of MBA (5 mL of 0.1 M, which corresponds to the degree of crosslinking of the polymer gel of 0.72 mol.% or 0.01 wt.% in relation to the acrylamide monomer) were added under stirring. The total volume of the reaction system was 100 mL. At the final stage of stirring, 0.5 mL of APS and 0.5 mL of SMB were added to the reaction mixture at a concentration of 0.1 M, respectively. After thorough mixing, the reaction mixture was poured onto rectangular and cylindrical substrates made of organic material and placed in an oven at 60 °C temperature, where in situ polymerization proceeded for 6 h, after which the samples were cooled to room temperature during the day. After the samples were removed from the substrates, they were repeatedly washed and dried in a thermostat to a constant weight at t < 50 °C to prevent thermal degradation of the polymer matrix.
2.3. Instruments and Measurements
The experiments on sorption of LRW radionuclides by a natural mineral and its percolated analogue were performed under static conditions by periodic stirring of the sorbent samples placed in 21 mL of LRW for 24 and 72 h. Then, the liquid and solid phases were separated in a separating funnel to prevent the sorption of RN on a paper filter. In the resulting filtrate, the specific activity of RN was determined, and the degree of binding (purification) θ and the distribution coefficient Kd of radionuclides between the liquid and solid phases were calculated according to Equations (1) and (2), respectively [15,16]:
where A0 and Aeq are the initial and equilibrium specific activity of RN before and after sorption, respectively; V is the volume of the liquid phase, mL; ms is the sorbent mass, g.
The activity of the radionuclides was measured on a gamma-spectrometer using a BE3820S coaxial germanium detector by Canberra Ind. (800 Research Parkway, Meriden, CT 06450, USA). The measured energy resolution of the detector was 1.7 keV at the half-height peak at an energy of 1332 keV (60Co).
The liquid radioactive waste from the research water-cooled reactor WWR-K of the Institute of Nuclear Physics (Almaty) with the following composition and radioactivity was used as model solution (Table 2).
For elemental analysis of leachates and water samples (if necessary, diluted with deionized water) the following methods were used:
- Inductively coupled plasma mass spectrometry (ICP MS) on the ELAN-9000 quadrupole mass spectrometer (PerkinElmer SCIEX, Waltham, MA, USA);
- Inductively coupled plasma optical-emission spectrometry (OES-ICP) on the OPTIMA-8000 double-view OE spectrometer (PerkinElmer Inc., Waltham, MA, USA).
Scanning electron microscopy (SEM) was performed using a Hitachi model TM 4000 Plus microscope equipped with an X-ray fluorescence energy dispersive analysis (EDS) attachment with a crystal detector. The samples were studied under low vacuum in the back-scattered electron mode.
X-ray diffraction analysis (XRD) was performed on a Bruker X-ray diffractometer with CuKα radiation. A thin dried film of a hybrid composition and powdered bentonite up to 10 µm in size were scanned in the 2θ angle range from 20° to 70° with a step of 0.02°, a scanning speed of 2 s/point and radiation parameters of 40 kV and 40 mA.
For the MS investigations, the samples were mixed with paraffin at a rate of 100 mg/cm2 and transferred into tablets. The Mössbauer spectra of the samples were recorded in the transmission geometry. The source of γ-quanta was 57Co in the chromium matrix. The spectra were recorded at 300 K on a nuclear gamma-resonance spectrometer MS1104Em. The reference sample α-Fe was used for spectra calibration. The Mössbauer spectra were processed using the software SpectrRelax by the methods of model fitting [17].
3. Results and Discussion
3.1. Characterization of Composite Polymeric Material
3.1.1. FT-IR Spectroscopy
In our previous publications [13,14], we provided a detailed analysis of the structure and composition of hybrid composites based on bentonite, its intercalated complex with copper ferrocyanide, impregnated into the volume of a three-dimensional polymer matrix of an acrylic acid–acrylamide copolymer. In this article, the main emphasis is primarily made on the identification of a hybrid composites obtained on the basis of an identical polymer matrix with the introduction of zeolite and its intercalated form with copper ferrocyanide and mixtures of natural minerals as well.
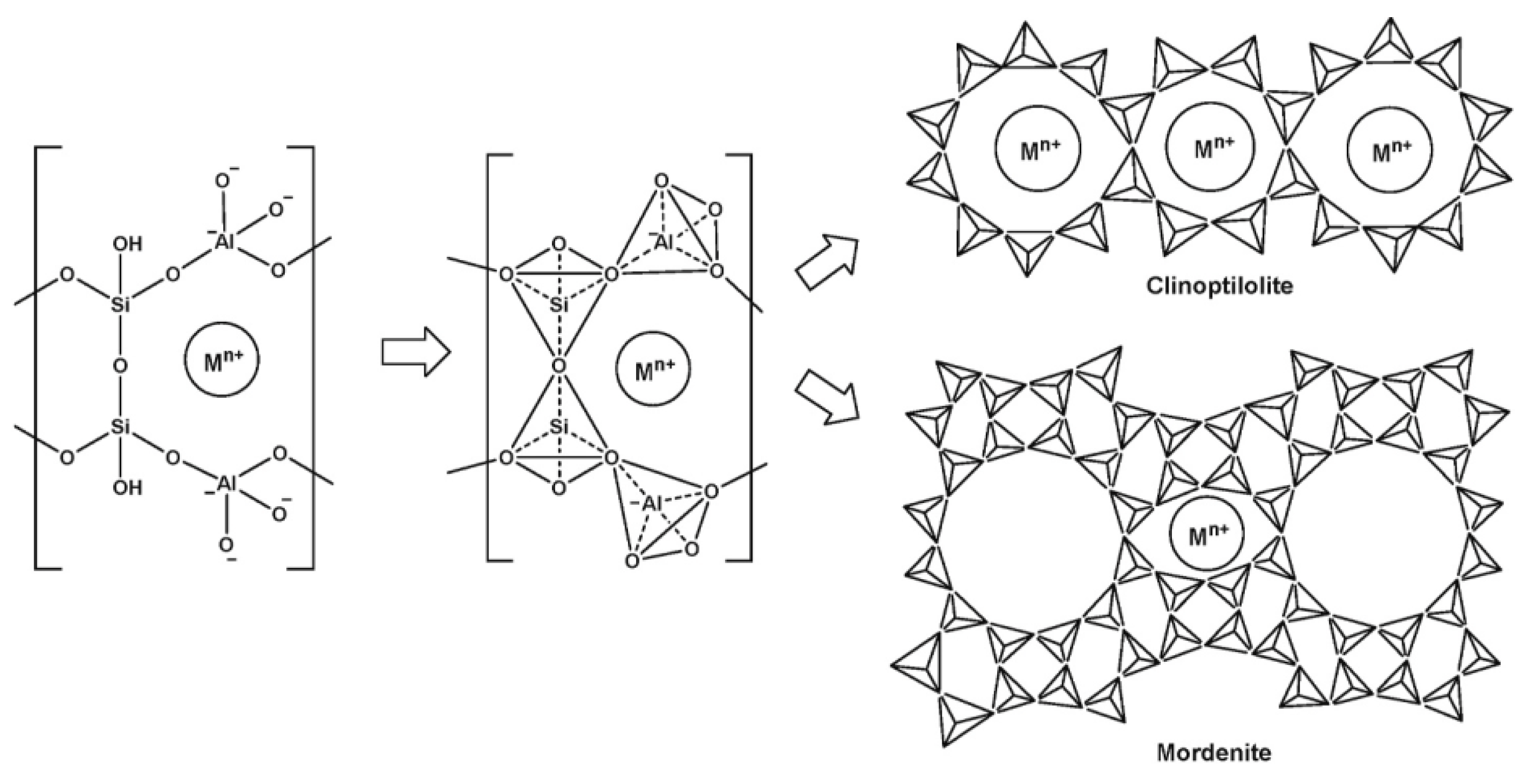
It is known that zeolites are nanoporous crystalline aluminosilicates containing water molecules and exchangeable cations such as K+, Na+, Ca2+ and Mg2+, which neutralize excess negative charges arising from the isomorphic substitution of Si4+ with Al3+. The primary structural unit of zeolites is conventionally taken to be the TO4 tetrahedron, where T is a silicon or aluminum atom. Tetrahedra, connecting with each other, form cuboctahedra (sodalite cells), from which the elementary cells of zeolite are built from three-, four-, five- or six-membered rings that make up its crystalline framework [18,19]. The cuboctahedra are connected to each other by channels, the accessibility of which is determined by the free cross-section of the entrance holes (windows) formed by rings of interconnected tetrahedra.
Each silicon ion has its +4 charge balanced by four tetrahedral oxygens and the silica tetrahedra are, therefore, electrically neutral. Each alumina tetrahedron has a residual charge of −1 since the trivalent aluminum is bonded to four oxygen anions [18]. Therefore, each alumina tetrahedron requires a +1 charge from an extra framework cation in the structure to maintain electrical neutrality [20]. These cations are usually sodium ions that are present in the synthesis medium [21,22]. Sodium ions can be easily exchanged by mono- or divalent cations. Several studies have mentioned the major role that these cations (Na+, K+, Li+, Mg2+, Ca2+, Zn2+, Mn2+) can play in increasing the affinity between the adsorbates and the adsorbents or in modifying the separation properties of the zeolites towards some metal ions [18,23].
Not only exchange cations, but also structural defects, hydroxyl groups and tricoordinated aluminum in the zeolite structure can act as active centers [22]. Ideally, the composition of zeolites can be expressed by the formula , where are the mono- and divalent cations, respectively.
The sorption properties of zeolite rocks largely depend on the mineral composition as well as the structural features of the mineral (Figure 1). Zeolites are characterized by a microporous structure, and zeolite-containing rock is a mixture of zeolite and impurity minerals; therefore, meso- and macropores are formed between the zeolite particles and the host rock, facilitating the interdiffusion of exchanging ions in the sorbent–sorbate system [18,19]. Layered aluminosilicates, such as montmorillonite, may be present as impurity minerals and galoysite as well.
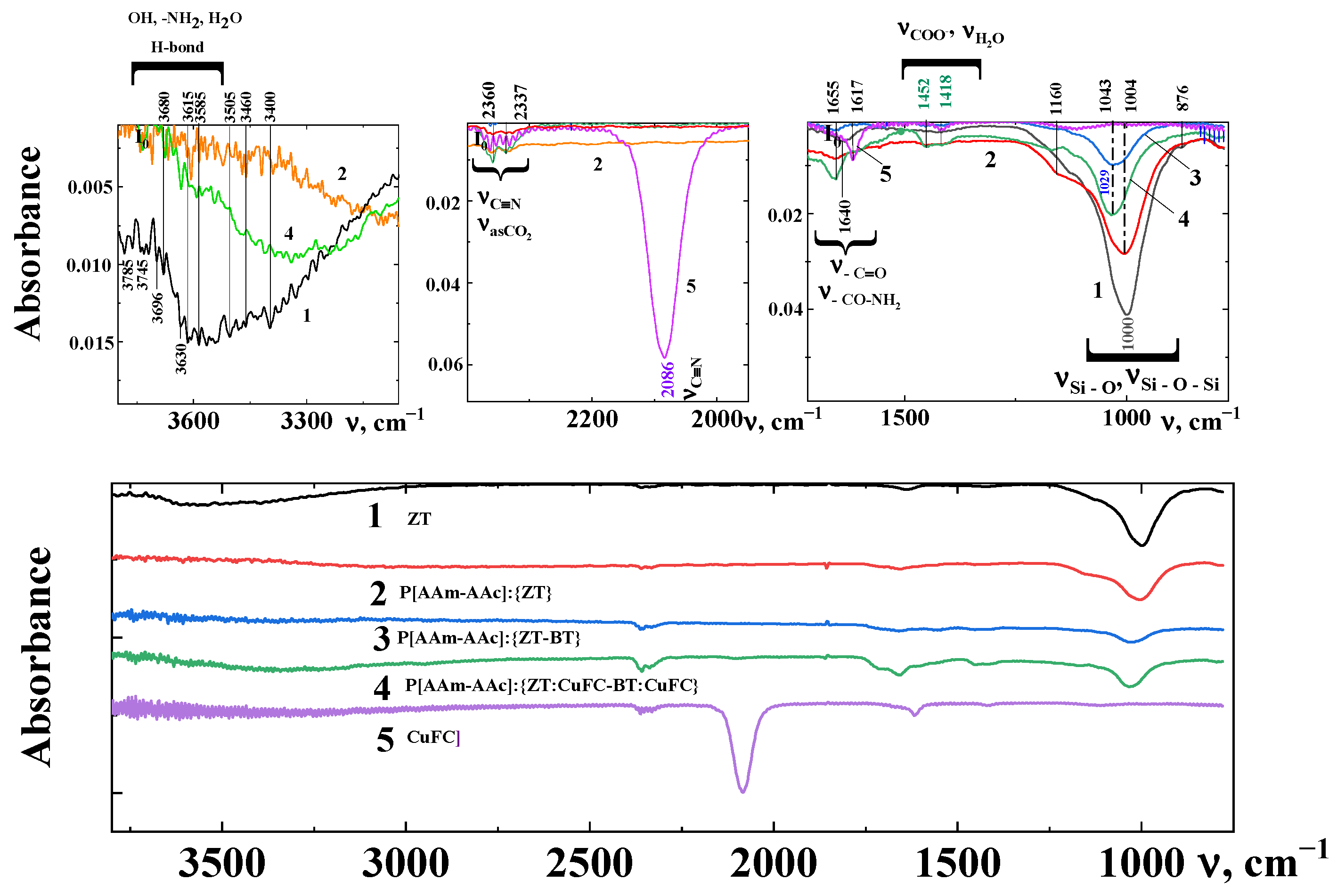
From the FT-IR spectra (Figure 2, line 1) of zeolites, one can observe a diffuse continuum in the region of 2900–3700 cm−1 of stretching vibrations of hydroxyl groups in crystallization adsorbed water associates. The broad band at 3400 cm−1 belongs to hydroxyl groups associated with hydrogen bonding with the oxygen of the aluminosilicate frame and bending vibrations of water molecules is located at 1640 cm−1 [25,26,27]. The position of both bands is influenced by the nature of the outer framework cation. The absorption band at 3630 cm−1 is attributed to structural hydroxyl groups directed towards vacant octahedral positions [28].
The difference of the absorption spectra in the regions of stretching and deformation vibrations of hydroxyl groups for the studied samples of hybrid composites is associated with a change in their chemical composition and with an increase in the aluminum content in octahedral positions. Thus, FT-IR analysis shows that the studied layered silicates have different relationships between the active centers on their surface associated with water in various forms and the position of aluminum in octahedral and tetrahedral positions, which was previously shown by us [14] using Mössbauer spectroscopy data.
The appearance of a shoulder at 1160 cm−1 (Figure 2, line 2) in the polymer–zeolite composition can be associated with the formation of relatively strong coordination bonds of the amino groups of the polymer with the functional centers of the basal surfaces of the zeolite Si-O- and Si-OH.
The FT-IR spectra of samples also evidence for absorption bands that is a characteristic of stretching vibrations of the Si–O bond in the tetrahedron (1000–1100 cm−1) and deformation vibrations of the Si-O-Al bond (700–800 cm−1), as well as absorption bands associated with the presence in the structure of various forms of water molecules (3400 cm−1, 2500 ÷ 3700 cm−1). Wide absorption peaks (1000–1200 cm−1) with maxima at 1043 cm−1 for zeolite–bentonite-containing polymer (Figure 2, line 3) composites correspond to the Si-O-Si stretching vibrations of the tetrahedra of the silicon–oxygen framework [25,26,27].
The layered silicates usually exhibit an intense band around 1000 cm−1 and a weaker band in the region of 1100 and 900 cm−1. The low-frequency component arises as a result of the reflection in the FT-IR spectra of Al → Si of substitution in the crystal lattice of natural minerals [29]. The hydroxyl groups with a high frequency stretching vibration band (ν = 3640 cm−1) are located in large cavities of the structure of zeolite and are accessible to adsorbed water molecules, while groups of the second type, represented in the IR spectra by a low-frequency band (ν = 2500–3400 cm−1), are less accessible to the adsorbate, and are probably located inside small cavities.
It can be stated that the presence of water and cations in the cavities of zeolites is explained by the replacement of Si4+ with Al3+ and the need to compensate the negative charge of the frame. Hydroxyl groups corresponding to the absorption band of 3745 cm−1 can be attributed to Si-OH groups localized on a structural defect, since this band is attributed to vibrations of hydroxyl groups at the ends of aluminum–silicon–oxygen chains or on amorphous silica. This band is the weakest and is observed in almost all spectra of natural zeolites regardless of the cations type [22,28,29].
Thus, the studied spectra are characterized by a typical set of absorption bands (3745, 3635 and 3580 cm−1), corresponding to vibrations in the Si-O-H group, localized in various structural positions of zeolite channels, which are preserved in hybrid compositions.
The CO2 molecule, widely used as a probe for the quantitative characterization of both basic and acidic properties of surface centers of zeolites and oxides by FT-IR, has the most intense absorption band of the asymmetric stretching vibration νass at 2360 cm−1. The integral absorption coefficient of this band is so large that in the case of zeolites, it is more convenient to make quantitative estimates of the overlap based on the intensity of the absorption bands of the main vibrations, for example, using the vibration band at a frequency of about 3714 cm−1.
The presence of copper hexaferrocyanide complexes in the intercalated zeolite is evidenced by the complex absorption bands in the region of 2000–2150 cm−1 (Figure 2, line 4) The absorption peak at 2086 cm−1 corresponds to the valence vibrations of nitrile groups (-C≡N) and most likely refers to the mixed hexaferrocyanide phase both of potassium and copper [28].
In the FT-IR spectra of the studied intercalated and percolated samples, the absorption bands at 2340 cm−1 and 2360 cm−1 were also recorded; those are characteristic of the C≡N valence vibrations and the intensity of which increases especially for intercalated and percolated ferrocyanides in the polymer–mineral blends (Figure 2, line 4) phases compared to pure copper ferrocyanide (Figure 2, line 5) [30].
3.1.2. XRD Analysis
The structural variability and mobility of the crystal lattice of natural aluminosilicates during their modification determines many aspects of their application [31,32,33]. Intercalation can be considered as methods these make it possible to change both the textural characteristics of aluminosilicates and their functional composition as well as to obtain a wide range of mesoporous structures. It should be assumed that for the intercalation of ferrocyanide complexes of transition metals into the bulk of natural aluminosilicates, first of all, it is necessary to take into account the dimension of the intercalated compounds with the inner size of the mineral matrix. That is the intercalation of new phases of transition metal ferrocyanide microcrystals into the volume of the mineral matrix should not undergo spatial restrictions for their placement in the interlayer space of clays (bentonite, montmorillonite) and in the “channeled windows” of zeolites. We described the first case in detail in article [14].
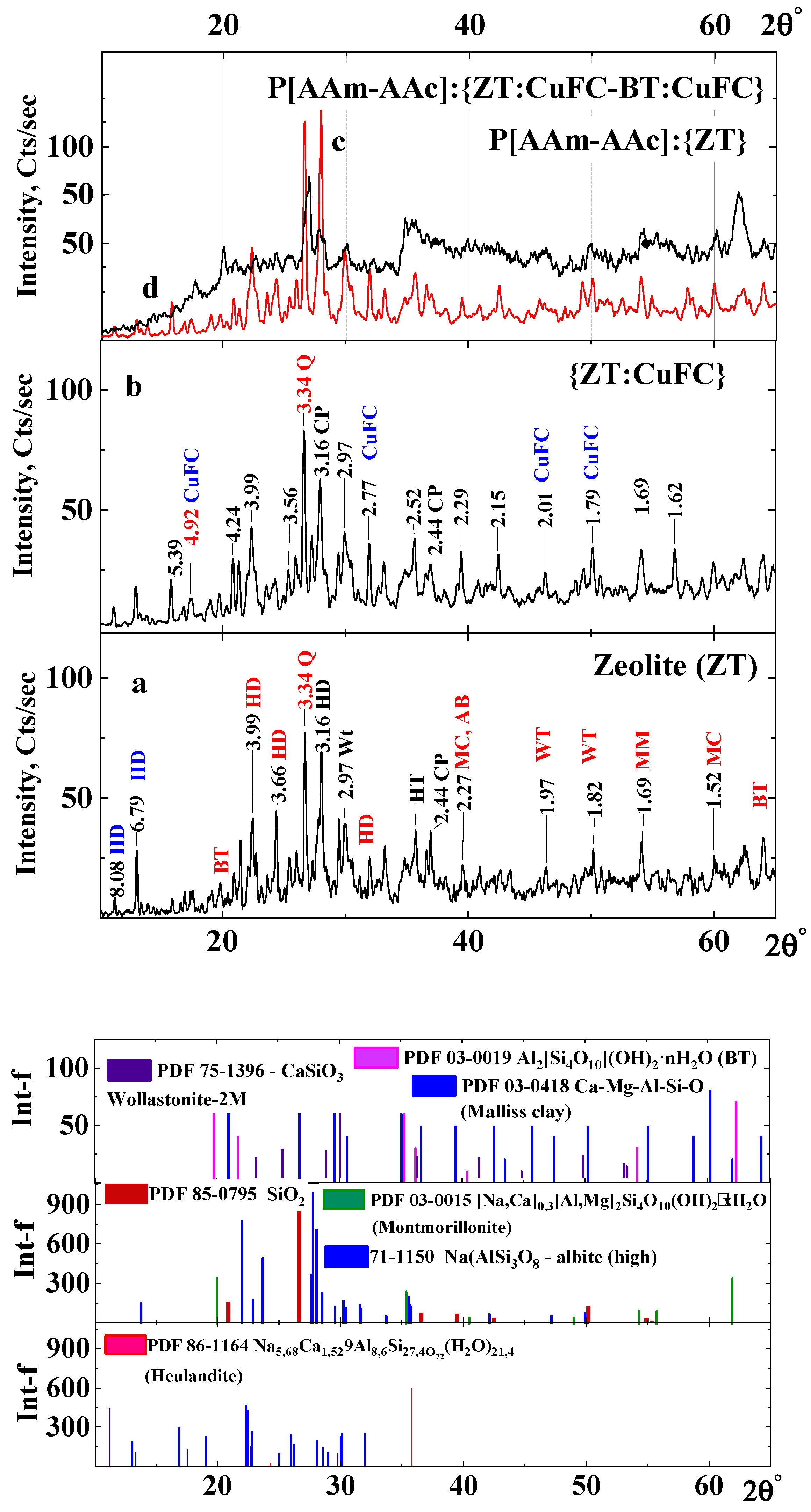
In general, the intensity of the static peak of the determined phase is a rather complex function and besides depends on the presence of other phases in the mineral sample. Multiphase often leads to overlapping information reflexes of the phases under study and instead of well-resolved peaks one has to deal with a multiplet. For example, the presence of various forms of silica in natural zeolite samples leads to partial or complete coincidence of their main diffraction reflections, as well as overlap with reflections of other mineral phases [34].
It can be seen (Figure 3) that quartz, being the main analytical peak, appears both in an individual sample of zeolite and in its forms intercalated with copper ferrocyanide (Figure 3a,b) [18,20,34]. The Bragg diffraction reflections obtained in diffraction patterns appear in angular dispersion and have a complex shape with significant asymmetry in the distribution of angular intensity and a significantly different functional character near the apex (monopics and dublets) and base area for each reflection. In addition to the siliceous substance, the clay minerals (bentonite, montmorillonite, albite, kyanite, mica) and quartz minerals (wollastonite) take part in the formation of zeolite rock. Diffraction lines of heulandite appear from the zeolite rock. It can be seen that the diffraction lines of predominantly heulandite are preserved when copper ferrocyanide microcrystals are intercalated into the zeolite body (2θ = 22.46° and 30.63°).
Previously, the radiographic identity of the framework of clinoptilolite and heulandite was shown, but they differ from each other in the topology [35,36] on the basis of generalized chemical analyses of heulandite and clinoptilolite samples that have established limits for changes in the Si/Al ratio. It means that this ratio for heulandite is in the range of 2.75 ÷ 3.25, while for clinoptilolite it is 4.25 ÷ 5.25, respectively. In addition, it is known that these minerals differ in their cation content. In clinoptilolite, the molecular sum of Na + K higher than (Ca + Mg), and vice versa in heulandite. According to X-ray fluorescence analysis data for the zeolite sample studied, the ratio Si/Al = 3.75, and the total amount of (Na+ + K+) = 4.05 and bivalent cations (Ca2+ + Mg2+) = 7.36. Based on this, it can be argued that the main mineral of the framework structure in the zeolite is heulandite. Despite the similarity of the X-ray data of the minerals, they differ in the size of the channels. In the structure of heulandite, the aluminum–silicon–oxygen tetrahedra are grouped into 4-, 5- and 6-membered rings, and in the structure of clinoptilolite into 3-, 4- and 5-membered rings. The dimensions of the channels formed by 10- and 8-membered tetrahedral rings of uclinoptilolite are somewhat larger than those of heulandite [18,20].
The next stage of modification of the zeolite and its intercalated with copper ferrocyanide (CuFC) form was percolation of macromolecules of the cross-linked P[AAm-AK] copolymer into their matrix by means of in situ polymerization. As a result of this procedure, there was a decrease or disappearance of some peaks characterizing predominantly clay rocks of a layered structure (albite, bentonite, etc.) and an increase in the intensity of the peak of quartz and heulandite at 26.64° and 27.96°, respectively. A significant amorphization of the structure (Table 3) of the percolated complexes of P[AAm-AAc]:{ZT:CuFC-BT:CuFC} is observed. The disappearance of the bands may be associated with the dissolution of some impurities of clay rocks in acrylic acid during the three-dimensional copolymerization with acrylamide in the bulk of the natural mineral, preserving the structures of quartz and heulandite.
At synthesis of the P[AAm-AAc]:{ZT-BT} hybrid composite, the amorphousness increases from 28.8% to 76.47% with a decreasie of the content of main mineral phases, which may be due to the amorphous nature of the polymer matrix as well. The polymer matrix formed in the interlayer space of bentonite and porous channels of zeolite can cause local tensile stresses in the natural minerals’ matrices.
Dealumination of the framework also occurs along with decationization during acid treatment of high-silica zeolites. Therefore, it was of interest to study how this affects the stability of zeolite particles in alkaline solutions, which is discussed in the next section of the article. Treatment of high-silica zeolites with acids leads to the sequential replacement of cations with hydronium ions. Longer treatment with acids leads to the removal of aluminum ions from the framework, each of which is replaced by four hydroxyl groups while maintaining a high degree of crystallinity [34].
The studies on acid decationation of zeolites show that this process is characterized by a complex mechanism [37,38]. Under the influence of concentrated solutions of acids, the crystal structure is destroyed with the simultaneous transition of cations and anion-forming atoms into the solution. However, the reactions with dilute acid solutions reveal two stages of the acid decationization process, namely, ion exchange and dealumination. Let us consider the corresponding schematic reactions of deionization of high-silicon zeolites:
- I.
- Ion exchange
- II.
- Dealumination
From the above schematic reactions of acid decationation of high-silicon zeolites, it is clear that at the stage of ion exchange, the hydrogen form of the original minerals can be obtained, which, by treating with solutions of the appropriate salt or alkali, can be converted into the original cationic form, that is, the hydrogen form is obtained without destroying the exchange centers. In this case, the proton reacts with zeolite water to form an oxonium cation. The reversibility of exchange reactions indicates the absence of interaction of the proton with the matrix of ion-exchange minerals.
In connection with the above, we can conclude that during in situ polymerization of a zeolite swollen in a solution of acrylamide and acrylic acid, the main framework of the zeolite matrix and ion exchange centers is preserved. The percentages of the main phases of zeolite and the degree of its amorphization at each stage of its modification were calculated based on X-ray diffraction analysis data.
The analysis results reveal the presence and co-existence of intercalated and percolated structures within the hybrid compositions (Figure 3c,d). The limited intercalation in P[AAm-AAc]:{ZT} is due to the rigid framework of zeolites, which can accommodate only a certain amount of polymer chains in their channels. Meanwhile in the presence of bentonite particles, the XRD patterns becomes more amorphous due to the layered structure of clay mineral. The XRD patterns for P[AAm-AAc]:{ZT:CuFC-BT:CuFC} show a decline in intensity both of quarts and heulandite that can be attributed to the disordered structure of bentonite.
Thus, the FT-IR spectra and XRD patterns of ZT and P[AAm-AAc]:{ZT} as well as P[AAm-AAc]:{ZT-BT} established that the zeolite and bentonite particles were relatively uniformly dispersed in the polymer matrix. The interfacial interaction between P[AAm-AAc] and ZT and BT was the binding formation between acrylamide and carboxylic groups of polymer matrix and the siloxane oxygen atoms or hydroxylated edge sites of minerals. Polymer impregnation into channels and holes of zeolite and interlayer of bentonite should also be possible [39,40]. The structural evaluation of BT and its composite with P[AAm-AAc] were provided in earlier studies [13,14], where similar features were reported for BT and P[AAm-AAc]:{BT}, with the difference of intercalation of clay layers by cross-linked polymer chains.
It should be noted [23] that not all phases present in a real sample fully satisfy the Debye condition and the shape of the resulting additive profile is the sum of lines most often described by different profile functions, which distinguishes the X-ray multiplet, for example, from the Mössbauer spectrum.
3.1.3. Mössbauer Spectroscopy
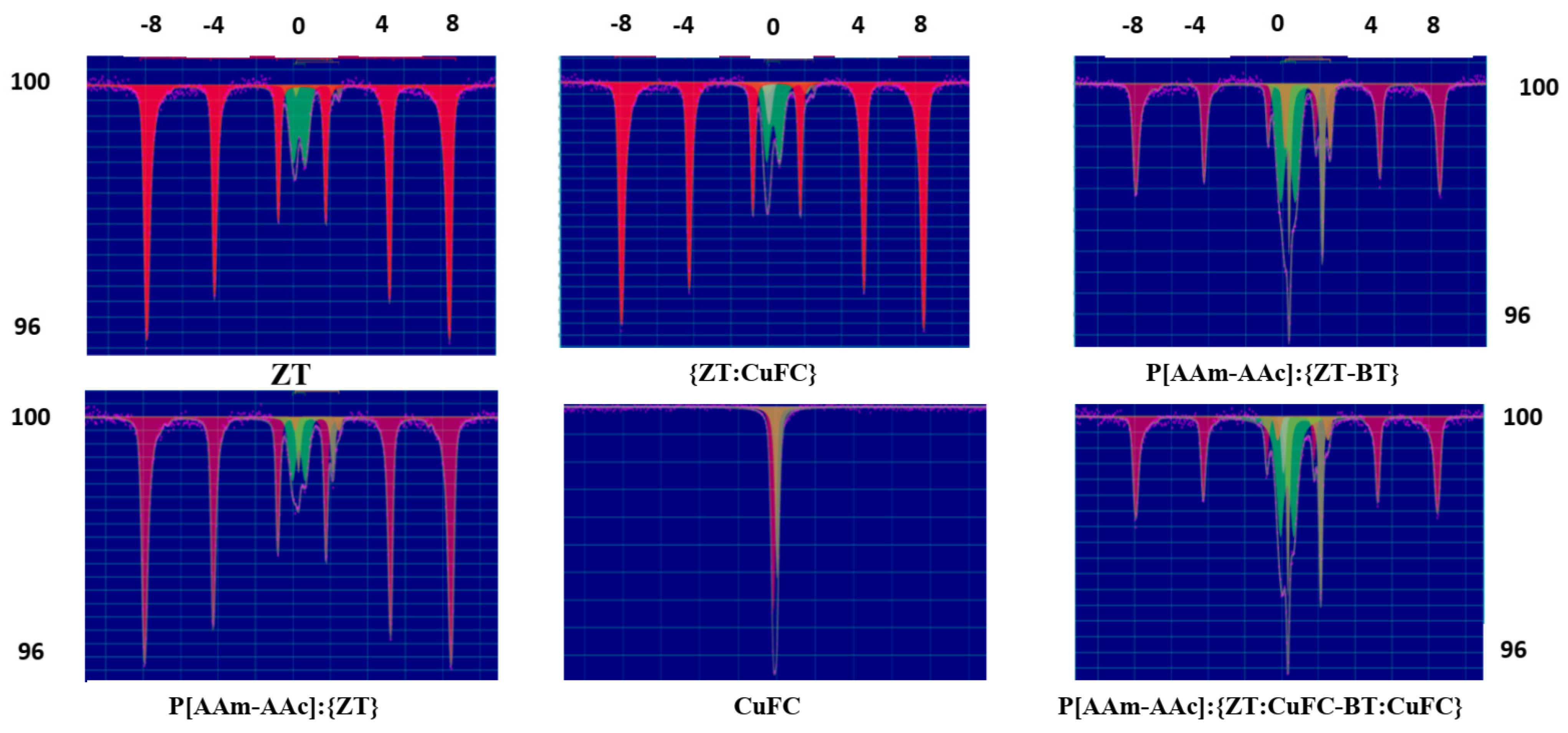
The problem of identifying subspectra with specific sites in minerals is not a trivial one, because most naturally occurring silicates are intermediate members of solid solution series, and therefore, include both iron and other metallic cations distributed more and less random over “equivalent” crystallographic sites [41]. In the last two decades, Mössbauer spectroscopy (MS) has proved to be a very suitable tool for the characterization of iron oxides and (oxy)hydroxides in soils and sediments” [41,42,43,44]. Because the various components commonly possess a small-particle morphology and occur mainly in close association with each other or with other minerals, the information obtained from MS is often much more direct, and in this regard, the crystalochemical structure of iron-containing zeolite minerals were studied by Mössbauer spectroscopy. Mössbauer spectra were recorded in the speed range of −11 ÷ + 11 mm/s at room temperature (Figure 4).
Two variants of multicomponent Mössbauer spectra of zeolite and its intercalated with copper ferrocyanide and percolated with a polymer matrix forms were obtained as a result of the measurements. All studied spectra have a sextet structure and the results of their parameterization are given in Table 4. In the spectra, a sextet is always distinguished relating to well-crystallized hematite and characterized by a hyperfine magnetic field value (H) of 508.7 kO, isomer shift δ = 0.37 mm/s and quadrupole splitting QS = −0.22 mm/s. The hematite phase is easily distinguished from other isomeric phases, such as maghemite or ε-hematite, by negative quadrupole splitting (QS). Three doublets are also observed with a chemical shift from 0.27 cm/sec to 1.13 mm/s and a quadrupole splitting from 0.64 mm/s to 2.10 mm/s, indicating that iron in the systems is presented in different valence states and can be included in the structure of several aluminosilicate mineral phases (quartz, heulandite, bentonite, montmorillonite, mica), while being distributed in some of them over non-equivalent structural positions [8]. The analysis of obtained data evidenced that the oxide phases in the zeolite are represented by hematite, which corresponds to its own sextets of lines with different values of the hyperfine field strengths on the iron nuclei. Due to the ultra-fine field on the iron nuclei in hematite, it can be easily distinguished from other iron oxides and hydroxides, even at room temperature.
According to Mössbauer spectra, more than 80% of the iron in zeolite is represented by aluminum-containing hematite. The remaining iron is attributed to aluminosilicate phases of other associated minerals. Consequently, the main part of the iron ions in the zeolite is in the trivalent state, occupying predominantly octahedral, and to a lesser extent, tetrahedral positions of the aluminosilicate framework.
The data for Fe2+ at M2 cluster tightly around QS = 2.37 mm/s and δ = 1.13 mm/s, suggesting that the local geometry about the M2 site is not radically affected by the broad compositional range of these samples. For the data for Fe2+ at Ml, one group of analyses clusters around Δ = 1.94 mm/s and δ = 1.15 mm/s. For Fe2+, lower quadrupole splitting generally implies more distortion around a site (the reverse is true for Fe3+), owing to some combination of the effects of metal-to-oxygen distances and angular components.
Mössbauer data for Fe3+ sites are less well defined and the wider range of values may be due to a problem with the fitting procedures and is not dependent on compositional effects. Fe3+ at M2 has a similar range of δ = 0.27–0.33 mm/s with a range mm/s of QS = 0.78–0.64 mm/s, respectively. The Fe3+ peaks are almost always less intense than the corresponding Fe2+ peaks in the same samples. In general, fitted Mössbauer doublets are less precise when peak areas are small. The samples in which Fe3+ Δ values lie between 0.60 mm/s and 1.0 mm/s probably represent spectra in which Fe3+ was present at both M2 and Ml, but in such small quantities that it was impossible to distinguish them [42,45]. The resultant single Fe3+ doublet in such samples has a QS that is the average of the quadrupole splitting values for the two separate sites.
It can be seen that In the case o” int’rcalation of copper ferrocyanide or percolation of the P[AAm-AAc] copolymer into the bulk of the crystalline zeolite matrix, a decrease in the total content of hematite and a noticeable increase in the content of Fe2+ and Fe3+ are observed. According to literature data [46,47], in the systems studied, Fe2+ ions occupy mainly the octahedral positions M1 and M2, which are characterized by trans- and cis-coordination of the arrangement of a pair of hydroxyl groups, respectively. The occupation of the M2 position by iron ions (II and III) leads to large lattice distortions. Thus, in Mössbauer spectra, such positions are assigned the most intense doublet with a large quadrupole splitting. Accordingly, the doublet with a lower quadrupole splitting is attributed to Fe2+ ions in the M1 position.
In the case of the mixed composition of P{aAm-aAc]:{ZT-BT}, the amount of divalent iron, occupying predominantly octahedral positions, increases significantly [14]. The occupation of the M2 position by these ions leads to large lattice distortions. Therefore, in Mössbauer spectra, such positions are assigned the most intense doublet with a large quadrupole splitting. Therefore, the doublet with a smaller contribution to the quadrupole splitting is attributed to Fe2+ ions in the M1 position. Their contribution to QS is attributed to Fe2+ ions in the M1 position. The doublets corresponding to Fe3+ ions are also attributed to different structural positions with a large quadrupole splitting: with a large QS to octahedral M2, with a smaller quadrupole splitting, they are classified as octahedral M1 or tetrahedral [45].
Thus, in the case of percolation of macromolecules of the P[AAm-AAc] copolymer and intercalation of CuFC into the volume of the zeolite mineral matrix, a redistribution of divalent and trivalent iron ions is observed. In the case of intercalation of copper ferrocyanide into the bulk of the zeolite, a decrease in the percentage of Fe2+ in hematite is observed with an increasing of the content of Fe3+ located in the octahedral structures of aluminosilicates impurities. In the case of a mixture of zeolite and bentonite (1:1) percolated with a polymer matrix, a significant decrease in the wing parts of the hematite sextet and an increase in the intensity of the peak characteristic of divalent iron in the octahedral structures of bentonite with δ = 1.33 mm/s and quadrupole splitting QS = 2.42 mm/s are observed [48,49].
Therefore, we can conclude that the main share of iron in the zeolite is accounted for by the share of aluminum-containing hematite. The remaining part of Fe2+ and Fe3+ belongs to the aluminosilicate phases of clay minerals. The parameters of the corresponding three doublets are in good agreement with literature data [49]. The main part of iron is in the trivalent state, occupying mainly octahedral and to a lesser extent tetrahedral positions. A small amount of iron ions is present in the lattices of clay minerals in the +2 oxidation state, occupying octahedral positions.
It is also clear that the intercalation of ferrocyanide ions into the volume of mineral matrices leads to the appearance of a singlet line with an isomeric shift δ = 0.07 mm/s and 0.09 mm/s for the ZT:CuFC and P[AAm-AAc]:{ZT:CuFC-BT:CuFC}, respectively. The Mössbauer spectrum for CuFC is shown for comparison (Figure 4). Characteristic for all systems is that quadrupole splitting is Δ = 0 mm/s. This may be due to the coexistence in these compounds of individual K4[Fe(CN)6] molecules and the copper ferrocyanide complex [14,48].
The results of the analysis of Mössbauer spectra show good agreement with the data of X-ray diffraction analysis, confirming he position that the degree of amorphization of percalated forms of natural minerals increases when a polymer matrix is incorporated into their structure. In this case, there is an increase in the Fe2+/Fe3+ ratio from 0.15 to 0.73 and 1.11 in the hybrid compositions P[AAm-AAc]:{ZT} and P[AAm-AAc]:{ZT-BT}, respectively. This indicates the loosening and partial destruction of the crystal lattice of natural mineral matrices during the process of in situ polymerization of acrylamide with acrylic acid, which promotes the free movement of iron ions (II and III) of different positions (octahedral positions M1, M2 and tetrahedron) within one zeolite matrix, and between the zeolite and bentonite matrices.
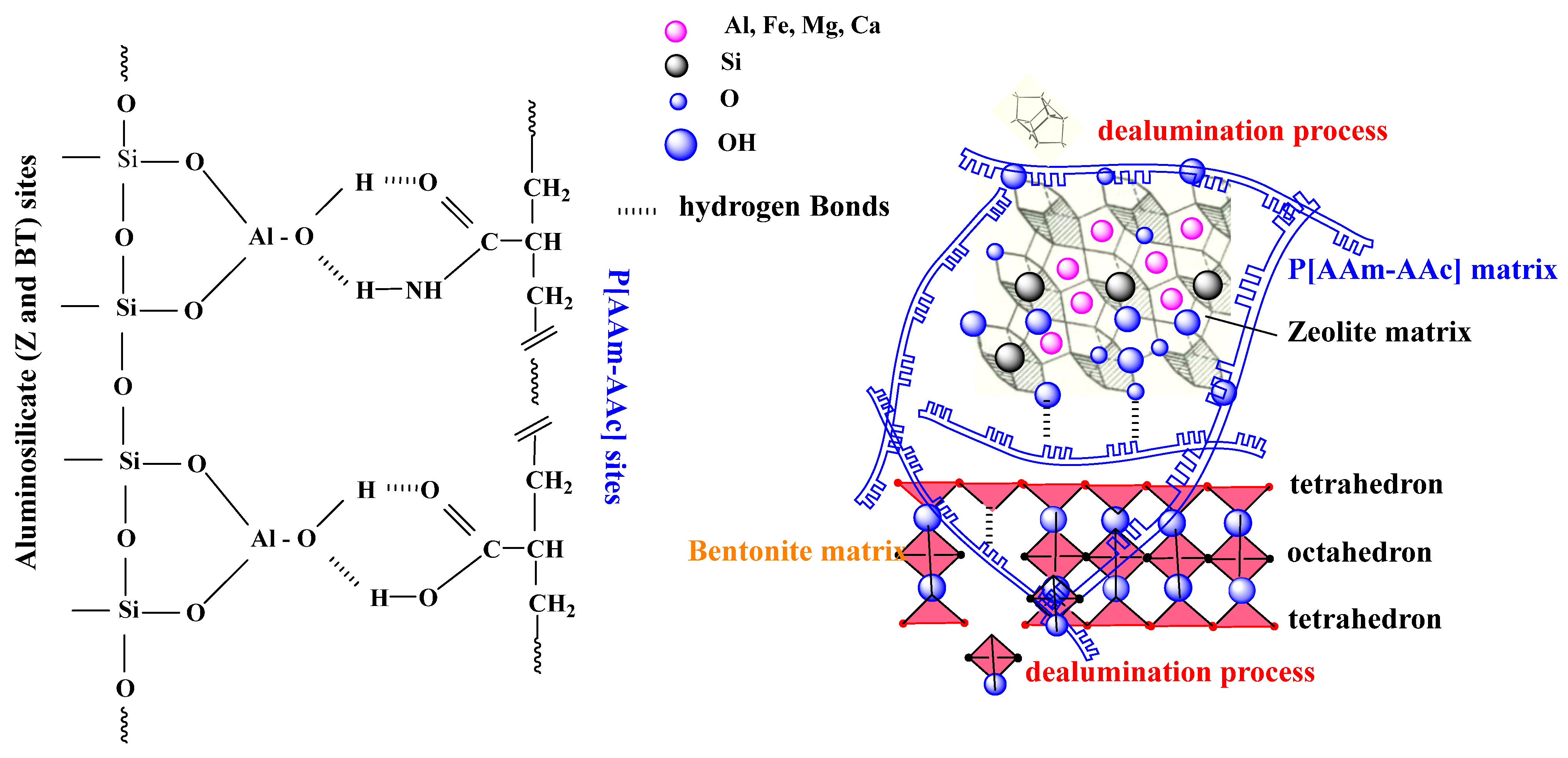
When layered silicates are modified by macromolecules of the P[AAm-AAc] copolymer, a process of partial disruption of the structure of the interlayer space, as well as the main layers of the octahedral and tetrahedral network of the mineral matrix, can occur. This may be reflected through the broadening of the doublet lines and a slight increase in the magnitude of the quadrupole splitting of Fe2+ ions and through the transition of some Fe3+ ions to a more distorted position. A decrease in the chemical shift, which is equivalent to an increase in the density of s-electrons near the iron nucleus, and a broadening of the splitting line, is associated with a decrease in the symmetry of the environment of iron ions. The following scheme for the formation of the hybrid composition P{AAm-AAc]:{ZT-BT} (Figure 5).
The dealumination process occurs without destruction of the framework structure and can be represented as the replacement of Al3+ by 3H+ with the formation of neutral structures of four hydroxyls in dealuminated ≡Si-OH zeolite instead of polar [AlO1/2]− Me+ groups.
We believe that the process of in situ polymerization in the bulk of the zeolite can also occur in the pores of the mineral matrix, which can lead to an increase the rigidity and adhesion forces between aluminosilicate particles as well. This may be due to the fact that polymerization will proceed inside the pores of zeolite particles with the release of the growing polymer chain into the interparticle space, where chain termination takes place. This means that the main polymer mass will be located inside the pores of the zeolite particles, where carboxyl and urea functional groups can form hydrogen bonds with the alumina centers of the natural mineral. In the case of in situ polymerization in the bulk of bentonite, a cross-linked polymer matrix is formed in the interlayer space of aluminosilicate units [14]; therefore, hybrid compositions are more elastic with a greater margin of compressive and tensile strength [50] compared to the zeolite–polymer composition, which show greater brittleness under compression and low degree of elongation.
In the case of in situ polymerization of acrylamide and acrylic acid in a mixture of two mineral sorbents, that is, zeolite and bentonite, an even relatively distribution of solid particles occurs in the volume of the cross-linking polymer. This can lead to the growth of active centers of sorption and exchange of metal ions in solution and in mineral sorbents. It should be noted that in situ polymerization in the presence of zeolite is complicated by the fact that the natural mineral particles have a density of 2.1 g/cm3, that is, higher than the density of water and aqueous solutions of a mixture of monomers. At the same time, it is difficult to keep the system from premature sedimentation of the zeolite before the start of the polymerization process, and therefore, the polymerization process must proceed at mixing, which can cause uneven distribution of solid particles in the volume of the polymer gel.
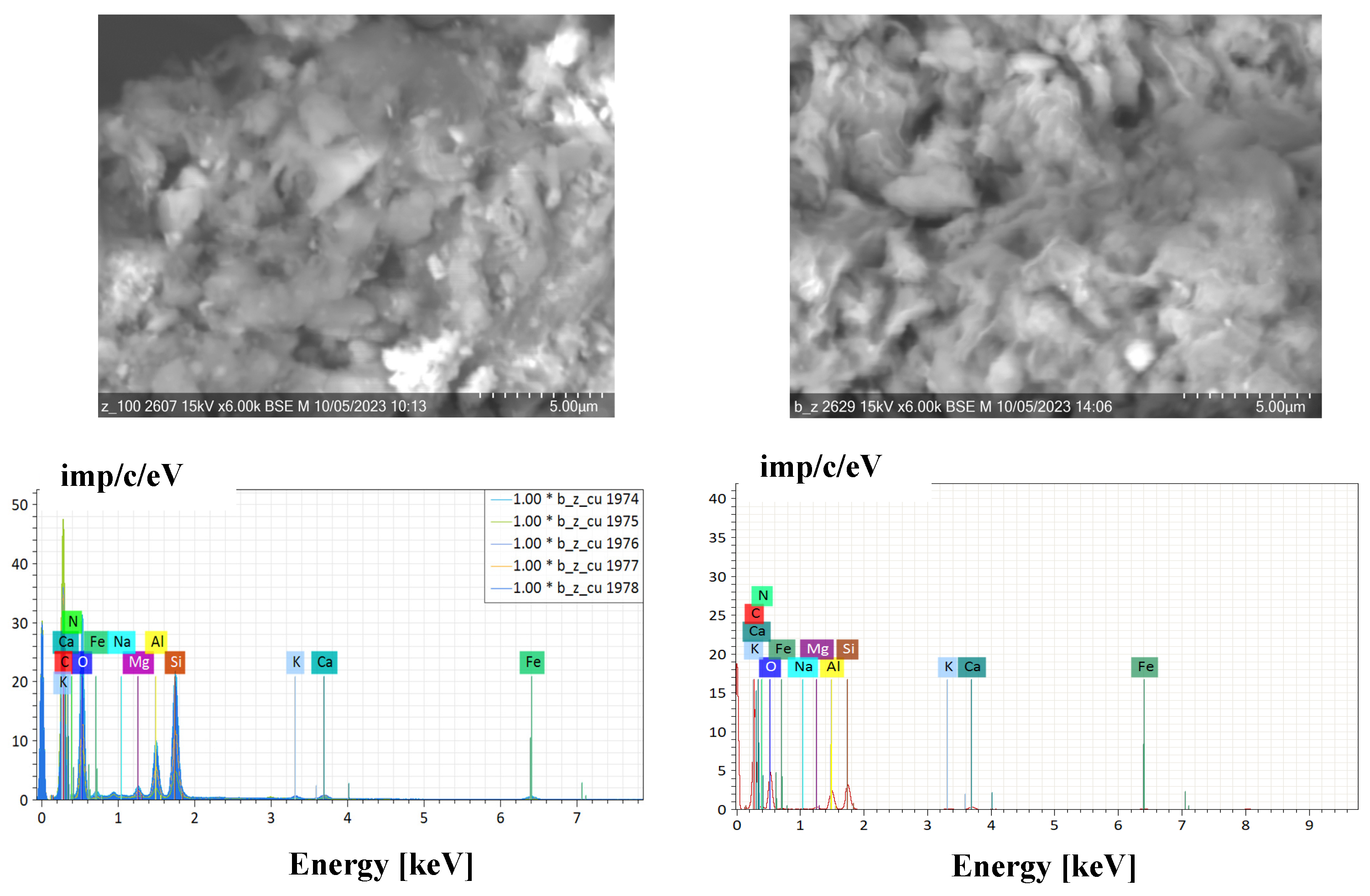
Microphotographs of the proposed composition are presented in Figure 6. The structure of the sample is heterogeneous, composed of various mineral phases, held together quite tightly and evenly distributed throughout the structure by a polymer binder, as can be seen from the texture. However, the surface morphology of the zeolite and bentonite is more irregularly heterogeneous and there is a presence of large clumps that cover the pores. There is no substantial difference in surface morphology of P[AAm-AAc]:{ZT} and P[AAm-AAc]:{ZT-BT} as a result of partial dealumination during in situ polymerization.
It can be concluded that the formation of a hybrid composition is not only due to adsorption interactions, but also the occurrence of chemical interactions between the functional surface groups of natural minerals and macromolecules of the cross-linked polymer matrix. The directed structural modification of mineral sorbents opens up a fundamentally new approach to the creation of polymer composite materials characterized by high physical and mechanical properties and adsorption characteristics in relation to radionuclides of artificial and natural origin.
Thus, the presence of an amorphous back-ground in all peaks also indicates the semi-crystalline structure of hybrid composites. The analysis results reveal the presence co-existence of intercalated, exfoliated and dealiminated structures within the different hybrid composites. The limited intercalation is due to the rigid framework of zeolites, which can accommodate only a certain amount of polymer chains in their channel.
3.1.4. Sorption of Radionuclides
In an earlier study [8], it was mentioned that the aggregation and coagulation of zeolite and clay particles under varying conditions of temperature and electrolytes lead to variations in the flow properties of these minerals. This is an undesired feature for their practical use as adsorbents [10,11,12]. The presence of a composite of a mineral and a polymer, in which the mineral dispersed in the polymer network, may enable the use of mineral itself as an adsorbent confined in an isolated and practically usable medium in aquatic solutions. In addition, the particles embedded in a network strengthen the gel and prevent its collapse in bad solvents. The enhancement in adsorptive features of the mineral can also be possible with additional surface modifications.
The chemical composition of the framework (the Si/Al ratio in the range of 3–5) modulates the ion-exchange properties of the zeolite expressed as cation-exchange capacity (CEC). The rather low Si/Al ratio and the consequently increased CEC render heulandite type zeolites advantageous for binding dissolved cations from aqueous solution. Of particular interest is the interaction of heulandite with metallic cations, which are sorbed by the zeolite and immobilized in the crystals, modifying the initial zeolite structure. This is crucial for environmental technology (for example, decontamination of wastewaters) and for certain industrial applications [51,52,53,54]. The dissolved metallic cations interact with the HEU-type zeolite crystals, and they are subsequently removed from the solution, through different sorption mechanisms including principally the so-called ion exchange. A typical ion-exchange mechanism is supposed to be a reversible chemical reaction wherein an ion from the solution is exchanged for a similarly charged ion which is initially bonded to a solid, behaving as ion-exchanger.
Researchers [55,56,57] have, therefore, been interested in immobilizing the adsorbents in polymer matrices that will provide mechanical support and minimize agglomeration. This results in a new branch of polymer composite materials, where the adsorbent/filler is embedded and disperse within a polymer matrix, thus increasing the effective surface area and improving regeneration as well as the operation of the technique.
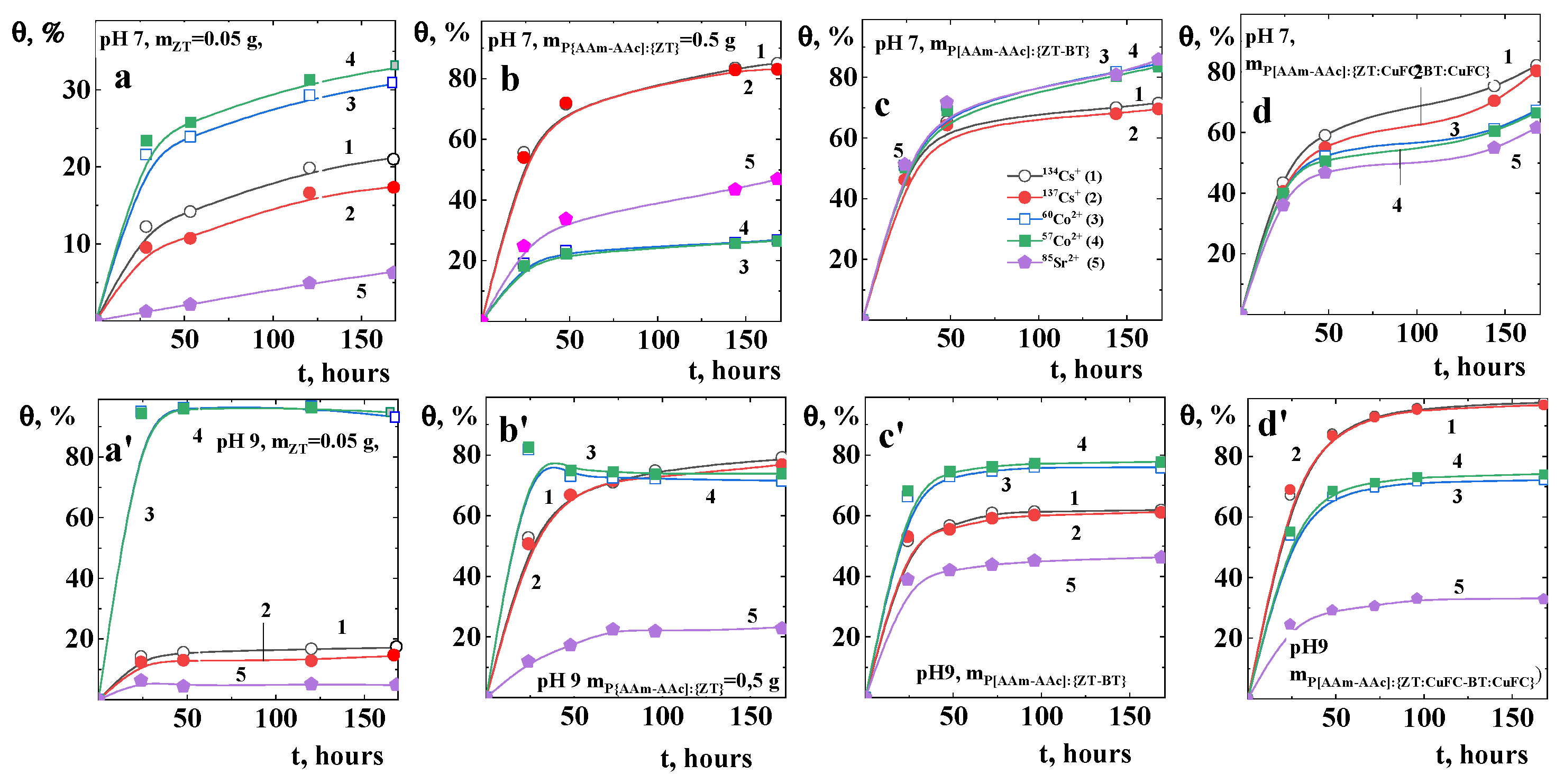
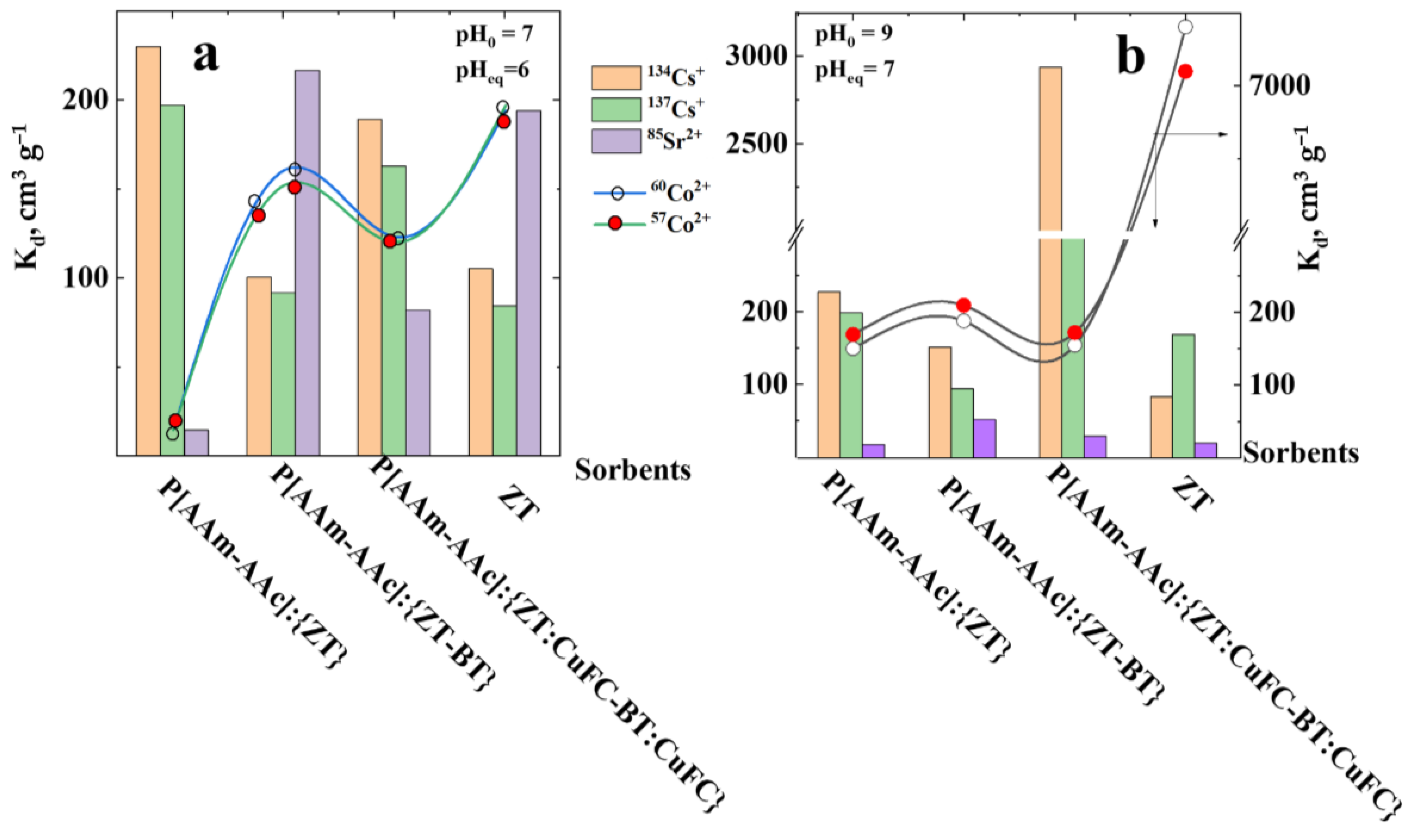
From Table 5 and Figure 7 and Figure 8, it can be seen that the degree of binding and distribution coefficient of radionuclides between the liquid medium and sorbents of various compositions and structures is both a function of the pH of the medium and the nature of the radionuclides themselves. Since the base sorbent is the natural mineral zeolite, further assessment of the influence of its component-structural transformations on the sorption of radionuclides was compared by its net mass in its pure form and as part of hybrid compositions. That is, the mass of zeolite and its mixtures with bentonite, their intercalated forms with copper ferrocyanide in the volume of the polymer matrix is equal to the mass of the original mineral sorbent (0.05 g). Kinetic studies have shown that the time to establish equilibrium state of sorption of radionuclides by zeolite and its hybrid polymer composites is 168 h, above which sorption proceeds relatively slowly and reaches a saturation plateau. It can be seen that both the percolation of P[AAm-AAc] macromolecules into the volume of the mineral matrix of both zeolite and its mixture with bentonite and their intercalated forms lead to a significant increase in the absorption of radionuclides. It was previously noted that the highest degree of sorption of cesium and strontium radioisotopes occurs in the alkaline region [56]. Zeolite preferentially sorbs cobalt radionuclides, while its percolated polymer matrix form preferentially sorbs alkali and alkaline earth metal ions. The hybrid composition P[AAm-AAc]{ZT-BT} actively sorbs divalent radioisotopes of cobalt and cesium. In the case of the P[AAm-AAc]{ZT:CuFC-BT:CuFC} system, preferential sorption of monovalent strontium radioisotopes is observed. With an increase in the pH of the environment, a significant decrease in the radiation background created by the isotopes 57Co2+ and 60Co2+ is observed with suppression of the sorption of cesium and strontium isotopes.
This indicates a change in the binding form of cobalt. At the initial stage, when adjusting the pH up to 9.0 of the medium by adding NaOH, both partial precipitation of cobalt hydroxide and ion exchange processes with mobile ions (Ca2+, Mg2+, Na+, etc.) of the mineral matrices of zeolite and bentonite occur. Over time, cobalt hydroxide can dissolve under the influence of protons of carboxyl groups and interact with carboxylate ions of the polymer matrix due to electrostatic interactions by the formation of a polymer–metal complex Co2+ ···2(-COO−), which can also be stabilized by donor-acceptor bonds with amide group of polyacrylamide [12].
It can be seen that in the case of binary mineral mixtures in the volume of the polymer matrix at pH ≈ 7, predominant sorption of alkali and alkaline earth metal ions is observed. In a more alkaline pH ≈ 9 environment, sorption and precipitation of cobalt hydroxide on the surface and pore space of sorbents predominates. In compositions with copper ferrocyanide intercalated into a matrix of mineral components, cesium ions are predominantly sorbed, as one would expect.
The high cesium sorption by the intercalated mineral matrix in the polymer network body can be explained by the fact that the main part of exchangeable ions of modified mineral matrix are potassium ions, for which ion exchanged with cesium ions proceeds more efficiently compared to strontium and cobalt ions. The maximum sorption capacity preferentially in the order of Cs+ ≥ Co2+ > Sr2+. The reason for such behavior was explained from the thermodynamic point of view [57] in accordance to which the sorption of Sr2+ indicate endothermic sorption process, whereas the sorption of Cs+ and Co2+ are exothermic in nature.
It is possible to present the following mechanism for binding of radionuclide ions with hybrid composition components: (A) complex formation with the -COOH groups of the polymer and ≡SiOH and ≡AlOH sites of mineral matrices with the release of protons, which explains the decrease in the pH of the medium; (B) ion-exchange process between sodium, calcium and magnesium ions of a natural mineral as well as cesium and strontium ions in external solution, respectively; (C) coordination binding of transition metal radionuclide ions (Co2+) with electron-donating oxygen and nitrogen atoms of the carboxyl and amide groups of the polymer matrix, respectively; and (D) incorporation into the cubic structure of copper hexaferrocyanide particles intercalated into clay, especially cesium ions, which is associated with the correspondence of the diameter of dehydrated Cs+ with the size of the entrance windows of transition metal hexaferrocyanides and the minimum solubility of cesium and strontium in hexaferrocyanide complexes [14].
4. Conclusions
Thus, polymer compositions based on zeolite and bentonite, as well as their intercalation with copper ferrocyanide in the bulk of a weakly cross-linked polymer matrix of acrylamide and acrylic acid, have been synthesized and identified. The high sorption capacity of the resulting compositions in relation to artificial radionuclides in comparison with individual mineral ion-exchange materials has been shown. It has been established that a process of partial dealumination of mineral matrices takes place during in situ polymerization that leads to an increase in their sorption capacity due to the growth of the internal cavity volume of the sorbents during percolation of macromolecules into their particles during the in situ polymerization process.
The results obtained in this investigation reveal that hybrid composites based on zeolite and bentonite as well as their intercalated with CuFC forms have a potential in purification of LRW effluent. An added advantage is that the P[AAm-AAc] polymer used in this study is hydrophilic enhancing the removal of metal ions due to the electrostatic and donor-acceptor mechanisms of interaction between them. Besides, zeolite and bentonite are readily available and cheap and environmentally benign, therefore alleviating problems of preventing the spread of LRW in the soil. The possibility of reuse renders this technique both economical and effective.
Author Contributions
Conceptualization, G.K.M.; methodology, D.A.Z., O.S.M.; investigation, D.A.Z., O.S.M., Y.R.N.; resources, G.K.M.; writing—original draft preparation, G.K.M.; writing—review and editing, G.K.M.; project administration, G.K.M. All authors have read and agreed to the published version of the manuscript.
Funding
The work was performed under financial support of the Ministry of Energy of the Republic of Kazakhstan within the framework of the Scientific-Technical Program BR 09158 958 “Development of nuclear physical methods and technologies for innovative modernization of economy of Kazakhstan”.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nisbet, A.F. Application of fertilizers and ameliorants to reduce soil to plant transfer of radiocaesium and radiostrontium in the medium to long term—A summary. Sci. Total Environ. 1997, 137, 173–182. [Google Scholar] [CrossRef]
- Sweeck, L.; Wauters, J.; Valcke, E.; Cremers, A.; Belli, M. (Eds.) Transfer of Radionuclides in Natural and Semi-Natural Enviroments; Elsevier Applied Science: London, UK, 1990; pp. 249–258. [Google Scholar]
- Myasoedov, B.F.; Novikov, A.P.; Pavlotskaya, F.I. Comprehensive geochemical studies of radionuclides behavior in aquatic and terrestrial ecosystems in the near impact zone of the PO “Mayak”. Methodology, objects and methods of research. Radiokhimiya [Radiochemistry] 1998, 40, 447–452. (In Russian) [Google Scholar]
- Fan, Q.; Yamaguchi, N.; Tanaka, M.; Tsukada, H.; Takahashi, Y. Relationship between the adsorption species of cesium and radiocesium interception potential in soils and minerals: An EXAFS study. J. Environ. Radioact. 2014, 138, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yasunari, T.J.; Stohl, A.; Hayano, R.S.; Burkhart, J.F.; Eckhardt, S.; Yasunari, T. Cesium-137 deposition and contamination of Japanese soils due to the Fukushima nuclear accident. Proc. Natl. Acad. Sci. USA 2011, 108, 19530–19534. [Google Scholar] [CrossRef] [PubMed]
- Aguila, B.; Banerjee, D.; Nie, Z.; Shin, Y.; Ma, S.; Thallapally, P.K. Selective removal of cesium and strontium using porous frameworks from high level nuclear waste. Chem. Commun. 2016, 52, 5940–5942. [Google Scholar] [CrossRef] [PubMed]
- Nilchi, A.; Saberi, R.; Moradi, M.; Azizpour, H.; Zarghami, R. Adsorption of cesium on copper hexacyanoferrate–PAN composite ion exchanger from aqueous solution. Chem. Eng. J. 2011, 172, 572–580. [Google Scholar] [CrossRef]
- S¸ims¸ek, S.; Ulusoy, U. UO22+, Tl+, Pb2+, Ra2+, Bi3+ and Ac3+ adsorption onto polyacrylamide-zeolite composite and its modified composition by Phytic Acid. J. Radioanal. Nucl. Chem. 2004, 261, 79–86. [Google Scholar] [CrossRef]
- Yudaev, P.; Butorova, I.; Stepanov, G.; Chistyakov, E. Extraction of Palladium(II) with a Magnetic Sorbent Based on Polyvinyl Alcohol Gel, Metallic Iron, and an Environmentally Friendly Polydentate Phosphazene Containing Extractant. Gels 2022, 8, 492. [Google Scholar] [CrossRef]
- Pefferkorn, E. Polyacrylamide at Solid/Liquid Interfaces. J. Colloid Interface Sci. 1999, 216, 197–220. [Google Scholar] [CrossRef]
- Luckham, P.F.; Rossi, S. The Colloidal and Rheological Properties of Bentonite Suspensions. Colloid Interface Sci. 1999, 82, 43–92. [Google Scholar] [CrossRef]
- Starodoubtsev, S.G.; Ryabova, A.A.; Dembo, A.T.; Dembo, K.A.; Aliev, I.I.; Wasserman, A.M.; Khokhlov, A.R. Composite gels of poly(acrylamide) with incorporated bentonite. Interaction with cationic surfactants, ESR and SAXS study. Macromolecules 2002, 35, 6362–6369. [Google Scholar] [CrossRef]
- Mamytbekov, G.K.; Zheltov, D.A.; Koztaeva, U.P.; Matienko, L.D. Hybrid Composite Materials for Immobilization of Radionuclides in Liquid Radioactive Waste. J. Miner Sci. Mater. 2023, 4, 1053. [Google Scholar] [CrossRef]
- Mamytbekov, G.K.; Zheltov, D.A.; Nurtazin, Y.R. Synthesis and Investigation of the Properties of Biphasic Hybrid Composites Based on Bentonite, Copper Hexacyanoferrate, Acrylamide and Acrylic Acid Hydrogel. Polymers 2023, 15, 2586. [Google Scholar] [CrossRef] [PubMed]
- Attallah, M.F.; Allan, K.F.; Mahmoud, M.R. Synthesis of poly(acrylic acid–maleic acid) /SiO2/Al2O3 as novel composite material for cesium removal from acidic solutions. J. Radioanal. Nucl. Chem. 2016, 307, 1231–1241. [Google Scholar] [CrossRef]
- Milyutin, V.V.; Nekrasova, N.A.; Belousov, P.E.; Krupskaya, V.V. Sorption of radionuclides 137Cs, 90Sr and 233U on various Natural Sorbents. Radiochemistry 2021, 63, 741–746. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, M.; Du, X.; Feng, S.; Miyamoto, N.; Kano, N. Removal of Cesium from Radioactive Waste Liquids Using Geomaterials. Appl. Sci. 2021, 11, 8407. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry, and Use; John Wiley and Sons: New York, NY, USA, 1974. [Google Scholar]
- Cheetham, A.K.; Day, P. Solid State Chemistry: Compounds; Clarendon Press: Oxford, UK, 1992. [Google Scholar]
- Chelishev, N.F.; Volodin, V.V.; Krukov, V.L. Ion Exchange Properties of Natural High-Silicon Zeolites; Nauka: Moscow, Russia, 1988; 128p. (In Russian)
- Tahraoui, Z.; Nouali, H.; Marichal, C.; Forler, P.; Klein, J.; Daou, T.J. Influence of the Compensating Cation Nature on the Water Adsorption Properties of Zeolites. Molecules 2020, 25, 944. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.I. Infrared Spectra of Minerals; Nedra: Moscow, Russia, 1976; 198p. (In Russian) [Google Scholar]
- Taylor, J.C.; Pecover, S.R. Quantitative Analysis of Phases in Zeolite Bearing Rocks from Full X-ray Diffraction Profiles. Aust. J. Phys. 1988, 41, 323–335. [Google Scholar] [CrossRef]
- Jimenez-Reyes, M.; Almazan-Sanchez, P.T.; Solache-Ríos, M. Radioactive waste treatments by using zeolites. A short review. J. Environ. Radioact. 2021, 233, 106610. [Google Scholar] [CrossRef]
- Plusnina, I.I. Infrared Spectra of Minerals; MSU: Moscow, Russia, 1976; Volume 175. (In Russian) [Google Scholar]
- Novikov, R.G.; Konopnitskii, R.; Tsyganenko, A.A. Distortions in IR Spectra Related to Registration Conditions: II. The Influence of Scattering. Opt. Spectrosc. 2018, 124, 655–659. [Google Scholar] [CrossRef]
- Dobrotvorskaia, A.N.; Gatilova, A.V.; Mursin, P.D.; Rudakova, A.V.; Shchepkin, D.N.; Tsyganenko, A.A. Effect of resonance dipole-dipole interaction on the infrared spectra of adsorbed CF4. Experimental investigation and theoretical modeling. J. Photochem. Photobiol. A Chem. 2017, 354, 4–10. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Applications in Coordination. In Organometallic and Bioinorganic Chemistry; Wiley: New York, NY, USA, 1997. [Google Scholar] [CrossRef]
- Jia, X.; Khan, W.; Wu, Z.; Choi, J.; Yip, A.C.K. Modern synthesis strategies for hierarchical zeolites: Bottom-up versus top-down strategies. Adv. Powder Technol. 2019, 30, 467–484. [Google Scholar] [CrossRef]
- Angell, C.L.; Howell, M.V. Infrared Spectroscopic Investigation of Zeolites and Adsorbed Molecules. IV. Acetonitrile. J. Phys. Chem. 1969, 73, 2551–2554. [Google Scholar] [CrossRef]
- Molinard, A.; Vansant, E.F. Controlled gas adsorption properties of various pillared clays. Adsorption 1995, 1, 49–59. [Google Scholar] [CrossRef]
- Burmistrova, N.N.; Ivanova, E.N.; Alekhina, L.B.; Konkova, T.V. Stability of intercalated montmorillonite clays. Adv. Chem. Chem. Technol. 2016, 30, 64–65. [Google Scholar]
- Tahraoui, Z.; Nouali, H.; Marichal, C.; Forler, P.; Klein, J.; Daou, T.J. Zeolite-Polymer Composite Materials as Water Scavenger. Molecules 2021, 26, 4815. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetsky, V.A. (Ed.) X-Ray Diffraction of Basic Types of Geological Minerals Layered and Framework Silicates; NEDRA Publishers: Saint Petersburg, Russia, 1983. (In Russian) [Google Scholar]
- Chelishchev, N.F.; Berenshtein, B.G.; Martynova, N.S. Comparative study of various cationic forms of clinoptilolite. Izv. Acad. Sci. USSR Inorg. Mater. 1975, 11, 704–708. [Google Scholar]
- Colella, C. Natural Zeolites in Environmentally Friendly Processes and Applications; Kiricsi, I., Pál-Borbély, G., Nagy, J.B., Karge, H.G., Eds.; Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1999; Volume 125, pp. 641–655. [Google Scholar] [CrossRef]
- Senderov, E.E.; Khitarov, N.I. Zeolites, Their Synthesis and Conditions of Formation in Nature; “Nauka” Publishing House: Moscow, Russia, 1970; 283p. [Google Scholar]
- Ulusoy, U.; Şimşek, S. Lead removal by polyacrylamide-bentonite and zeolite composites: Effect of phytic acid immobilization. J. Hazard. Mater. 2005, 127, 163–171. [Google Scholar] [CrossRef]
- Ginzburg, V.V.; Singh, C.; Balazs, A.C. Theoretical phase diagrams of polymer/clay composites: The role of grafted organic modifiers. Macromolecules 2000, 33, 1089–1099. [Google Scholar] [CrossRef]
- Alexandre, M.; Dubois, P. Polymer-layered silicate nanocomposites: Preparation, properties and uses of a new class of materials. Mater. Sci. Eng. 2000, 28, 1–63. [Google Scholar] [CrossRef]
- Dyar, D. A Review of Mossbauer Data on trioctahedral micas: Evidence for tetrahedral Fe3+ and cation ordering. Am. Mineral. 1987, 72, 102–112. [Google Scholar]
- Vandenberghe, R.; Barrero, C.; da Costa, G.; Van San, E.; De Grave, E. Mössbauer characterization of iron oxides and (oxy)hydroxides: The present state of the art. Hyperfine Interact. 2000, 126, 247–259. [Google Scholar] [CrossRef]
- Lutoev, V.P.; Silaev, V.I.; Ponomarenko, A.N.; Brik, A.B.; Dodchenko, N.A.; Yushin, A.; Yu Shevchuk, S.S. Transformation of structure of natural oxides/oxyhydroxides of iron as a result of influence of external factors. Vestn. Instituta Geol. Komi NC UrO RAN 2013, 217, 20–25. [Google Scholar]
- Lutoev, V.P.; Kochergin, A.V.; Lysyuk AYu Silaev, V.I.; Golubev, E.A.; Suetin, V.P. Phase Composition and Structural State of Natural Iron Oxide Pigments. Dokl. Akad. Nauk. 2009, 425, 372–377. [Google Scholar] [CrossRef]
- Rosenson, I.; Bauminger, E.K.; Heller-Kallai, L. Miissbauer spectra of ion in 1:1 phylosilicates. American Mineralogist 1979, 64, 893–901. [Google Scholar]
- Mackenzie, K.J.D.; Berezowski, R.M. Thermal and Mössbauer studies of iron-containing hydrous silicates. V. Berthierine. Thermochim. Acta 1984, 74, 291–312. [Google Scholar] [CrossRef]
- Kuzmann, E.; Nagy, S.; Vértes, A.; Weiszburg, T.G.; Garg, V.K. Geological and Mineralogical Applications of Mössbauer Spectroscopy. In Nuclear Methods in Mineralogy and Geology; Vértes, A., Nagy, S., Süvegh, K., Eds.; Plenum: New York, NY, USA, 1998; pp. 285–376. [Google Scholar]
- Klencsár, Z.; Wang, J.; Ge, R.; Zhou, W.; Liu, D.; Rykov, A.I.; Zhang, T. Further development of the database of the Mössbauer Effect Data Center. Hyperfine Interact. 2020, 241, 30. [Google Scholar] [CrossRef]
- Bowen, L.H. Mössbauer Effect Reference and Data Index; Stevens, J., Ed.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1978; p. 76. [Google Scholar]
- Cardile, C.M. Structural site occupation of iron within 2:1 dioctahedral phyllosilicates studied by57Fe Mössbauer spectroscopy. Hyperfine Interact 1988, 41, 767–770. [Google Scholar] [CrossRef]
- Yamada, H.; Tamura, K.; Watanabe, Y.; Iyi, N.; Morimoto, K. Geomaterials: Their application to environmental remediation. Sci. Technol. Adv. Mater. 2011, 12, 064705. [Google Scholar] [CrossRef]
- Galarneau, A.; Di Renzo, F.; Fajula, F.; Vedrine, J. (Eds.) Zeolites and Mesoporous Materials at the Dawn of the 21st Century: Proceedings of the 13th International Zeolite Conference, Montpellier, France, 8–13 July 2001; Elsevier: Amsterdam, The Netherlands, 2001; Volume 135, pp. 13–27. [Google Scholar]
- Misaelides, P.; Godelitsas, A.; Harissopoulos, S.; Anousis, I. Interaction of granitic biotite with selected lanthanides and actinides. J. Radioanal. Nucl. Chem. 2001, 247, 325–328. [Google Scholar] [CrossRef]
- Motsa, M.M.; Msagati TA, M.; Thwala, J.M.; Mamba, B.B. Polypropylene–zeolite polymer composites for water purification: Synthesis, characterisation and application. Desalination Water Treat. 2013, 53, 2604–2612. [Google Scholar] [CrossRef]
- Li, W.A.; Peng, Y.C.; Ma, W.; Huang, X.Y.; Feng, M.L. Rapid and selective removal of Cs+ and Sr2+ ions by two zeolite-type sulfides via ion exchange method. Chem. Eng. J. 2022, 442, 136377. [Google Scholar] [CrossRef]
- Ma, B.; Oh, S.; Shin, W.S.; Choi, S.-J. Removal of Co2+, Sr2+ and Cs+ from aqueous solution by phosphate-modified montmorillonite (PMM). Desalination 2011, 276, 336–346. [Google Scholar] [CrossRef]
- Ambrosino, F.; Esposito, A.M.; Mancini, F.; Verde, G.L.; Sabbarese, C.; Caputo, D.; Pugliese, M. Zeolites identification for wastewater radionuclides removal in the decommissioning of a former Italian nuclear power plant. Eur. Phys. J. Plus 2023, 138, 909. [Google Scholar] [CrossRef]
Figure 1.
Basic zeolite structure [24].
Figure 1.
Basic zeolite structure [24].

Figure 2.
FT-IR spectra of ZT (1), P[AAm-AAc]:{ZT} (2), P[AAm-AAc]:{ZT-BT} (3), P[AAm-AAc]:{ZT:CuFC-BT:CuFC} (4) and CuFC (5).
Figure 2.
FT-IR spectra of ZT (1), P[AAm-AAc]:{ZT} (2), P[AAm-AAc]:{ZT-BT} (3), P[AAm-AAc]:{ZT:CuFC-BT:CuFC} (4) and CuFC (5).

Figure 3.
XDR spectra of ZT (a), {ZT:CuFC} (b), P[AAm-AAc]:{ZT-BT} (c) and P[AAm-AAc]:{ZT:CuFC-BT:CuFC} (d).
Figure 3.
XDR spectra of ZT (a), {ZT:CuFC} (b), P[AAm-AAc]:{ZT-BT} (c) and P[AAm-AAc]:{ZT:CuFC-BT:CuFC} (d).

Figure 4.
Mössbauer Spectra of ZT, {ZT:CuFC}, P[AAm-AAc]:{ZT-BT}, P[AAm-AAc]:{ZT}, CuFC and P[AAm-AAc]:{ZT:CuFC-BT:CuFC}.
Figure 4.
Mössbauer Spectra of ZT, {ZT:CuFC}, P[AAm-AAc]:{ZT-BT}, P[AAm-AAc]:{ZT}, CuFC and P[AAm-AAc]:{ZT:CuFC-BT:CuFC}.

Figure 5.
Principle scheme of hybrid composite formation.

Figure 6.
SEM images of P[AAm-AAc]:{ZT} and P[AAm-AAc]:{ZT-BT} hybrid composites.

Figure 7.
Kinetic of RN sorption by ZT (a,a′), P[AAm-AAc]:[ZT} (b,b′), P[AAm-AAc]:[ZT-BT} (c,c′) and P[AAm-AAc]:[ZT:CuFC-BT:CuFC} (d,d′) at pH ≈ 7 (a–d) and pH ≈ 9 (a′–d′).
Figure 7.
Kinetic of RN sorption by ZT (a,a′), P[AAm-AAc]:[ZT} (b,b′), P[AAm-AAc]:[ZT-BT} (c,c′) and P[AAm-AAc]:[ZT:CuFC-BT:CuFC} (d,d′) at pH ≈ 7 (a–d) and pH ≈ 9 (a′–d′).

Figure 8.
Variation of RN distribution coefficient on ZT and its hydride composites at pH ≈ 7 (a) and pH ≈ 9 (b).
Figure 8.
Variation of RN distribution coefficient on ZT and its hydride composites at pH ≈ 7 (a) and pH ≈ 9 (b).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical composition of the studied zeolite, mass %.
| Oxides | ω, % | Oxides | ω, % |
|---|---|---|---|
| SiO2 | 57.9 | K2O | 1.7 |
| Al2O3 | 15.0 | P2O5 | 0.1 |
| Fe2O3 | 5.5 | TiO2 | 0.6 |
| CaO | 5.1 | SrSO4 | 0.3 |
| MgO | 2.3 | MnO2 | 0.1 |
| Na2O | 2.4 | Total | 100 |
Copper vitriol CuSO4·5H2O and potassium hexaferrocyanide K4[Fe(CN)6]·3H2O of chemically pure grade were used with no further purification.
Table 2.
Composition and radioactivity of a model liquid radioactive waste solution.
| RN | 134Cs+ | 137Cs+ | 60Co2+ | 57Co2+ | 85Sr2+ |
|---|---|---|---|---|---|
| Radioactivity, Bq·L−1 | 1931 ± 193 | 2444 ± 244 | 11,423 ± 1142 | 1351 ± 135 | 5657 ± 565 |
All experiments were always performed in duplicates.
Table 3.
Change in the degree of amorphization of minerals during in situ polymerization.
| ω, % Mass | Zeolite | Bentonite | P[AAm-AAc]:{ZT-BT} |
|---|---|---|---|
| wmorphous phase, % | 28.8 | 28.5 | 76.5 |
| wQuarts, % | 20.0 | 21.2 | 3.3 |
| wAlbite, % | 17.7 | 3.1 | 10.1 |
| wKyanite, % | 16.4 | - | - |
| wAlumosilicate, % | 13.1 | 30.00 | 4.0 |
| wMontmorillonite, % | 4.0 | 17.2 | 6.1 |
| Total | 100 | 100 | 100 |
Table 4.
Mössbauer parameters of zeolite and its composites.
| Sampe | S | A, % | δ, mm/s | QS, mm/s | Г, mm/s | H, kOe | Fe Position | Fe2+/Fe3+ |
|---|---|---|---|---|---|---|---|---|
| ZT | S1 | 83.7 | 0.37 | −0.22 | 0.21 fix | 508,7 | Fe2O3-Al | 0.15 |
| D1 | 1.2 | 1.15 | 1.94 | 0.23 | Fe2+, M1 | Fe2+VI, ZT | ||
| D2 | 0.9 | 1.30 | 2.34 | 0.21 | Fe2+, M2 | Fe2+VI, ZT | ||
| D3 | 14.3 | 0.33 | 0.64 | 0.51 | Fe3+, M2 | Fe3+VI, ZT | ||
| P[AAm-AAc]:{ZT} | S1 | 80.8 | 0.37 | −0.22 | 0.21 fix | 507,4 | Fe2O3-Al | 0.73 |
| D1 | 6.9 | 1.22 | 1.84 | 0.28 | Fe2+, M1 | Fe2+VI, ZT | ||
| D2 | 1.2 | 1.29 | 2.34 | 0.26 | Fe2+, M2 | Fe2+VI, ZT | ||
| D3 | 11.1 | 0.32 | 0.67 | 0.46 | Fe3+, M2 | Fe3+ VI, ZT | ||
| {ZT:CuFC} | S1 | 78.2 | 0.37 | −0.22 | 0.21 fix | 509,1 | Fe2O3-Al | 0.18 |
| D1 | 2.1 | 1.04 | 2.10 | 0.52 | Fe2+, M1 | Fe2+VI, ZT | ||
| D2 | 0.8 | 1.13 | 2.68 | 0.21 | Fe2+, M2 | Fe2+VI, ZT | ||
| D3 | 16.0 | 0.30 | 0.70 | 0.49 | Fe3+, M2 | Fe3+VI, ZT | ||
| C1 | 3 | 0.07 | 0 | 0.40 | [Fe(CN)6]4− | |||
| P[AAm-AAc]:{ZT-BT} | S1 | 43.6 | 0.37 | −0.22 | 0.21 fix | 508,7 | Fe2O3-Al | 1.10 |
| D1 | 21.7 | 1.23 | 1.80 | 0.22 | Fe2+, M1 | Fe2+VI, ZT, BT | ||
| D2 | 7.8 | 1.33 | 2.42 | 0.28 | Fe2+, M2 | Fe2+VI, ZT, BT | ||
| D3 | 26.8 | 0.27 | 0.78 | 0.58 | Fe3+, M2 | Fe3+IV, ZT, BT | ||
| P[AAm-AAc]:{ZT:CuFC-BT:CuFC} | S1 | 38.8 | 0.37 | −0.22 | 0.21 fix | 508.6 | Fe2O3-Al | 0.75 |
| D1 | 21.9 | 1.23 | 1.80 | 0.21 | Fe2+, M1 | Fe2+VI, ZT, BT | ||
| D2 | 2.9 | 1.15 | 2.73 | 0.30 | Fe2+, M2 | Fe2+VI, ZT, BT | ||
| D3 | 32.9 | 0.29 | 0.73 | 0.51 | Fe3+, M2 | Fe3+IV, ZT, BT | ||
| C1 | 3.4 | 0.09 | 0 | 0.29 | [Fe(CN)6]4− | - | - | |
| CuFC | C2 | 52.8 | −0.17 | 0 | 0.30 | - | - | - |
| C1 | 47.2 | 0.02 | 0 | 0.27 | [Fe(CN)6]4− | - | - |
Table 5.
The order of absorption of radionuclides by ZT and its hydride–polymer composites at pH ≈ 7 and pH ≈ 9.
Table 5.
The order of absorption of radionuclides by ZT and its hydride–polymer composites at pH ≈ 7 and pH ≈ 9.
| Sorbent | pH ≈ 7 | pH ≈ 9 |
|---|---|---|
| ZT | 57Co2+ ≈ 60Co2+ > 134Cs+ > 137Cs+ > 85Sr2+ | 60Co2+ > 57Co2+ > 137Cs+ > 134Cs+ > 85Sr2+ |
| P[AAm-A>Ac]:{ZT} | 134Cs+ > 137Cs+ > 85Sr2+ > 57Co2+ ≈ 60Co2+ | 134Cs+ > 137Cs+ > 57Co2+ > 60Co2+ > 85Sr2+ |
| P[AAm-AAc]:{ZT-BT} | 58Sr2+ > 60Co2+ > 57Co2+ > 134Cs+ > 137Cs+ | 57Co2+ > 60Co2+ > 134Cs+ > 137Cs+ > 85Sr2+ |
| P[AAm-AAc]:{ZT:CuFC-BT:CuFC} | 134Cs+ > 137Cs+ > 57Co2+ ≈ 60Co2+ > 85Sr2+ | 134Cs+ > 137Cs+ > 57Co2+ > 60Co2+ > 85Sr2+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mamytbekov, G.K.; Zheltov, D.A.; Milts, O.S.; Nurtazin, Y.R. Polymer–Zeolite Composites: Synthesis, Characterization and Application. Colloids Interfaces 2024, 8, 8. https://doi.org/10.3390/colloids8010008
AMA Style
Mamytbekov GK, Zheltov DA, Milts OS, Nurtazin YR. Polymer–Zeolite Composites: Synthesis, Characterization and Application. Colloids and Interfaces. 2024; 8(1):8. https://doi.org/10.3390/colloids8010008
Chicago/Turabian StyleMamytbekov, Galymzhan Kulamkadyrovich, Dmitry Anatol’evich Zheltov, Olga Sergeevna Milts, and Yernat Rashidovich Nurtazin. 2024. "Polymer–Zeolite Composites: Synthesis, Characterization and Application" Colloids and Interfaces 8, no. 1: 8. https://doi.org/10.3390/colloids8010008