
PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants



 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Basic Concepts
2.1.1. Machine Learning
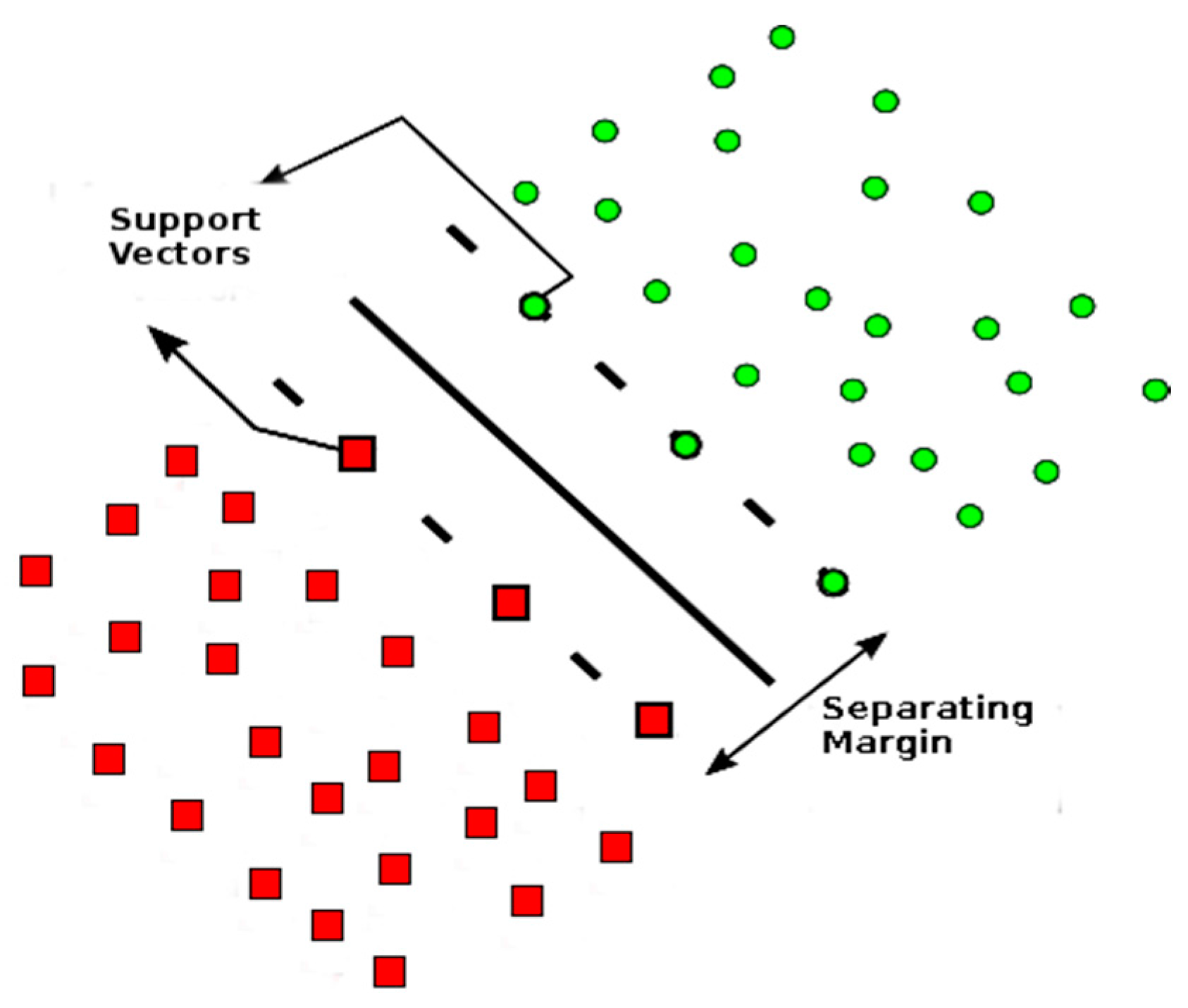
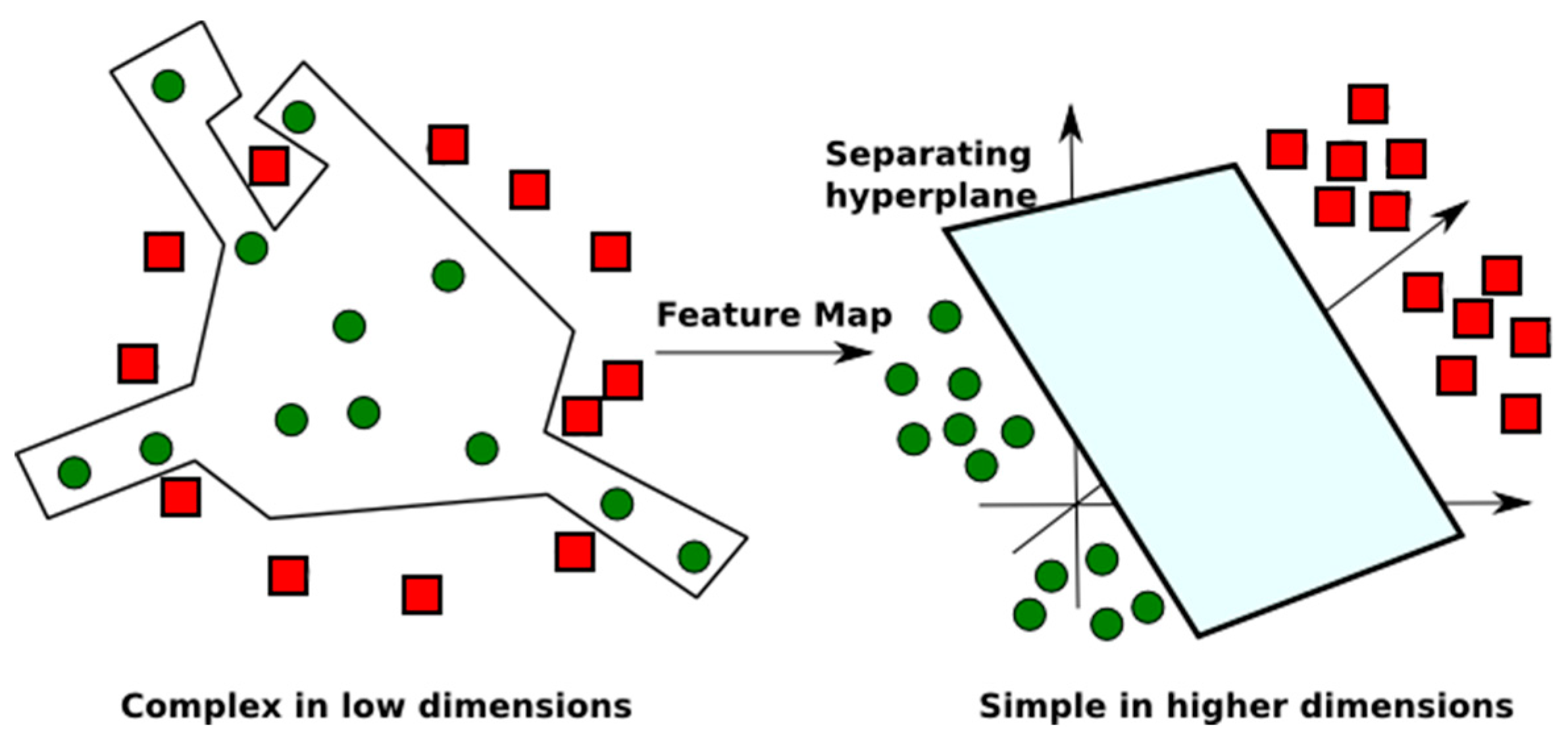
2.1.2. Support Vector Machine
2.1.3. Training Features
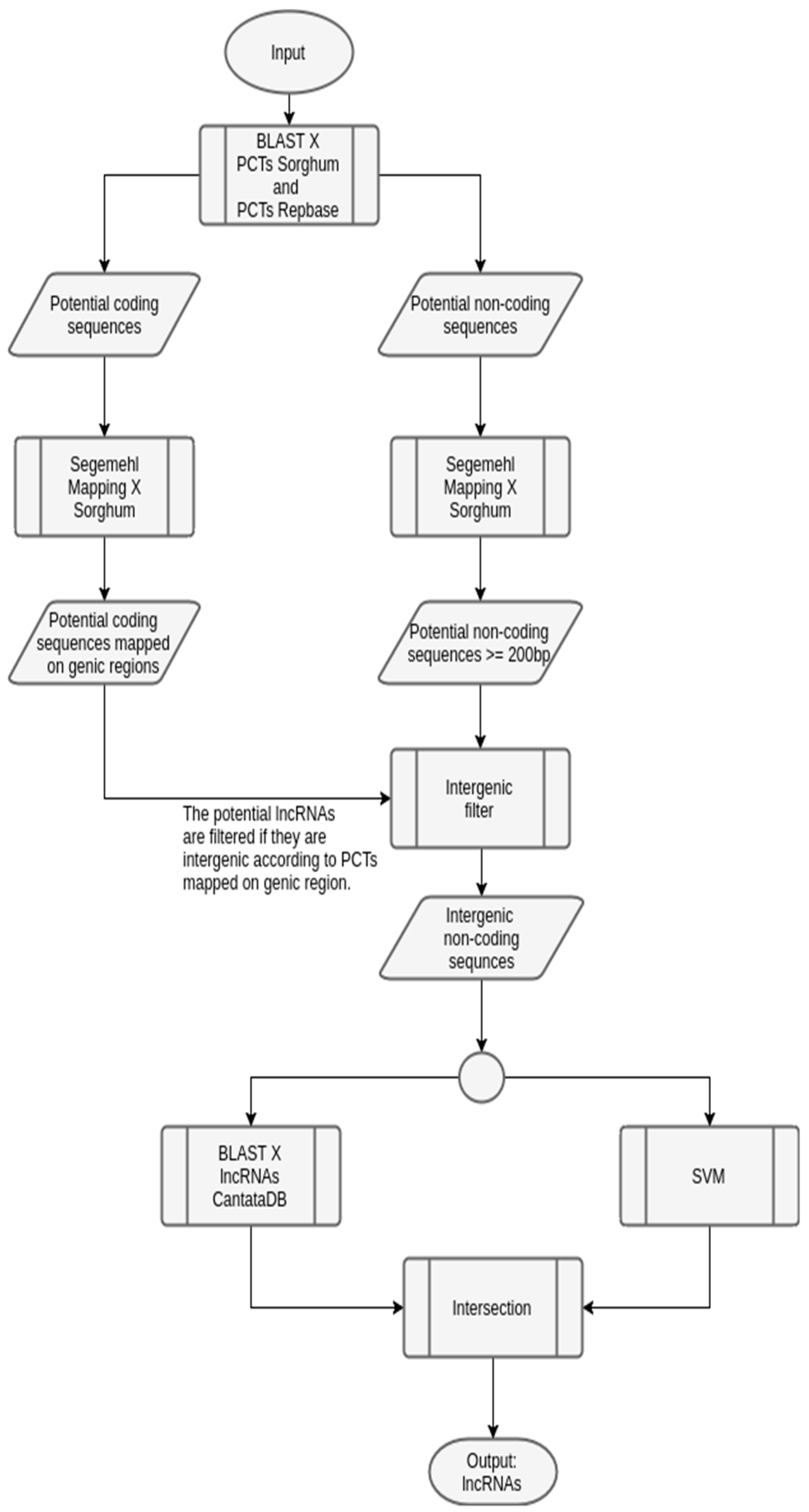
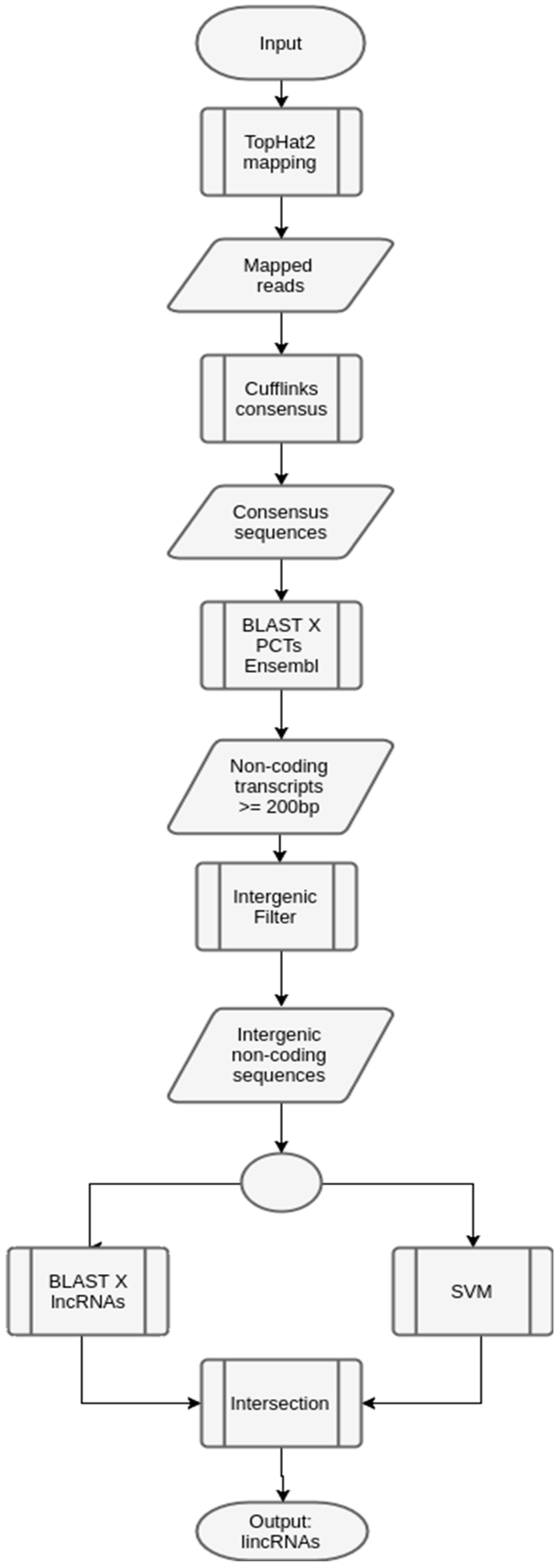
2.2. Workflow to Predict LincRNAs in Plants
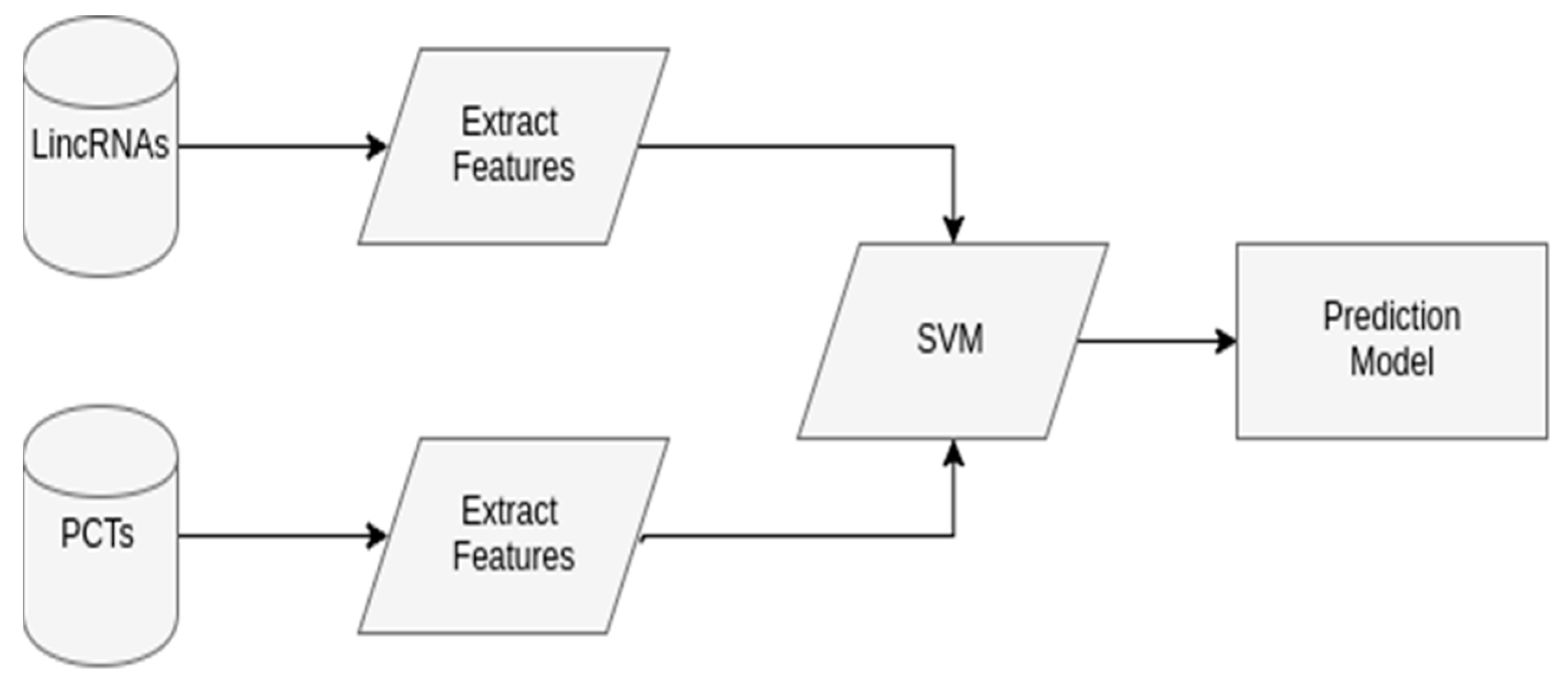
2.2.1. SVM Model to Predict LincRNAs
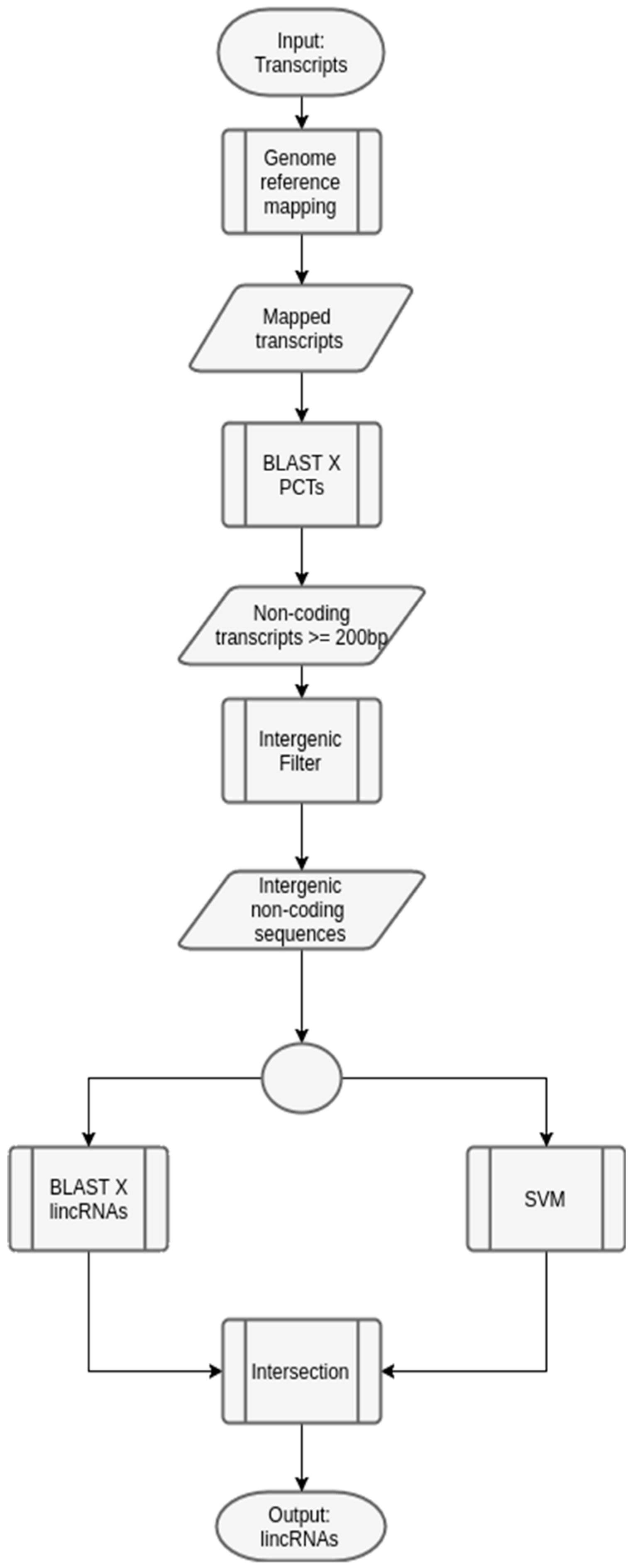
2.2.2. Generic Workflow to Predict LincRNAs in Plants
3. Results
3.1. Case Study 1: Sugarcane
3.2. Case Study 2: Maize
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bernal, A.; Ear, U.; Kyrpides, N. Genomes OnLine Database (GOLD): A monitor of genome projects worldwide. Nucleic Acids Res. 2001, 29, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Sabin, L.R.; Delás, M.J.; Hannon, G.J. Dogma derailed: The many influences of RNA on the genome. Mol. Cell 2013, 49, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Delneri, D.; O’Keefe, R. Non-coding RNAs in Saccharomyces cerevisiae: What is the function? Biochem. Soc. Trans. 2012, 40, 907. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Shiekhattar, R. Noncoding RNAs and enhancers: Complications of a long-distance relationship. Trends Genet. 2011, 27, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.E.; Pedrazzini, T.; Chang, C.P.; Dorn, G.W., 2nd; Thum, T.; Heymans, S.; Cardiolinc network. Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. LincRNAs: Genomics, evolution and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through Support Vector Machine. PLoS Genet. 2006, 2, e29. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and Support Vector Machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Arrial, R.; Togawa, R.C.; Brígido, M.d.M. Outlining a strategy for screening non-coding RNAs on a transcriptome through support vector machine. In Advances in Bioinformatics and Computational Biology; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4643, pp. 149–152. [Google Scholar]
- Wang, C.; Ding, C.; Meraz, R.F.; Holbrook, S.R. PSoL: A positive sample only learning algorithm for finding non-coding RNA genes. Bioinformatics 2006, 22, 2590–2596. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Hofacker, I.L.; Stadler, P.F. SnoReport: Computational identification of snoRNAs with unknown targets. Bioinformatics 2008, 24, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Tafer, H.; Kehr, S.; Hertel, J.; Hofacker, I.L.; Stadler, P.F. RNASnoop: Efficient target prediction for H/ACA snoRNAs. Bioinformatics 2010, 26, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Bartschat, S.; Kehr, S.; Tafer, H.; Stadler, P.F.; Hertel, J. SnoStrip: A snoRNA annotation pipeline. Bioinformatics 2014, 30, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, H.; Zhang, L.; Meng, J. LncRScan-SVM: A tool for predicting long non-coding RNAs using Support Vector Machine. PLoS ONE 2015, 10, e0139654. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.N.; Zhang, S.W. lncRNA-MFDL: Identification of human long non-coding RNAs by fusing multiple features and using deep learning. Mol. Biosyst. 2015, 11, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Achawanantakun, R.; Chen, J.; Sun, Y.; Zhang, Y. LncRNA-ID: Long non-coding RNA IDentification using balanced random forests. Bioinformatics 2015, 31, 3897–3905. [Google Scholar] [CrossRef] [PubMed]
- Pian, C.; Zhang, G.; Chen, Z.; Chen, Y.; Zhang, J.; Yang, T.; Zhang, L. LncRNApred: Classification of long non-coding RNAs and protein-coding transcripts by the Ensemble Algorithm with a new hybrid feature. PLoS ONE 2016, 11, e0154567. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Chen, X.; Jiang, P.; Song, X.; Wang, H.; Sun, H. iSeeRNA: Identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics 2013, 14 (Suppl. S2), 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Wang, Q.; Lv, Y.; Wang, S.; Chen, X.; Li, X. Computational identification of human long intergenic non-coding RNAs using a GA–SVM algorithm. Gene 2014, 533, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Niu, Q.W.; Wu, H.W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Evans, M.M. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Norvig, P. AI a Modern Approach; Pearson: Harlow, UK, 2010; Volume 3. [Google Scholar]
- Why are Support Vectors Machines called so? Available online: https://onionesquereality.wordpress.com/2009/03/22/why-are-support-vectors-machines-called-so/ (accessed on 26 January 2016).
- Haykin, S. A comprehensive foundation. In Neural Networks and Learning Machines, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2009. [Google Scholar]
- Big Data Optimization at SAS. Available online: http://www.maths.ed.ac.uk/~prichtar/Optimization_and_Big_Data/slides/Polik.pdf (accessed on 4 December 2016).
- SVM—Support Vector Machines. Available online: https://www.dtreg.com/solution/view/20 (accessed on 6 January 2016).
- Refaeilzadeh, P.; Tang, L.; Liu, H. Cross-validation. In Encyclopedia of Database Systems; Springer: New York, NY, USA, 2009; pp. 532–538. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Dimitriadou, E.; Hornik, K.; Leisch, F.; Meyer, D.; Weingessel, A. R Package Version 1.5. E1071: Misc Functions of the Department of Statistics (E1071); TU Wien: Vienna, Austria, 2011. [Google Scholar]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.W. Prediction of long non-coding RNAs using Machine Learning Techniques. Doctorate Dissertation, Department of Computer Science, University of Brasilia, Brasilia, Brasil, 2016. [Google Scholar]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thiebaut, F.; Rojas, C.; Grativol, C.; Calixto, E.; Motta, M.; Ballesteros, H.; Peixoto, B.; de Lima, B.; Vieira, L.M.; Walter, M.E.M.T.; et al. Sugarcane sRNAome upon pathogenic infection: The starring role of miR408. 2017; Submitted. [Google Scholar]
- Szczesniak, M.; Rosikiewicz, W.; Makałowska, I. Cantatadb: A collection of plant long non-coding RNAs. Plant Cell Physiol. 2016, 57, e8. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Chang, C.C.; Lin, C.J. A Practical Guide to Support Vector Classification; Department of Computer Science National Taiwan University: Taipei, Taiwan, 2003. [Google Scholar]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.; Lawrence, C.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res. 2008, 36 (Suppl. S1), D959–D965. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermueller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Ensembl. Available online: http://www.ensembl.org/index.html (accessed on 21 January 2016).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.M.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Number of Sequences |
|---|---|
| Raw Input | 168,767 |
| Filtered input (transcripts not annotated as PCTs) | 63,389 |
| Transcripts mapped on sorghum gene regions | 9488 |
| Non-coding transcripts mapped on sorghum | 4425 |
| Non-coding transcripts mapped on intergenic region (using the sorghum genome as reference) | 2432 |
| LincRNAs predicted by the SVM model | 1689 |
| LincRNAs annotated by BLAST | 97 |
| LincRNAs annotated by BLAST and predicted by SVM model | 67 |
| Library | id 9 (Control) | id 10 | id 11 (Control) | id 12 |
|---|---|---|---|---|
| Raw input | 7,158,821 | 5,032,782 | 8,277,629 | 6,327,933 |
| Mapped Reads | 4,528,477 | 3,161,660 | 8,321,623 | 3,662,784 |
| Consensus sequences | 156,565 | 155,472 | 157,562 | 155,578 |
| Non-coding transcripts ≥ 200 bp | 2988 | 2731 | 3168 | 2792 |
| Intergenic non-coding transcripts ≥ 200 bp | 2776 | 2536 | 2938 | 2592 |
| lincRNAs predicted by SVM | 2743 | 2512 | 2904 | 2567 |
| lincRNAs annotated by BLAST | 513 | 420 | 543 | 446 |
| lincRNAs annotated by BLAST and predicted by SVM | 507 | 418 | 539 | 444 |
| Library | id 29 | id 30 | id 31 (Control) | id 32 (Control) |
|---|---|---|---|---|
| Raw input | 17,555,365 | 15,322,340 | 17,149,619 | 15,497,154 |
| Mapped Reads | 11,904,702 | 10,246,454 | 11,847,394 | 10,745,811 |
| Consensus sequences | 161,476 | 160,962 | 162,208 | 161,167 |
| Non-coding transcripts ≥ 200 bp | 3985 | 3760 | 4405 | 3999 |
| Intergenic non-coding transcripts ≥ 200 bp | 3554 | 3381 | 3866 | 3534 |
| lincRNAs predicted by SVM | 3494 | 3330 | 3814 | 3486 |
| lincRNAs annotated by BLAST | 737 | 692 | 877 | 765 |
| lincRNAs annotated by BLAST and predicted by SVM | 732 | 687 | 870 | 759 |
| Set of Libraries | Number of LincRNAs Differentially Expressed |
|---|---|
| H. seropedicae (libraries id 9, id 10, id 11 and id 12) | 267 |
| A. brasilense (libraries id 29, id 30, id 31 and id 32) | 476 |
| H. seropedicae and A. brasilense (all the libraries: id 9, id 10, id 11, id 12, id 29, id 30, id 31 and id 32) | 1164 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, L.M.; Grativol, C.; Thiebaut, F.; Carvalho, T.G.; Hardoim, P.R.; Hemerly, A.; Lifschitz, S.; Ferreira, P.C.G.; Walter, M.E.M.T. PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants. Non-Coding RNA 2017, 3, 11. https://doi.org/10.3390/ncrna3010011
Vieira LM, Grativol C, Thiebaut F, Carvalho TG, Hardoim PR, Hemerly A, Lifschitz S, Ferreira PCG, Walter MEMT. PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants. Non-Coding RNA. 2017; 3(1):11. https://doi.org/10.3390/ncrna3010011
Chicago/Turabian StyleVieira, Lucas Maciel, Clicia Grativol, Flavia Thiebaut, Thais G. Carvalho, Pablo R. Hardoim, Adriana Hemerly, Sergio Lifschitz, Paulo Cavalcanti Gomes Ferreira, and Maria Emilia M. T. Walter. 2017. "PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants" Non-Coding RNA 3, no. 1: 11. https://doi.org/10.3390/ncrna3010011
APA StyleVieira, L. M., Grativol, C., Thiebaut, F., Carvalho, T. G., Hardoim, P. R., Hemerly, A., Lifschitz, S., Ferreira, P. C. G., & Walter, M. E. M. T. (2017). PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants. Non-Coding RNA, 3(1), 11. https://doi.org/10.3390/ncrna3010011