
Swelling Dynamics of a DNA-Polymer Hybrid Hydrogel Prepared Using Polyethylene Glycol as a Porogen

Abstract
:
1. Introduction
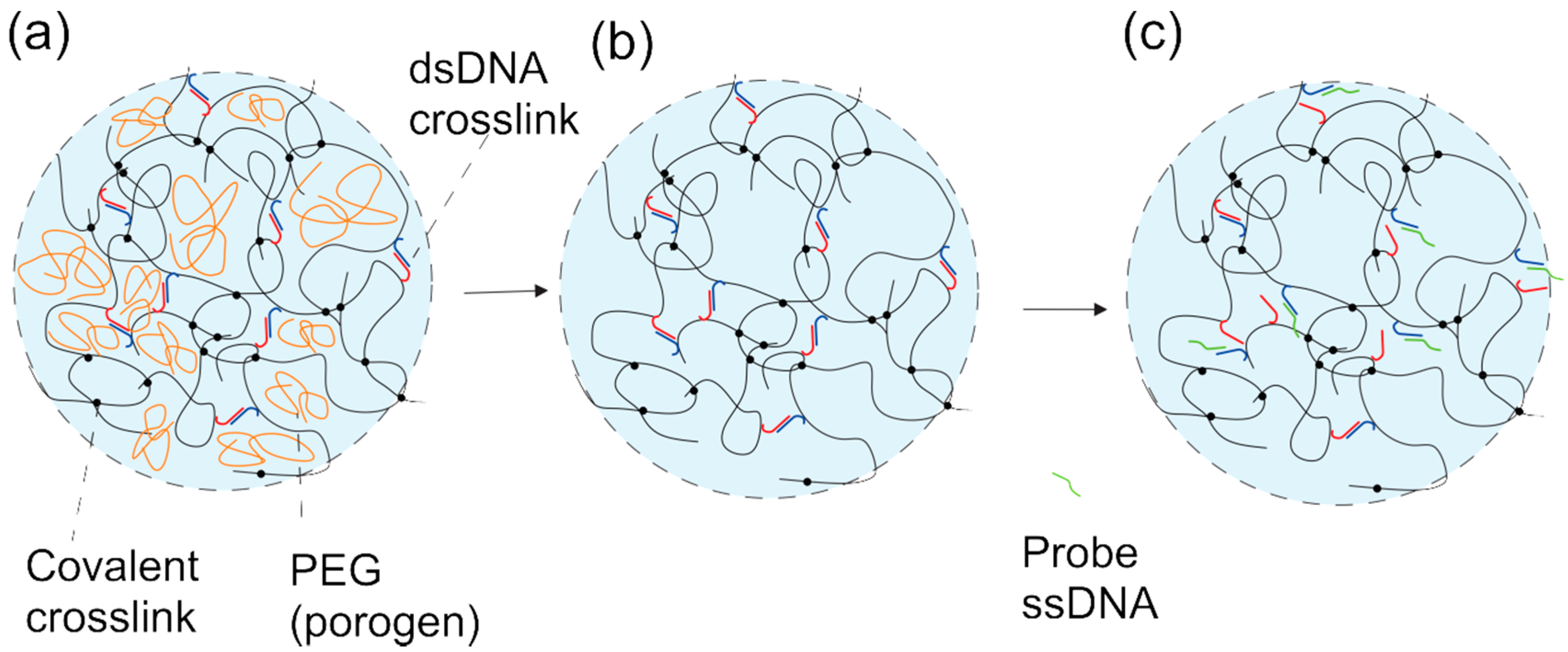
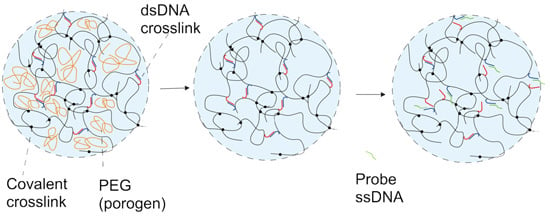
2. Results and Discussion
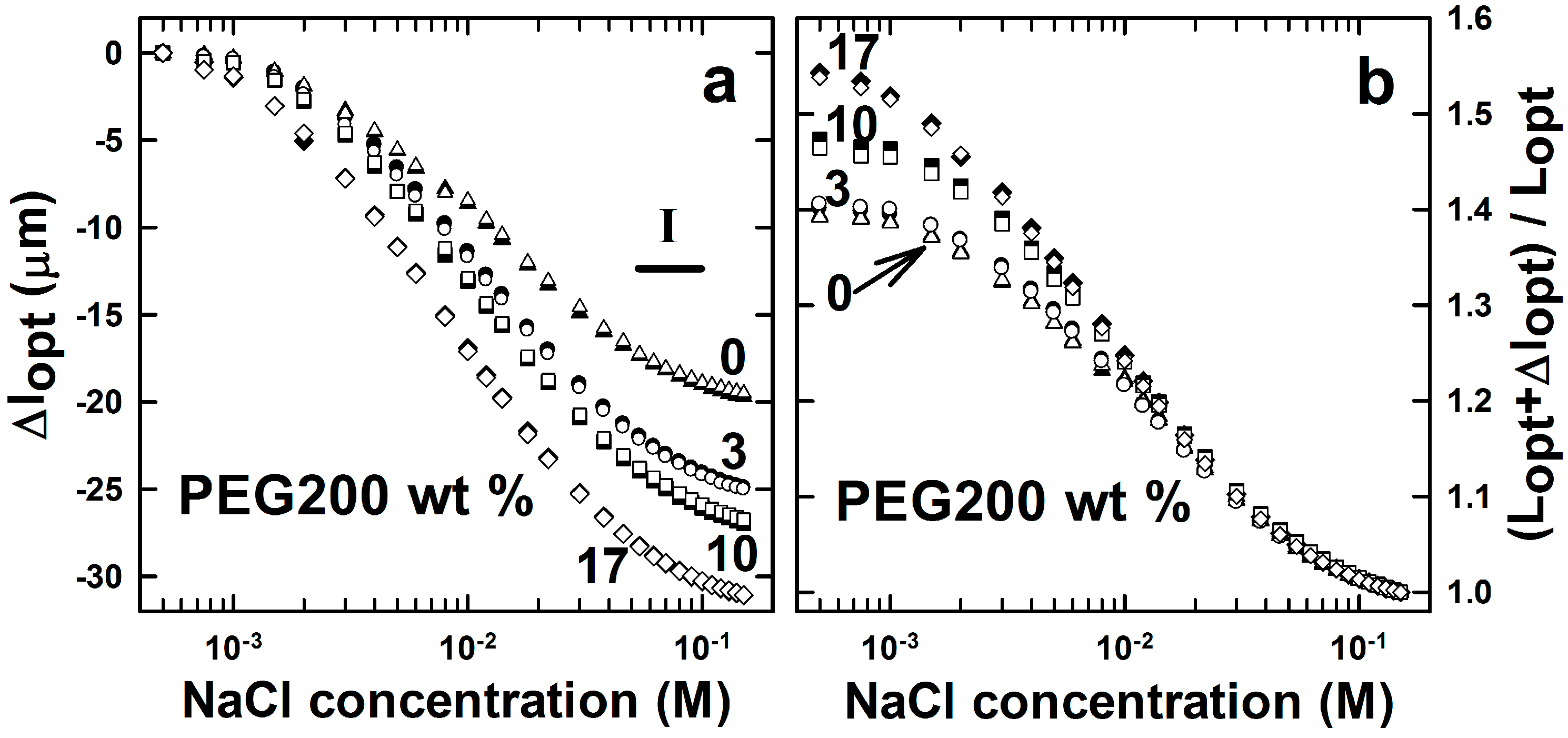
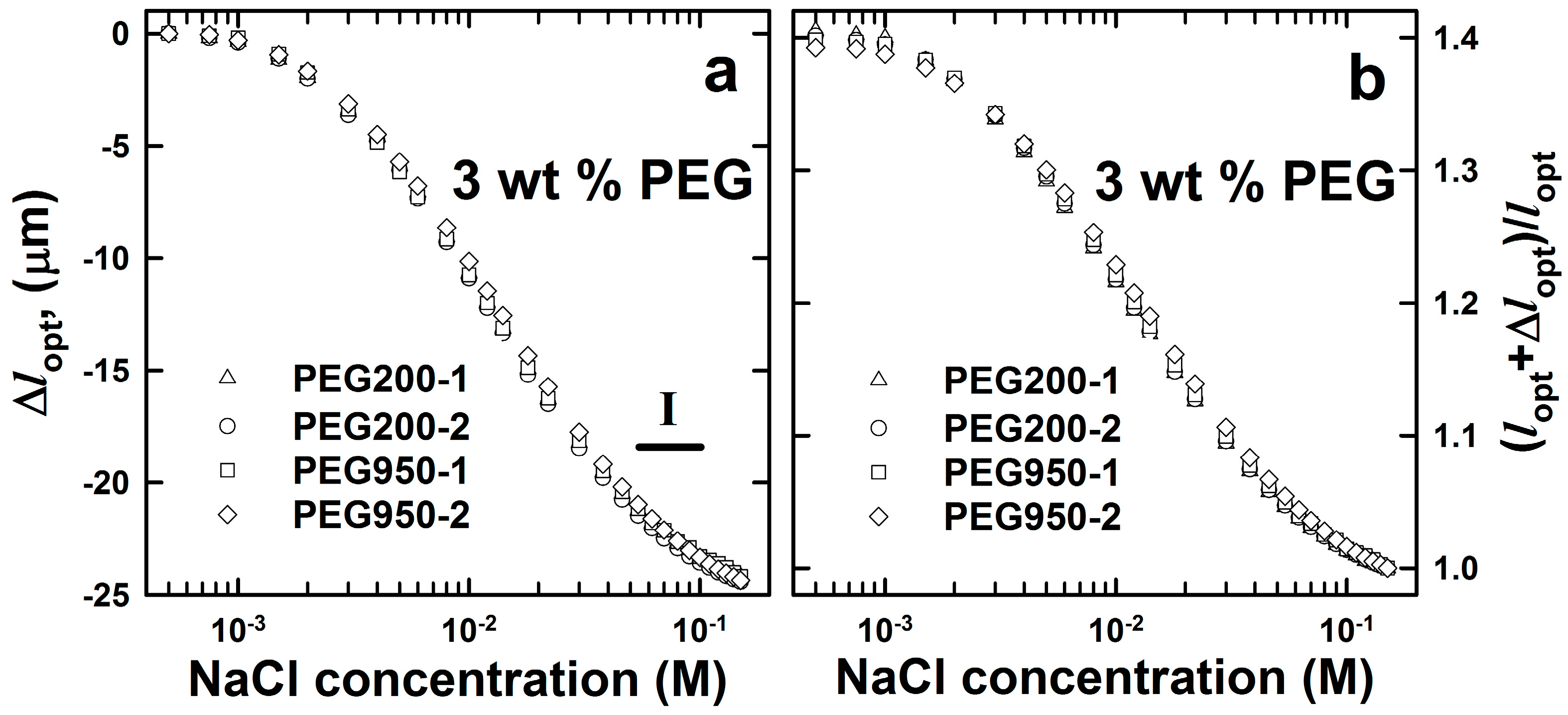
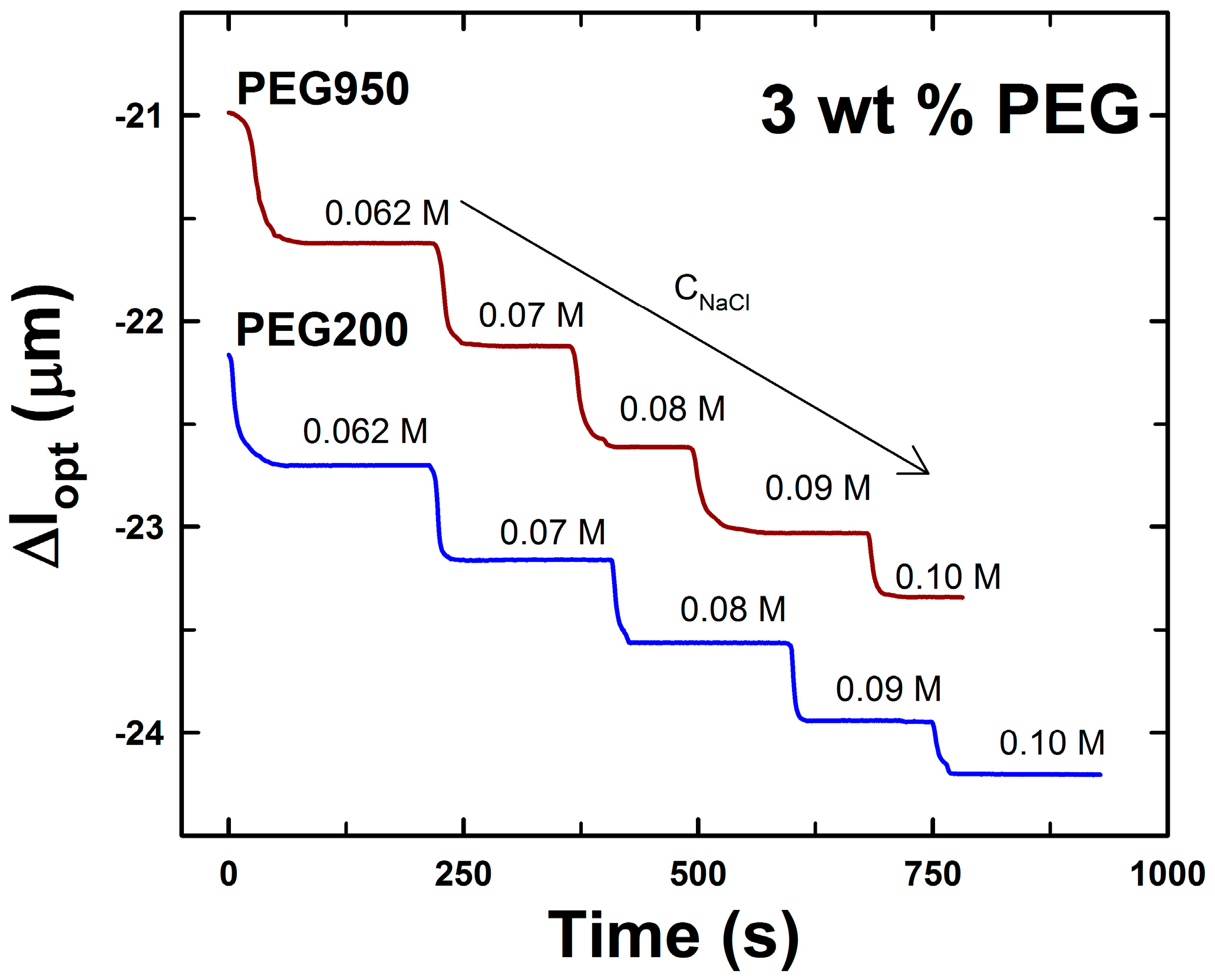
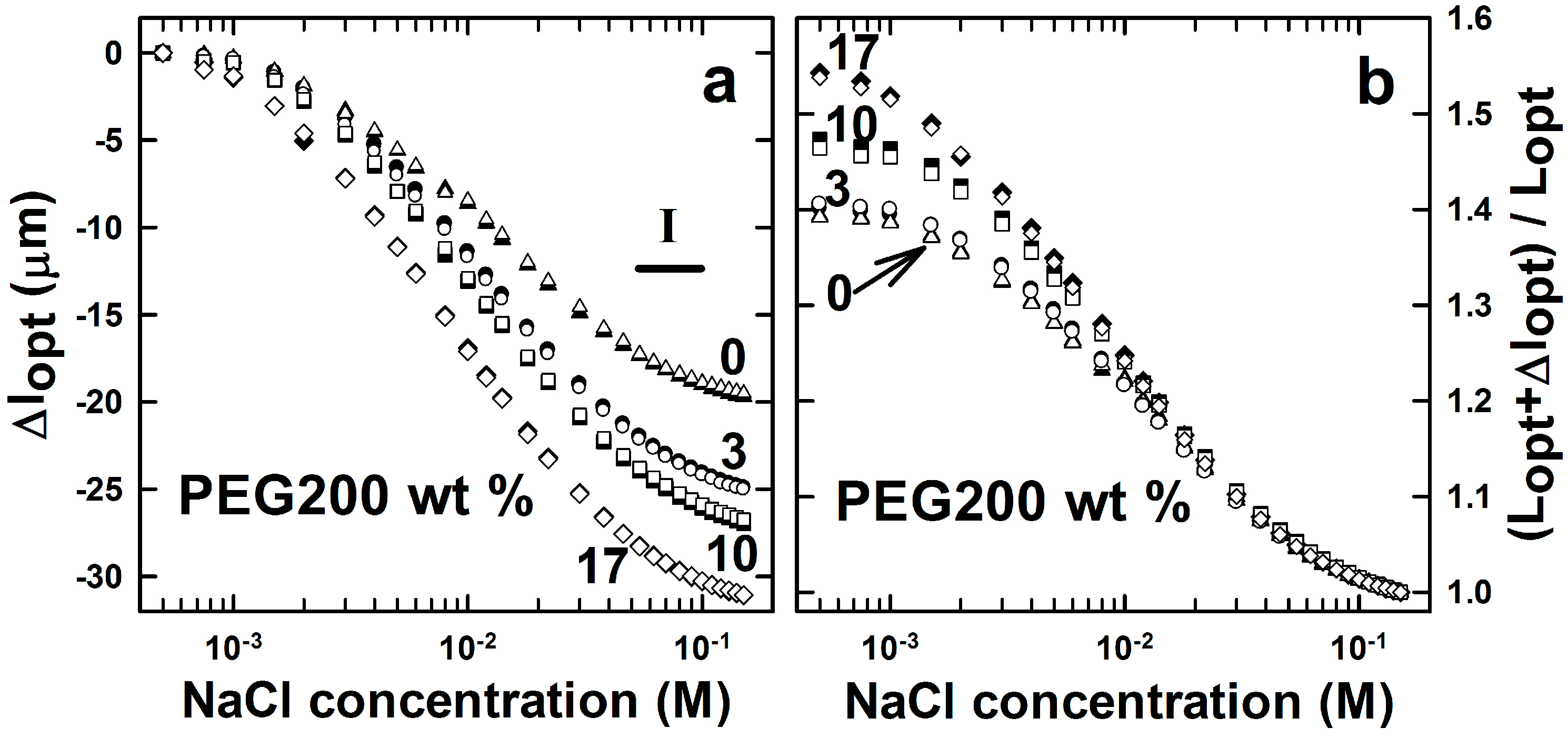
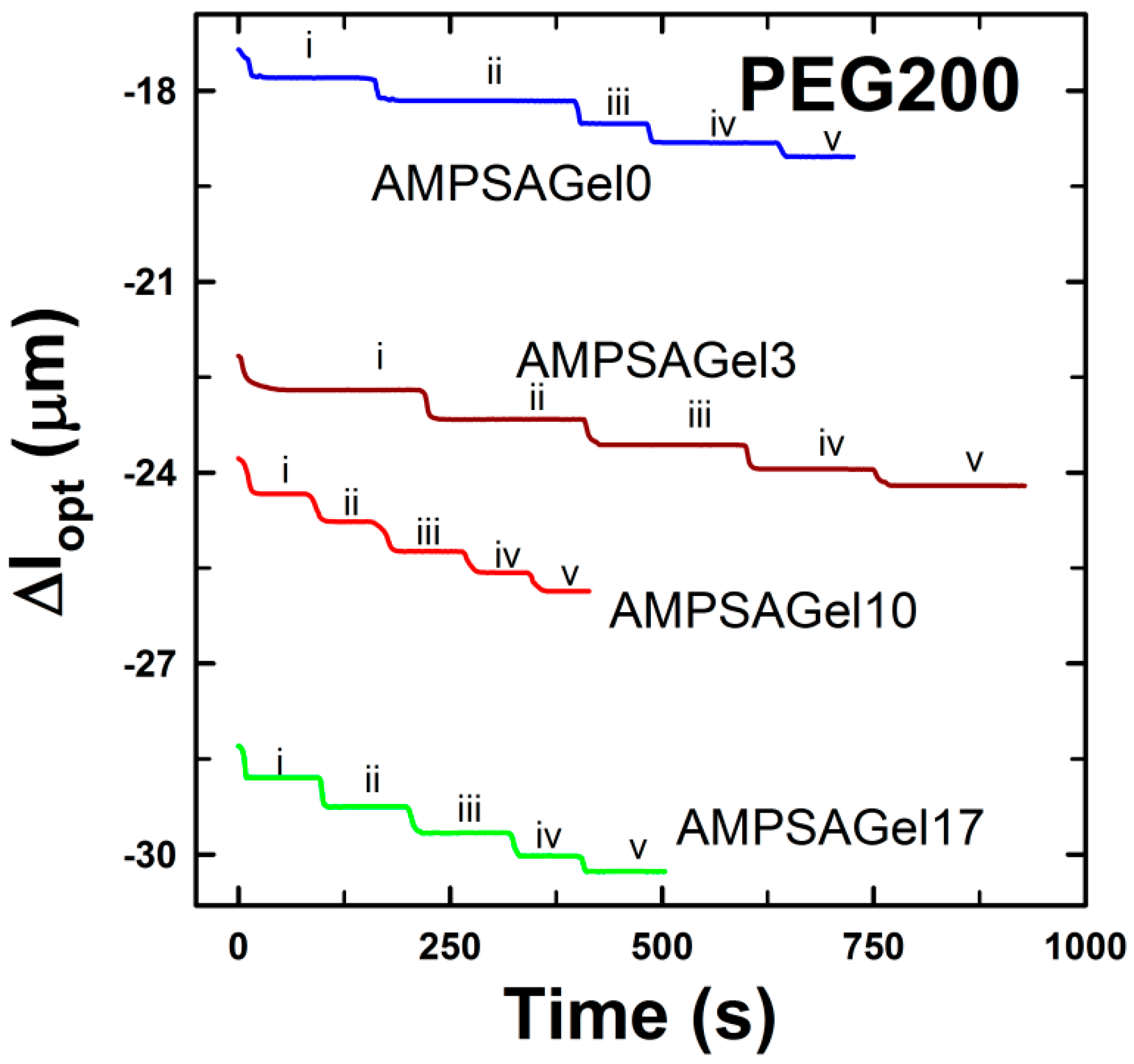
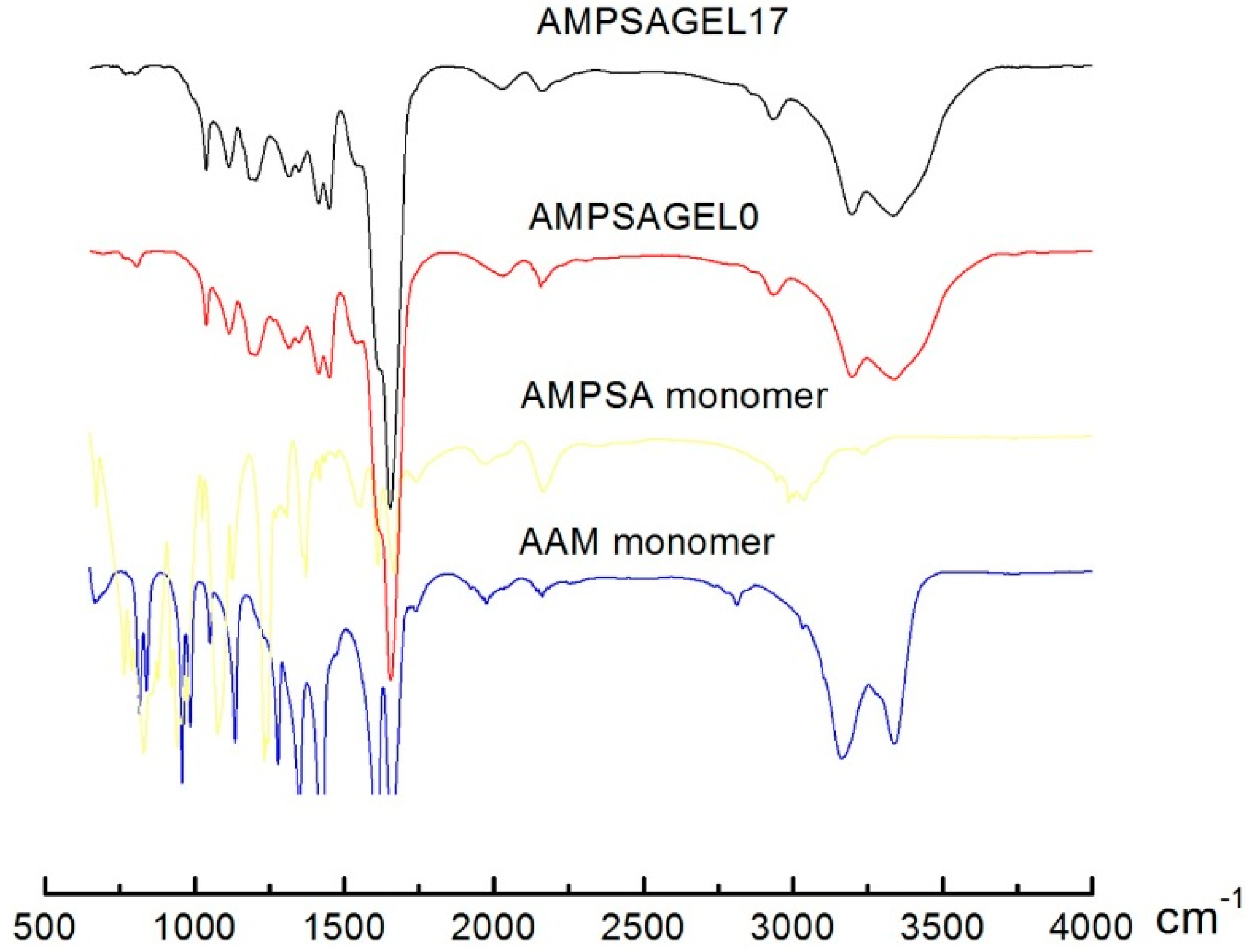
2.1. AMPSA-co-AAm Hydrogels Synthesized with Various Fractions of Porogens
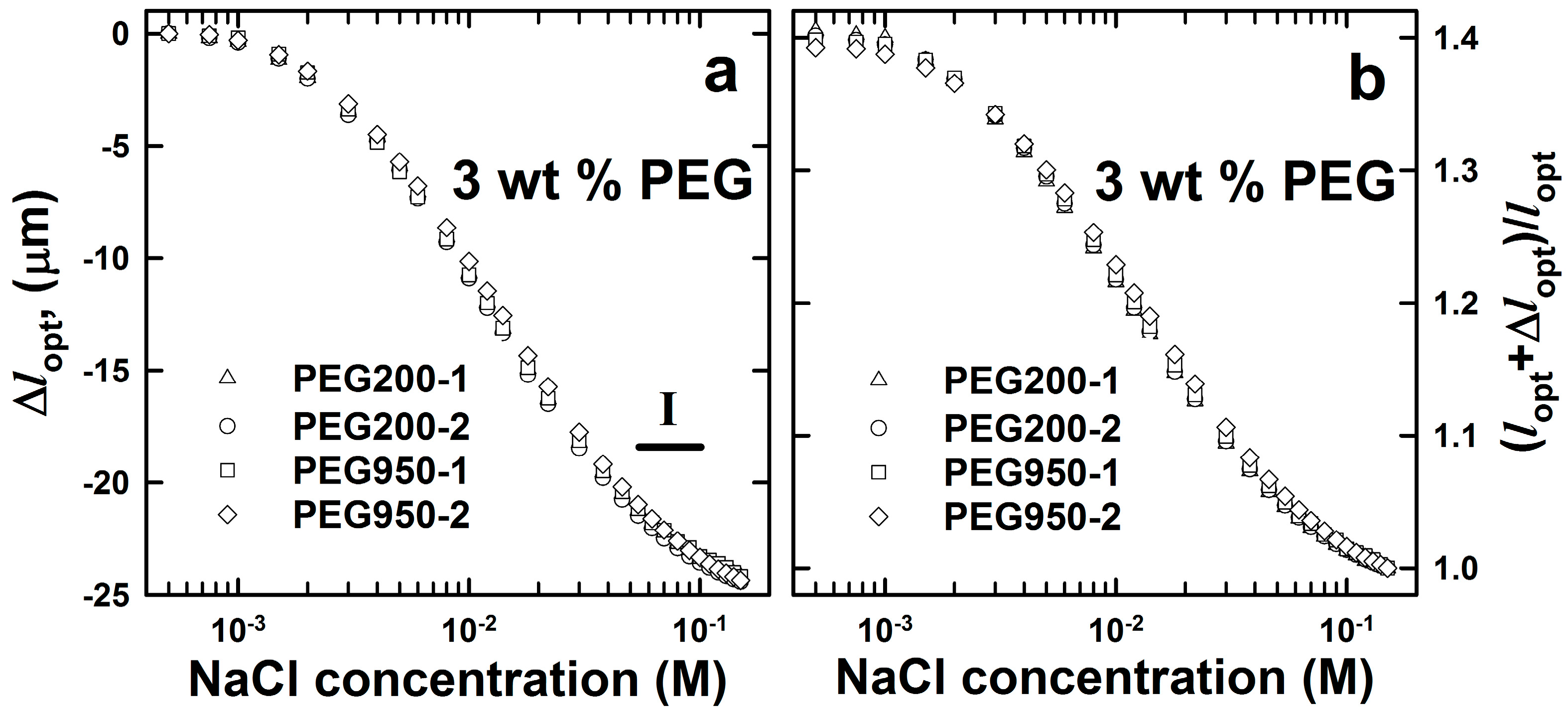
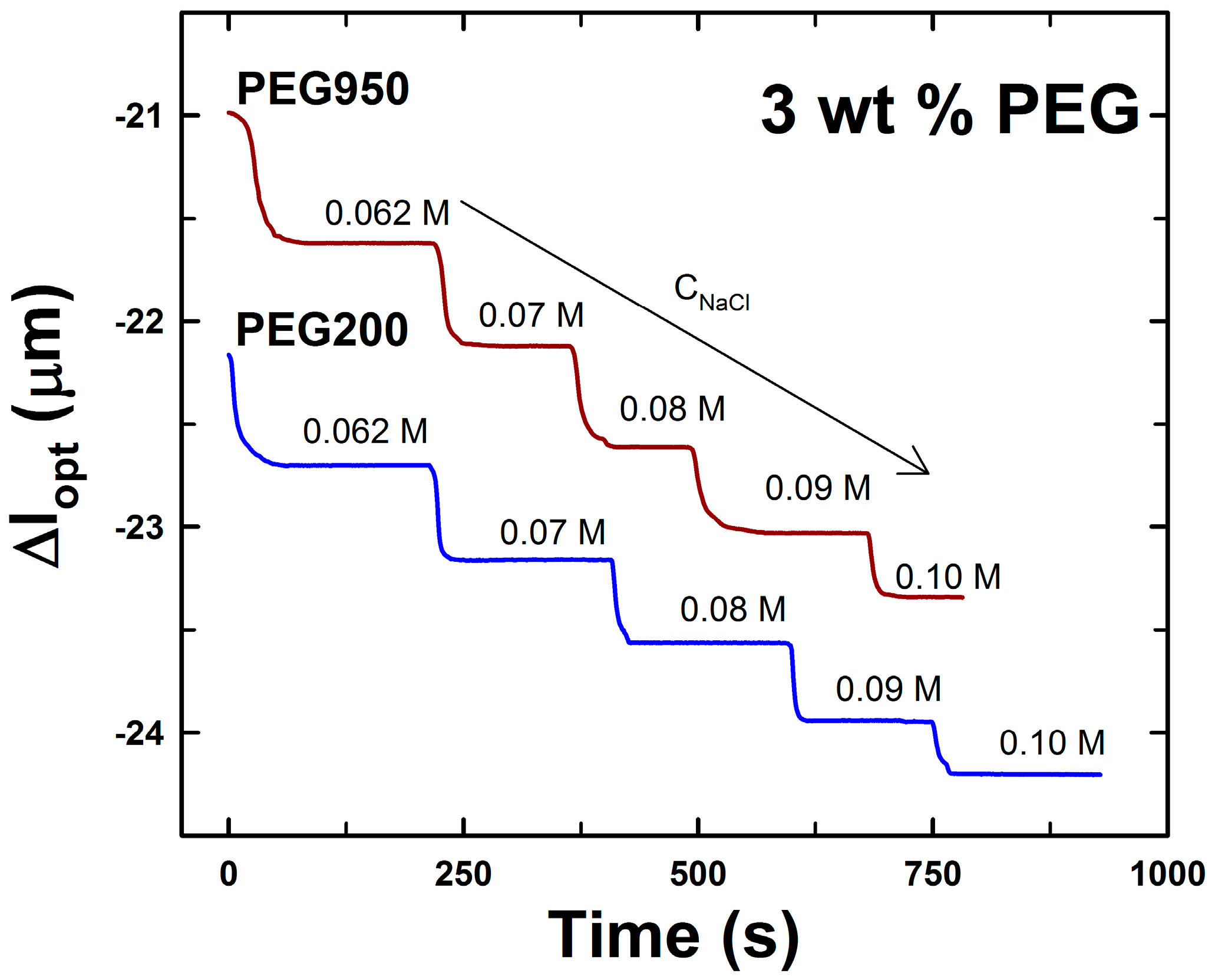
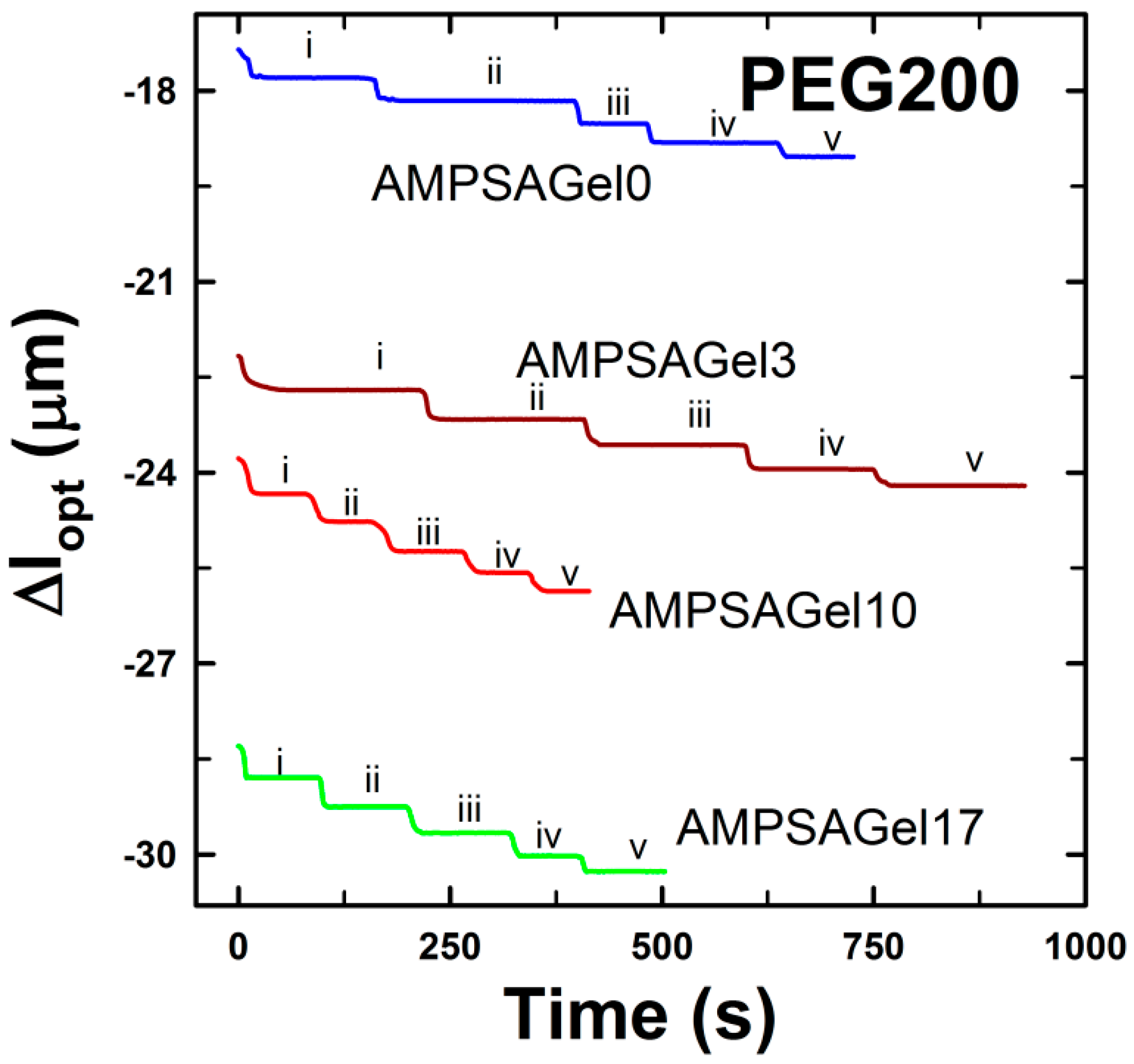
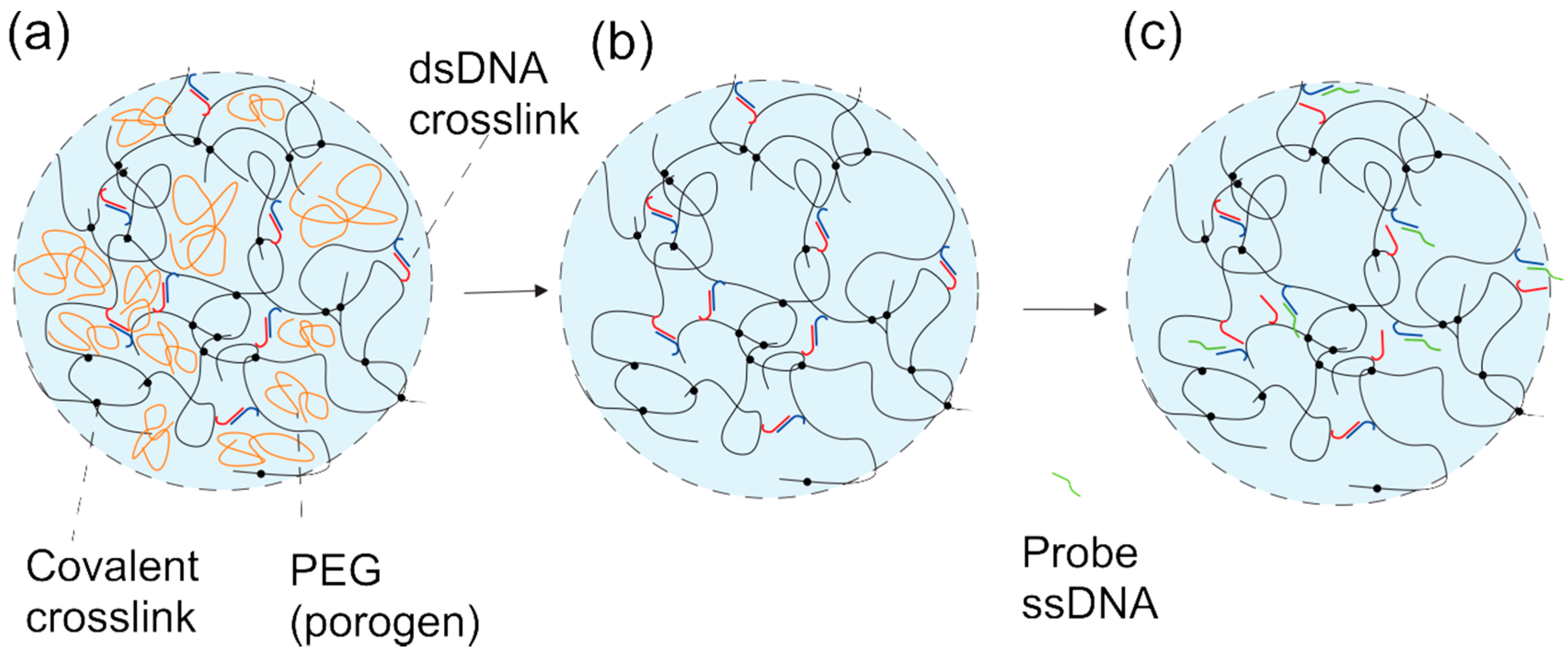
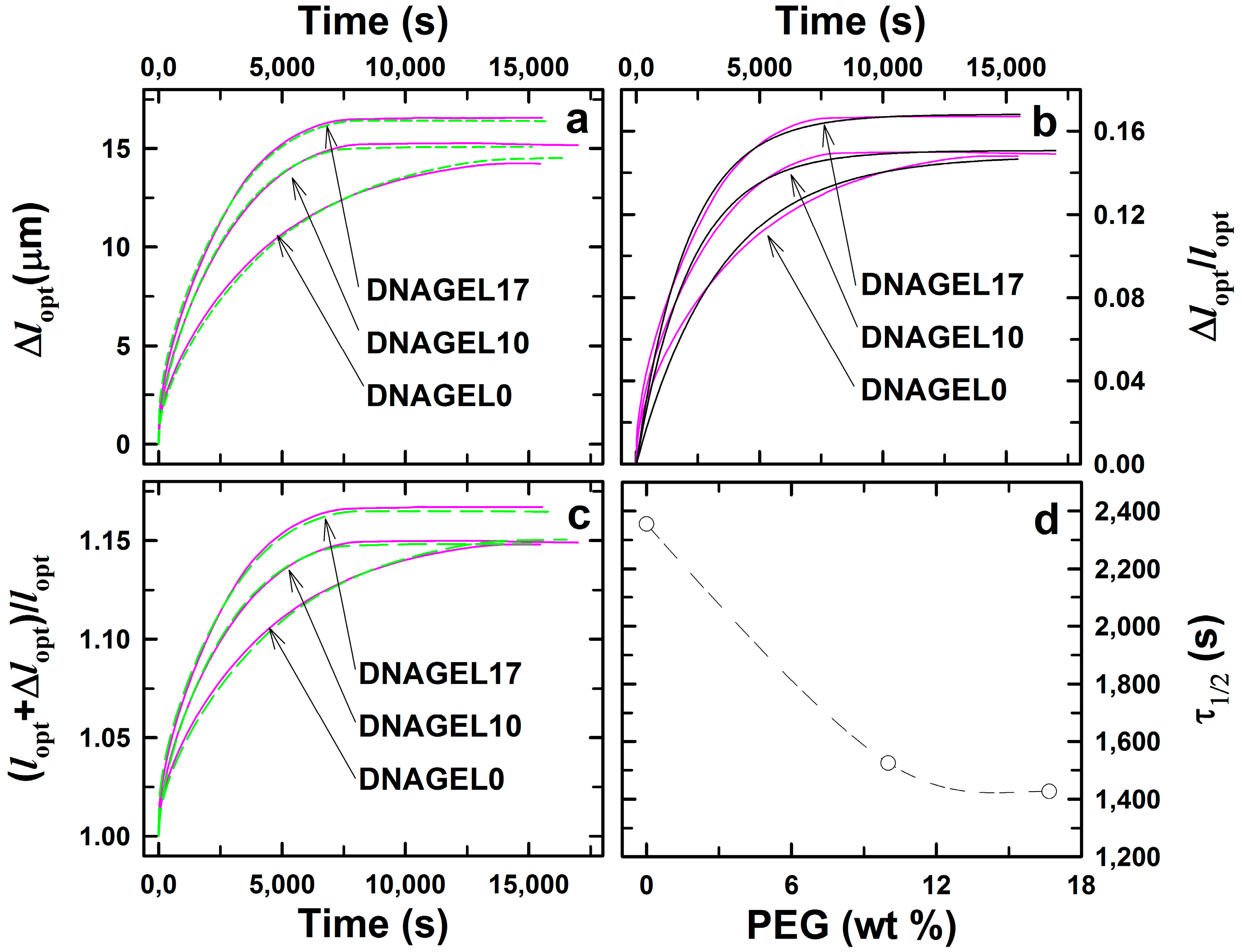
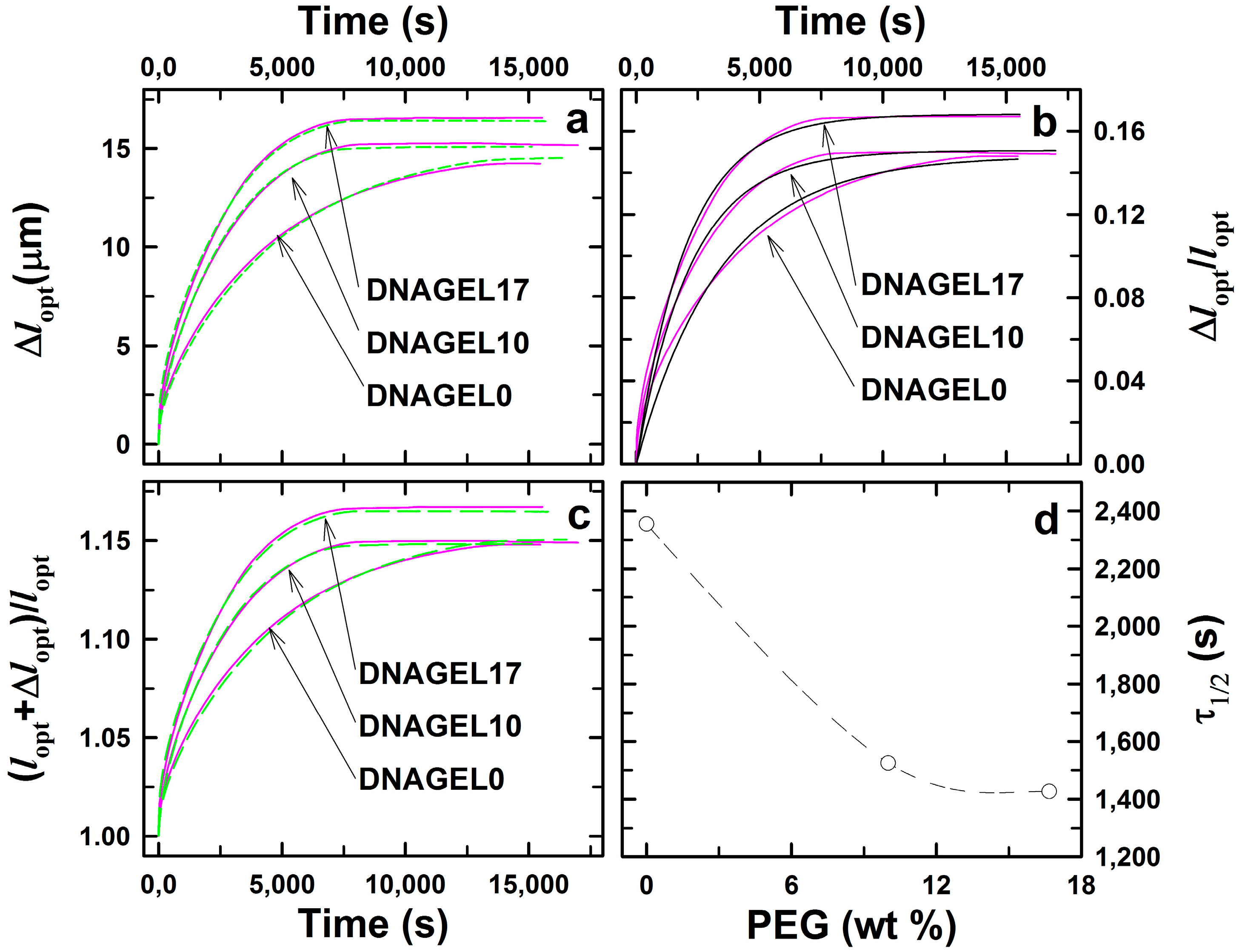
2.2. DNA-g-AAm Hydrogels with Altered Pore Size Distribution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Base Pair Sequence |
|---|---|
| Sensing (blue) | 5′-CTG ATC TAA GTA ACT ACT AG-3′ |
| Blocking (red) | 5′-CTC AGT CAC TAG TAG TTA CT-3′ |
| Probe (green) | 5′-CTA GTA GTT ACT TAG ATC-3′ |
3. Conclusions
4. Experimental Section

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ricka, J.; Tanaka, T. Swelling of ionic gels: Quantitative performance of the donnan theory. Macromolecules 1984, 17, 2916–2921. [Google Scholar] [CrossRef]
- Brannon-Peppas, L.; Peppas, N.A. Dynamic and equilibrium swelling behaviour of pH-sensitive hydrogels containing 2-hydroxyethyl methacrylate. Biomaterials 1990, 11, 635–644. [Google Scholar] [CrossRef]
- Brannon-Peppas, L.; Peppas, N.A. Time-dependent response of ionic polymer networks to pH and ionic strength changes. Int. J. Pharm. 1991, 70, 53–57. [Google Scholar] [CrossRef]
- Beebe, D.J.; Moore, J.S.; Bauer, J.M.; Yu, Q.; Liu, R.H.; Devadoss, C.; Jo, B.-H. Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 2000, 404, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Nakamae, K.; Hoffman, A.S.; Kanzaki, Y. Stimuli-sensitivities of hydrogels containing phosphate groups. Macromol. Chem. Phys. 1994, 195, 1111–1120. [Google Scholar] [CrossRef]
- Yoshida, R.; Uchida, K.; Kaneko, Y.; Sakai, K.; Kikuchi, A.; Sakurai, Y.; Okano, T. Comb-type grafted hydrogels with rapid deswelling response to temperature changes. Nature 1995, 374, 240–242. [Google Scholar] [CrossRef]
- Lee, W.F.; Yuan, W.Y. Thermoreversible hydrogels X: Synthesis and swelling behavior of the (N-isopropylacrylamide-co-sodium 2-acrylamido-2-methylpropyl sulfonate) copolymeric hydrogels. J. Appl. Polym. Sci. 2000, 77, 1760–1768. [Google Scholar] [CrossRef]
- Schröder, U.P.; Oppermann, W. Properties of polyelectrolyte gels. In Physical Properties of Polymeric Gels; Wiley & Sons: Chichester, UK, 1996; pp. 19–38. [Google Scholar]
- Park, T.G.; Hoffman, A.S. Sodium chloride-induced phase transition in nonionic poly(N-isopropylacrylamide) gel. Macromolecules 1993, 26, 5045–5048. [Google Scholar] [CrossRef]
- Grimshaw, P.E.; Nussbaum, J.H.; Grodzinsky, A.J.; Yarmush, M.L. Kinetics of electrically and chemically induced swelling in polyelectrolyte gels. J. Chem. Phys. 1990, 93, 4462–4472. [Google Scholar] [CrossRef]
- Sun, S.; Mak, A.F.T. The dynamical response of a hydrogel fiber to electrochemical stimulation. J. Polym. Sci. B Polym. Phys. 2001, 39, 236–246. [Google Scholar] [CrossRef]
- Mamada, A.; Tanaka, T.; Kungwatchakun, D.; Irie, M. Photoinduced phase transition of gels. Macromolecules 1990, 23, 1517–1519. [Google Scholar] [CrossRef]
- Eeckman, F.; Moes, A.J.; Amighi, K. Surfactant induced drug delivery based on the use of thermosensitive polymers. J. Controll. Release 2003, 88, 105–116. [Google Scholar] [CrossRef]
- Holtz, J.H.; Asher, S.A. Polymerized colloidal crystal hydrogel films as intelligent chemical sensing materials. Nature 1997, 389, 829–832. [Google Scholar] [CrossRef]
- Miyata, T.; Asami, N.; Uragami, T. A reversibly antigen-responsive hydrogel. Nature 1999, 399, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, V.L.; Sharma, A.C.; Goponenko, A.V.; Das, S.; Lednev, I.K.; Wilcox, C.S.; Finegold, D.N.; Asher, S.A. High ionic strength glucose-sensing photonic crystal. Anal. Chem. 2003, 75, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.M.; Gu, H.W.; Fu, D.G.; Gao, P.; Lam, J.K.; Xu, B. Enzymatic formation of supramolecular hydrogels. Adv. Mater. 2004, 16, 1440–1444. [Google Scholar] [CrossRef]
- Kim, H.; Cohen, R.E.; Hammond, P.T.; Irvine, D.J. Live lymphocyte arrays for biosensing. Adv. Funct. Mater. 2006, 16, 1313–1323. [Google Scholar] [CrossRef]
- Yang, Z.; Ho, P.-L.; Liang, G.; Chow, K.H.; Wang, Q.; Cao, Y.; Guo, Z.; Xu, B. Using β-lactamase to trigger supramolecular hydrogelation. J. Am. Chem. Soc. 2007, 129, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Tierney, S.; Sletmoen, M.; Skjak-Braek, G.; Stokke, B.T. Interferometric characterization of swelling of covalently crosslinked alginate gel and changes associated with polymer impregnation. Carbohydr. Polym. 2010, 80, 828–832. [Google Scholar] [CrossRef]
- Tierney, S.; Falch, B.M.H.; Hjelme, D.R.; Stokke, B.T. Determination of glucose levels using a functionalized hydrogel-optical fiber biosensor: Toward continuous monitoring of blood glucose in vivo. Anal. Chem. 2009, 81, 3630–3636. [Google Scholar] [CrossRef] [PubMed]
- Tierney, S.; Volden, S.; Stokke, B.T. Glucose sensors based on a responsive gel incorporated as a fabry-perot cavity on a fiber-optic readout platform. Biosens. Bioelectron. 2009, 24, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Skjaervold, N.K.; Solligard, E.; Hjelme, D.R.; Aadahl, P. Continuous measurement of blood glucose validation of a new intravascular sensor. Anesthesiology 2011, 114, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Gawel, K.; Stokke, B.T. Toehold of dsdna exchange affects the hydrogel swelling kinetics of a polymer-dsDNA hybrid hydrogel. Soft Matter 2011, 7, 1741–1746. [Google Scholar] [CrossRef]
- Gawel, K.; Stokke, B.T. Logic swelling response of DNA-polymer hybrid hydrogel. Soft Matter 2011, 7, 4615–4618. [Google Scholar] [CrossRef]
- Tierney, S.; Stokke, B.T. Development of an oligonucleotide functionalized hydrogel integrated on a high resolution interferometric readout platform as a label-free macromolecule sensing device. Biomacromolecules 2009, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Gawel, K.; Gao, M.; Stokke, B.T. Impregnation of weakly charged anionic microhydrogels with cationic polyelectrolytes and their swelling properties monitored by a high resolution interferometric technique. Transformation from a polyelectrolyte to polyampholyte hydrogel. Eur. Polym. J. 2012, 48, 1949–1959. [Google Scholar] [CrossRef]
- Gao, M.; Gawel, K.; Stokke, B.T. High resolution interferometry as a tool for characterization of swelling of weakly charged hydrogels subjected to amphiphile and cyclodextrin exposure. J. Colloid Interface Sci. 2013, 390, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Tierney, S.; Hjelme, D.R.; Stokke, B.T. Determination of swelling of responsive gels with nanometer resolution. Fiber-optic based platform for hydrogels as signal transducers. Anal. Chem. 2008, 80, 5086–5093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Moore, J.S. Fast pH- and ionic strength-responsive hydrogels in microchannels. Langmuir 2001, 17, 4758–4763. [Google Scholar] [CrossRef]
- Bouklas, N.; Huang, R. Swelling kinetics of polymer gels: Comparison of linear and nonlinear theories. Soft Matter 2012, 8, 8194–8203. [Google Scholar] [CrossRef]
- Reynaldo, L.P.; Vologodskii, A.V.; Neri, B.P.; Lyamichev, V.I. The kinetics of oligonucleotide replacements. J. Mol. Biol. 2000, 297, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Luan, G.Y.; Guo, Q.P.; Liang, J.X. A new class of homogeneous nucleic acid probes based on specific displacement hybridization. Nucleic Acids Res. 2002, 30, e5. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Ouldridge, T.E.; Sulc, P.; Schaeffer, J.M.; Yurke, B.; Louis, A.A.; Doye, J.P.K.; Winfree, E. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Res. 2013, 41, 10641–10658. [Google Scholar] [CrossRef] [PubMed]
- Livshits, M.A.; Mirzabekov, A.D. Theoretical analysis of the kinetics of DNA hybridization with gel-immobilized oligonucleotides. Biophys. J. 1996, 71, 2795–2801. [Google Scholar] [CrossRef]
- Kaneko, Y.; Sakai, K.; Kikuchi, A.; Yoshida, R.; Sakurai, Y.; Okano, T. Influence of freely mobile grafted chain length on dynamic properties of comb-type grafted poly(N-isopropylacrylamide) hydrogels. Macromolecules 1995, 28, 7717–7723. [Google Scholar] [CrossRef]
- Bin Imran, A.; Seki, T.; Takeoka, Y. Recent advances in hydrogels in terms of fast stimuli responsiveness and superior mechanical performance. Polym. J. 2010, 42, 839–851. [Google Scholar] [CrossRef]
- Annabi, N.; Nichol, J.W.; Zhong, X.; Ji, C.D.; Koshy, S.; Khademhosseini, A.; Dehghani, F. Controlling the porosity and microarchitecture of hydrogels for tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Toita, S.; Sawada, S.; Akiyoshi, K.; Stokke, B.T. Cyclodextrin triggered dimensional changes of polysaccharide nanogel integrated hydrogels at nanometer resolution. Soft Matter 2013, 9, 5178–5185. [Google Scholar] [CrossRef]
- Prot, V.; Sveinsson, H.M.; Gawel, K.; Gao, M.; Skallerud, B.; Stokke, B.T. Swelling of a hemi-ellipsoidal ionic hydrogel for determination of material properties of deposited thin polymer films: An inverse finite element approach. Soft Matter 2013, 9, 5815–5827. [Google Scholar] [CrossRef]
- Caykara, T.; Bulut, M.; Dilsiz, N.; Akyuz, Y. Macroporous poly(acrylamide) hydrogels: Swelling and shrinking behaviors. J. Macromol. Sci. Pure Appl. Chem. 2006, 43, 889–897. [Google Scholar] [CrossRef]
- Caykara, T.; Bulut, M.; Demirci, S. Preparation of macroporous poly(acrylamide) hydrogels by radiation induced polymerization technique. Nucl. Instrum. Methods Phys. Res. Sect. B 2007, 265, 366–369. [Google Scholar] [CrossRef]
- Gemeinhart, R.A.; Chen, J.; Park, H.; Park, K. pH-sensitivity of fast responsive superporous hydrogels. J. Biomater. Sci. Polym. Ed. 2000, 11, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Yurke, B.; Langrana, N.A. Mechanical properties of a reversible, DNA-crosslinked polyacrylamide hydrogel. J. Biomech. Eng. Trans. Asme 2004, 126, 104–110. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Gawel, K.; Stokke, B.T. Swelling Dynamics of a DNA-Polymer Hybrid Hydrogel Prepared Using Polyethylene Glycol as a Porogen. Gels 2015, 1, 219-234. https://doi.org/10.3390/gels1020219
Gao M, Gawel K, Stokke BT. Swelling Dynamics of a DNA-Polymer Hybrid Hydrogel Prepared Using Polyethylene Glycol as a Porogen. Gels. 2015; 1(2):219-234. https://doi.org/10.3390/gels1020219
Chicago/Turabian StyleGao, Ming, Kamila Gawel, and Bjørn Torger Stokke. 2015. "Swelling Dynamics of a DNA-Polymer Hybrid Hydrogel Prepared Using Polyethylene Glycol as a Porogen" Gels 1, no. 2: 219-234. https://doi.org/10.3390/gels1020219