Intramolecular Spin State Locking in Iron(II) 2,6-Di(pyrazol-3-yl)pyridine Complexes by Phenyl Groups: An Experimental Study

 ,
,
Abstract
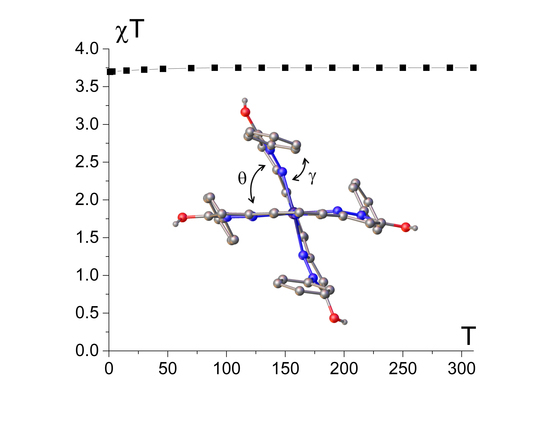
:
1. Introduction
2. Results and Discussion
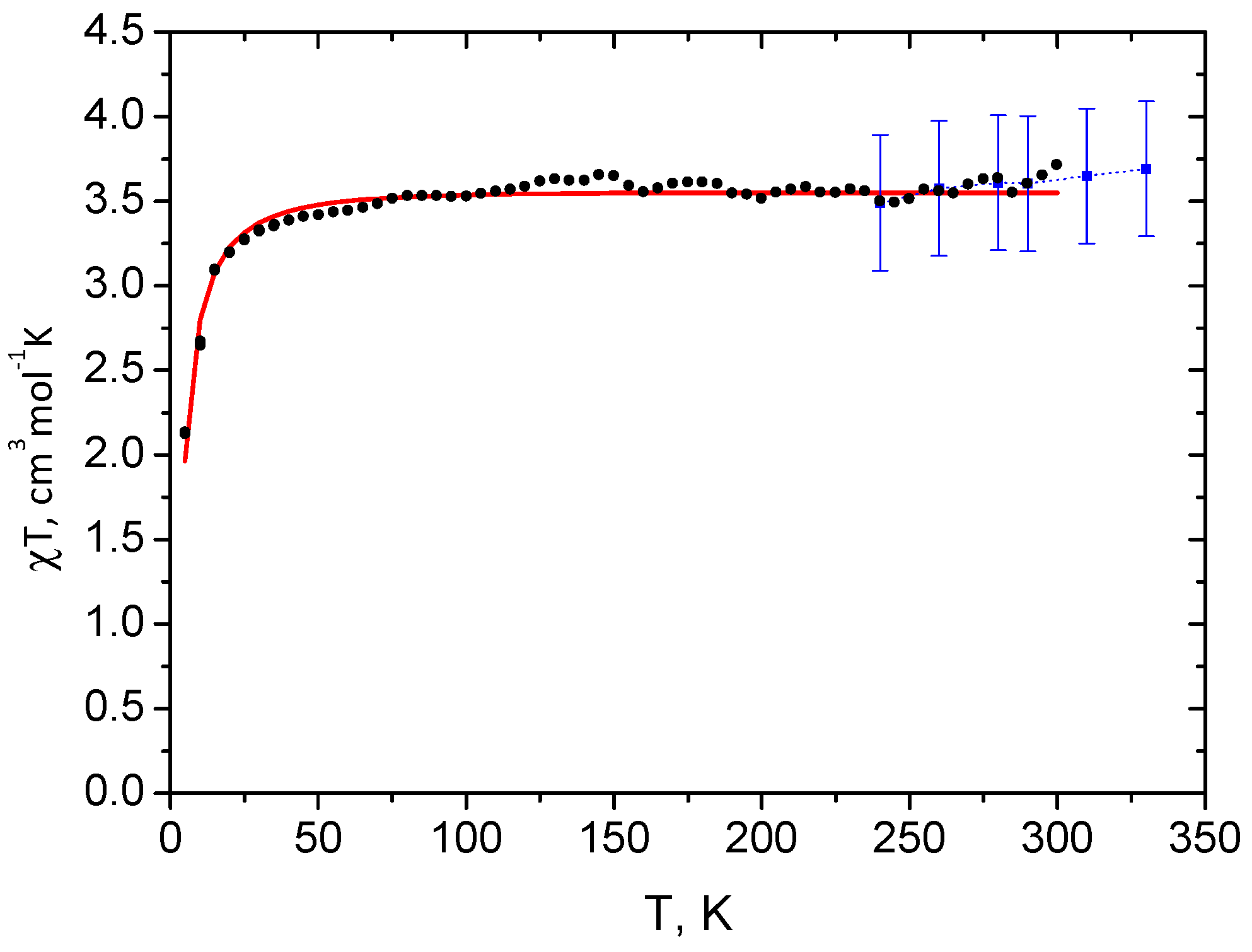
2.1. Spin State from Magnetometry
2.2. Spin State from NMR
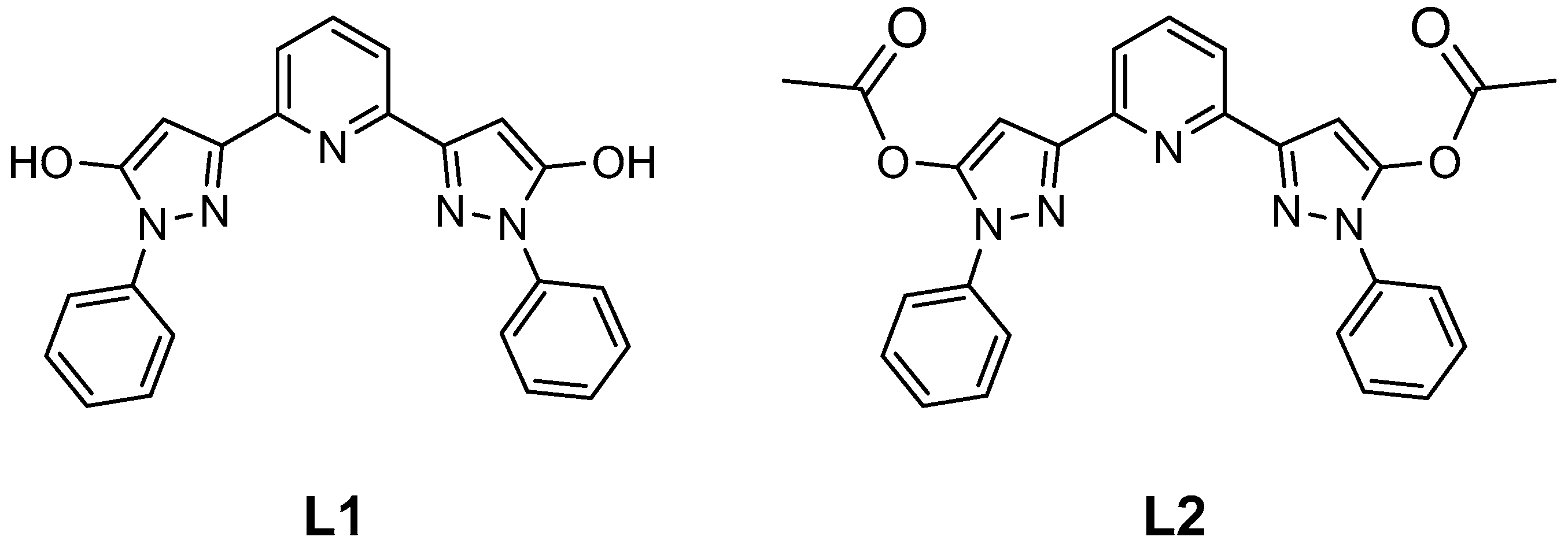
2.3. Spin State and Molecular Geometry from X-Ray Diffraction
2.4. Molecular Geometry from NMR
2.5. Distortion of Molecular Geometry from Symmetry Measures
2.6. Distortion of Molecular Geometry by Secondary Interactions
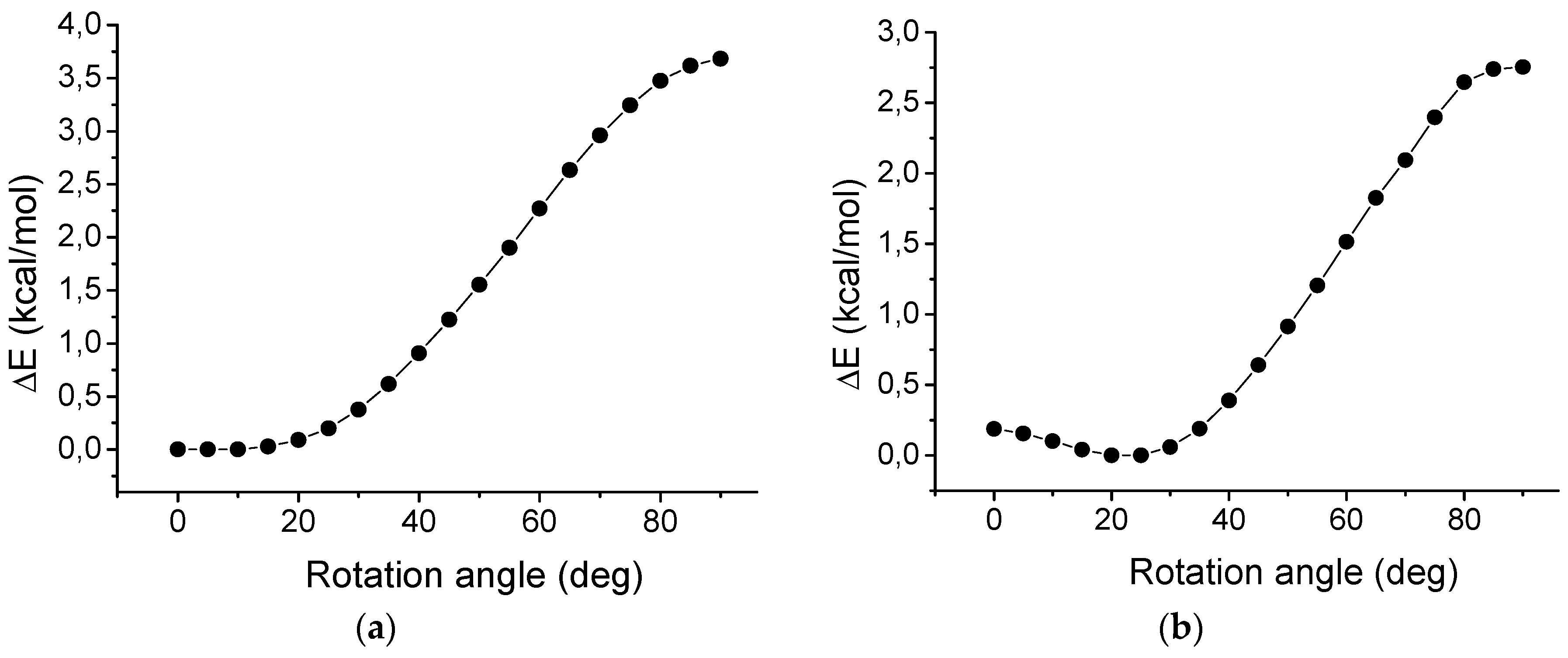
2.7. Rotation of Phenyl Groups
2.8. Stacking Interactions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Gutlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Holmes, S.M.; Halcrow, M.A. Spin-state switches in molecular materials chemistry. J. Mat. Chem. C 2015, 3, 7775–7778. [Google Scholar] [CrossRef] [Green Version]
- Molnar, G.; Rat, S.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Spin Crossover Nanomaterials: From Fundamental Concepts to Devices. Adv. Mater. 2017, 30, 1703862. [Google Scholar] [CrossRef] [PubMed]
- Senthil Kumar, K.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Halcrow, M.A. Structure: Function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119–4142. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. The synthesis and coordination chemistry of 2,6-bis(pyrazolyl)pyridines and related ligands—Versatile terpyridine analogues. Coord. Chem. Rev. 2005, 249, 2880–2908. [Google Scholar] [CrossRef]
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Kulmaczewski, R.; Mohammed, R.; Dudley, S.; Barrett, S.A.; Little, M.A.; Deeth, R.; Halcrow, M.A. A Unified Treatment of the Relationship between Ligand Substituents and Spin State in a Family of Iron(II) Complexes. Angew. Chem. Int. Ed. 2016, 55, 4327–4331. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef] [PubMed]
- Canton, S.E.; Zhang, X.; Lawson Daku, M.L.; Liu, Y.; Zhang, J.; Alvarez, S. Mapping the Ultrafast Changes of Continuous Shape Measures in Photoexcited Spin Crossover Complexes without Long-Range Order. J. Phys. Chem. C 2015, 119, 3322–3330. [Google Scholar] [CrossRef]
- Kershaw Cook, L.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin state behavior of iron(II)/dipyrazolylpyridine complexes. New insights from crystallographic and solution measurements. Coord. Chem. Rev. 2015, 289–290, 2–12. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Thorp-Greenwood, F.L.; Comyn, T.P.; Cespedes, O.; Chastanet, G.; Halcrow, M.A. Unexpected Spin-Crossover and a Low-Pressure Phase Change in an Iron(II)/Dipyrazolylpyridine Complex Exhibiting a High-Spin Jahn–Teller Distortion. Inorg. Chem. 2015, 54, 6319–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, G.A.; Costa, J.S.; Roubeau, O.; Teat, S.J.; Aromi, G. Local Coordination Geometry and Spin State in Novel FeII Complexes with 2,6-Bis(pyrazol-3-yl)pyridine?Type Ligands as Controlled by Packing Forces: Structural Correlations. Chem. Eur. J. 2012, 18, 11703–11715. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. The Effect of Ligand Design on Metal Ion Spin State—Lessons from Spin Crossover Complexes. Crystals 2016, 6, 58. [Google Scholar] [CrossRef]
- Roberts, T.D.; Little, M.A.; Kershaw Cook, L.J.; Barrett, S.A.; Tuna, F.; Halcrow, M.A. Iron(II) complexes of 2,6-di(1-alkylpyrazol-3-yl)pyridine derivatives—The influence of distal substituents on the spin state of the iron centre. Polyhedron 2013, 64, 4–12. [Google Scholar] [CrossRef]
- Bartual-Murgui, C.; Vela, S.; Roubeau, O.; Aromi, G. Designed intramolecular blocking of the spin crossover of an Fe(II) complex. Dalton Trans. 2016, 45, 14058–14062. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, J.M.; Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Control of the spin state of Fe(II) 2,6-di(pyrazol-1-yl)pyridine complexes by distal ligand substitution. Inorg. Chem. Commun. 2002, 5, 328–332. [Google Scholar] [CrossRef]
- Petzold, H.; Djomgoue, P.; Horner, G.; Lochenie, C.; Weber, B.; Ruffer, T. Bis-meridional Fe2+ spincrossover complexes of phenyl and pyridyl substituted 2-(pyridin-2-yl)-1,10-phenanthrolines. Dalton Trans. 2018, 47, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, J.; Evans, D.J.; Kilner, C.A.; Halcrow, M.A. A structural, magnetic and Mossbauer spectroscopic study of an unusual angular Jahn–Teller distortion in a series of high-spin iron(II) complexes. Dalton Trans. 2005, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.D.; Little, M.A.; Kershaw Cook, L.J.; Halcrow, M.A. Iron(II) complexes of 2,6-di(1H-pyrazol-3-yl)-pyridine derivatives with hydrogen bonding and sterically bulky substituents. Dalton Trans. 2014, 43, 7577–7588. [Google Scholar] [CrossRef] [PubMed]
- Polezhaev, A.V.; Chen, C.-H.; Kinne, A.S.; Cabelof, A.C.; Lord, R.L.; Caulton, K.G. Ligand Design toward Multifunctional Substrate Reductive Transformations. Inorg. Chem. 2017, 56, 9505–9514. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ishida, T. Spin-Crossover Temperature Predictable from DFT Calculation for Iron(II) Complexes with 4-Substituted Pybox and Related Heteroaromatic Ligands. ACS Omega 2018, 3, 6737–6747. [Google Scholar] [CrossRef]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Piguet, C. Paramagnetic Susceptibility by NMR: The “Solvent Correction” Removed for Large Paramagnetic Molecules. J. Chem. Educ. 1997, 74, 815. [Google Scholar] [CrossRef]
- Petzold, H.; Djomgoue, P.; Horner, G.; Speck, J.M.; Ruffer, T.; Schaarschmidt, D. 1H NMR spectroscopic elucidation in solution of the kinetics and thermodynamics of spin crossover for an exceptionally robust Fe2+ complex. Dalton Trans. 2016, 45, 13798–13809. [Google Scholar] [CrossRef] [PubMed]
- Petzold, H.; Djomgoue, P.; Horner, G.; Heider, S.; Lochenie, C.; Weber, B.; Ruffer, T.; Schaarschmidt, D. Spin state variability in Fe2+ complexes of substituted (2-(pyridin-2-yl)-1,10-phenanthroline) ligands as versatile terpyridine analogues. Dalton Trans. 2017, 46, 6218–6229. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.A.; Denisov, G.L.; Kiskin, M.A.; Nelyubina, Y.V.; Novikov, V.V. Probing Spin Crossover in a Solution by Paramagnetic NMR Spectroscopy. Inorg. Chem. 2018, 56, 14759–14762. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A.; Kilner, C.A.; Thornton-Pett, M. Bis{2,6-bis[3-(2,4,6-trimethylphenyl)-pyrazol-1-yl-kN2]pyridine-jN}zinc(II) diperchlorate bis(nitromethane) solvate. Acta Cryst. C 2000, 56, 1425–1426. [Google Scholar] [CrossRef]
- Solanki, N.K.; Leech, M.A.; McInnes, E.J.L.; Mabbs, F.E.; Howard, J.A.K.; Kilner, C.A.; Rawson, J.M.; Halcrow, M.A. A crystallographic and EPR study of the fluxional Cu(II) ion in [CuL2][BF4]2(L = 2,6-dipyrazol-1-ylpyridine). J. Chem. Soc.-Dalton Trans. 2002, 1295–1301. [Google Scholar] [CrossRef]
- Balde, C.; Desplanches, C.; Le Gac, F.; Guionneau, P.; Letard, J.-F. The role of iron(II) dilution in the magnetic and photomagnetic properties of the series [FexZn1−x(bpp)2](NCSe)2. Dalton Trans. 2014, 43, 7820–7829. [Google Scholar] [CrossRef] [PubMed]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. Phi: A Powerful New Program for the Analysis of Anisotropic Monomeric and Exchange-Coupled Polynuclear D- and F-Block Complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, J.; Kilner, C.A.; Halcrow, M.A. Structural diversity in iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridine and 2,6-di(3-methylpyrazol-1-yl)pyridine. Dalton Trans. 2006, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Luchinat, C.; Parigi, G.; Ravera, E. Solution NMR of Paramagnetic Molecules, 2nd ed.; Applications to Metallobiomolecules and Models; Elsevier: New York, NY USA, 2015. [Google Scholar]
- Pavlov, A.A.; Nelyubina, Y.V.; Kats, S.V.; Penkova, L.V.; Efimov, N.N.; Dmitrienko, A.O.; Vologzhanina, A.V.; Belov, A.S.; Voloshin, Y.Z.; Novikov, V.V. Polymorphism in a Cobalt-Based Single-Ion Magnet Tuning Its Barrier to Magnetization Relaxation. J. Phys. Chem. Lett. 2016, 7, 4111–4116. [Google Scholar] [CrossRef] [PubMed]
- Novikov, V.V.; Pavlov, A.A.; Belov, A.S.; Vologzhanina, A.V.; Savitsky, A.; Voloshin, Y.Z. Transition Ion Strikes Back: Large Magnetic Susceptibility Anisotropy in Cobalt(II) Clathrochelates. J. Phys. Chem. Lett. 2016, 5, 3799–3803. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.A.; Savkina, S.A.; Belov, A.S.; Nelyubina, Y.V.; Efimov, N.N.; Voloshin, Y.Z.; Novikov, V.V. Trigonal Prismatic Tris-pyridineoximate Transition Metal Complexes: A Cobalt(II) Compound with High Magnetic Anisotropy. Inorg. Chem. 2017, 56, 6943–6951. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Luchinat, C.; Parigi, G.; Pierattelli, R. NMR Spectroscopy of Paramagnetic Metalloproteins. ChemBioChem 2005, 6, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, R.; Kilner, C.A.; Halcrow, M.A. Iron(II) complexes with a terpyridine embrace packing motif show remarkably consistent cooperative spin-transitions. Chem. Commun. 2007, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Lopez, M.C.; Clemente-Leon, M.; Gimenez-Saiz, C. Unravelling the spin-state of solvated [Fe(bpp)2]2+ spin-crossover complexes: Structure–function relationship. Dalton Trans. 2018, 47, 10453–10462. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.C.; Jakubikova, E. Ray-Dutt and Bailar Twists in Fe(II)-Tris(2,2′-bipyridine): Spin States, Sterics, and Fe–N Bond Strengths. Inorg. Chem. 2018, 57, 5585–5596. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. Relationships between Temperature, Magnetic Moment, and Continuous Symmetry Measures in Spin Crossover Complexes. J. Am. Chem. Soc. 2003, 125, 6795–6802. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.-L.; Liu, Z.; Wei, W.-J.; Xing, Y.-H.; Liu, J.; Zhang, R.; Hou, Y.-N.; Wang, X.; Bai, F.-Y. Synthesis, structure, spectroscopy of four novel supramolecular complexes and cytotoxicity study by application of multiple parallel perfused microbioreactors. New J. Chem. 2014, 38, 3258–3268. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Money, V.A.; Carbonera, C.; Elhaik, J.; Halcrow, M.A.; Howard, J.A.K.; Letard, J.-F. Interplay between Kinetically Slow Thermal Spin-Crossover and Metastable High-Spin State Relaxation in an Iron(II) Complex with Similar T1/2 and T(LIESST). Chem. Eur. J. 2007, 13, 5503–5514. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E. Understanding Substituent Effects in Noncovalent Interactions Involving Aromatic Rings. Acc. Chem. Res. 2013, 46, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Sherrill, C.D. Effects of Heteroatoms on Aromatic Interactions: Benzene-Pyridine and Pyridine Dimer. J. Phys. Chem. A 2009, 113, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pan, H.-R.; Yang, Q.; Fu, H.-Y.; Chen, H.; Li, R.-X. A family of novel cationic ruthenium pincer complexes: Synthesis, characterization and catalytic activity in the transfer hydrogenation of ketones. Inorg. Chem. Comm. 2011, 14, 1422–1427. [Google Scholar] [CrossRef]
- Pelascini, F.; Wesolek, M.; Peruch, F.; Cian, A.D.; Kyritsakas, N.; Lutz, P.J.; Kress, J. Iron complexes of terdentate nitrogen ligands: Formation and X-ray structure of three new dicationic complexes. Polyhedron 2004, 23, 3193–3199. [Google Scholar] [CrossRef]
- Constable, E.C.; Baum, G.; Bill, E.; Dyson, R.; van Eldik, R.; Fenske, D.; Kaderli, S.; Morris, D.; Neubrand, A.; Neuburger, M.; et al. Control of Iron(II) Spin States in 2,2′:6′,2″-Terpyridine Complexes through Ligand Substitution. Chem. Eur. J. 1999, 5, 498–508. [Google Scholar] [CrossRef]
- Brauchli, S.Y.; Constable, E.C.; Harris, K.; Haussinger, D.; Housecroft, C.E.; Rosel, P.J.; Zampese, J.A. Towards catenanes using pi-stacking interactions and their influence on the spin-state of a bis(2,2′:6′,2″-terpyridine)iron(II) domain. Dalton Trans. 2010, 39, 10739–10748. [Google Scholar] [CrossRef] [PubMed]
- Dell’Amico, D.B.; Calderazzo, F.; Englert, U.; Labella, L.; Marchetti, F. The first crystallographically established bis-qtpy (qtpy = 2,2′6′,2′′6′′,2′′′-quaterpyridine) metal complex. J. Chem. Soc.-Dalton Trans. 2001, 357–358. [Google Scholar] [CrossRef]
- Barrios, L.A.; Bartual-Murgui, C.; Peyrecave-Lleixa, E.; Le Guennic, B.; Teat, S.J.; Roubeau, O.; Aromi, G. Homoleptic versus Heteroleptic Formation of Mononuclear Fe(II) Complexes with Tris-Imine Ligands. Inorg. Chem. 2016, 55, 4110–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, M.; Michael, B.; Malkin, V.G. Calculation of NMR and EPR Parameters: Theory and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Bertini, I.; Luchinat, C. NMR of Paramagnetic Substances. Coord. Chem. Rev. 1996, 150, 1–296. [Google Scholar]
- Bertini, I.; Luchinat, C.; Parigi, G. Magnetic susceptibility in paramagnetic NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 249–273. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Kim, M.-C.; Sim, E.; Benali, A.; Heinonen, O.; Burke, K. Benchmarks and Reliable DFT Results for Spin Gaps of Small Ligand Fe(II) Complexes. J. Chem. Theory Comput. 2018, 14, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, M.; Buhl, M.; Malkin, V.G. Calculation of NMR and EPR Parameters: Theory and Applications; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2004. [Google Scholar]
- van Wullen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.A.; Halcrow, M.A. Anion-dependent spin crossover in solution for an iron(II) complex of a 1H-pyrazolyl ligand. RSC Adv. 2014, 4, 11240–11243. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe(L1)2OTf2 | Zn(L1)2(ClO4)2 | Fe(L2)2OTf2 | Fe(L2)2(BF4)2 | |
|---|---|---|---|---|
| M-N (1), Å | 2.132 (3) | 2.116 (2) | 2.130 (5) | 2.145 (4) |
| M-N (1A), Å | 2.140 (4) | 2.115 (2) | 2.131 (5) | 2.144 (4) |
| M-N (2), Å | 2.213 (4) | 2.171 (2) | 2.263 (5) | 2.207 (5) |
| M-N (2A), Å | 2.234 (3) | 2.171 (2) | 2.220 (5) | 2.229 (5) |
| M-N (4), Å | 2.231 (4) | 2.172 (2) | 2.206 (5) | 2.223 (4) |
| M-N (4A), Å | 2.171 (4) | 2.189 (2) | 2.235 (5) | 2.230 (5) |
| θ° | 67.5 | 69.4 | 66.7 | 69.8 |
| ϕ° | 176.1 | 178.9 | 177.4 | 175.5 |
| S(Oh) | 6.12 | 5.30 | 6.02 | 6.33 |
| S(itp) | 10.49 | 11.08 | 10.98 | 10.52 |
| S(ebcT) | 9.015 | 9.011 | 9.402 | 8.849 |
| γ° | 50.1/42.7 | 56.3/57.0 | 47.8/60.3 | 49.9/59.4 |
| 46.2/60.5 | 66.4/58.9 | 50.3/55.7 | 53.1/63.4 | |
| β° | 10.7/10.6 | 7.7/6.9 | 9.0/8.0 | 4.4/13.1 |
| 15.5/16.2 | 9.0/5.9 | 10.9/7.8 | 7.3/16.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelyubina, Y.; Polezhaev, A.; Pavlov, A.; Aleshin, D.; Savkina, S.; Efimov, N.; Aliev, T.; Novikov, V. Intramolecular Spin State Locking in Iron(II) 2,6-Di(pyrazol-3-yl)pyridine Complexes by Phenyl Groups: An Experimental Study. Magnetochemistry 2018, 4, 46. https://doi.org/10.3390/magnetochemistry4040046
Nelyubina Y, Polezhaev A, Pavlov A, Aleshin D, Savkina S, Efimov N, Aliev T, Novikov V. Intramolecular Spin State Locking in Iron(II) 2,6-Di(pyrazol-3-yl)pyridine Complexes by Phenyl Groups: An Experimental Study. Magnetochemistry. 2018; 4(4):46. https://doi.org/10.3390/magnetochemistry4040046
Chicago/Turabian StyleNelyubina, Yulia, Alexander Polezhaev, Alexander Pavlov, Dmitrii Aleshin, Svetlana Savkina, Nikolay Efimov, Teimur Aliev, and Valentin Novikov. 2018. "Intramolecular Spin State Locking in Iron(II) 2,6-Di(pyrazol-3-yl)pyridine Complexes by Phenyl Groups: An Experimental Study" Magnetochemistry 4, no. 4: 46. https://doi.org/10.3390/magnetochemistry4040046