
Precise Control over the Rheological Behavior of Associating Stimuli-Responsive Block Copolymer Gels

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
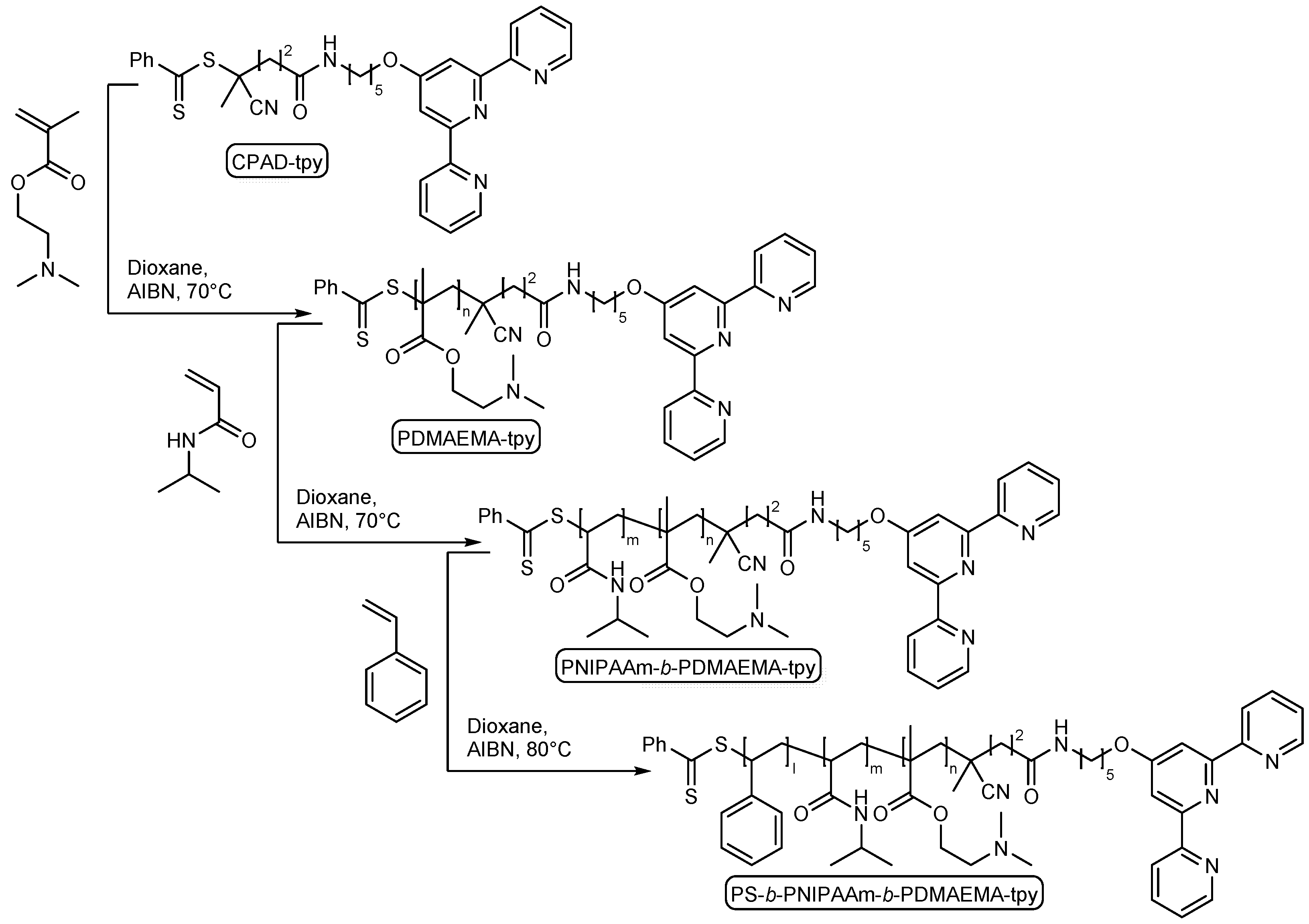
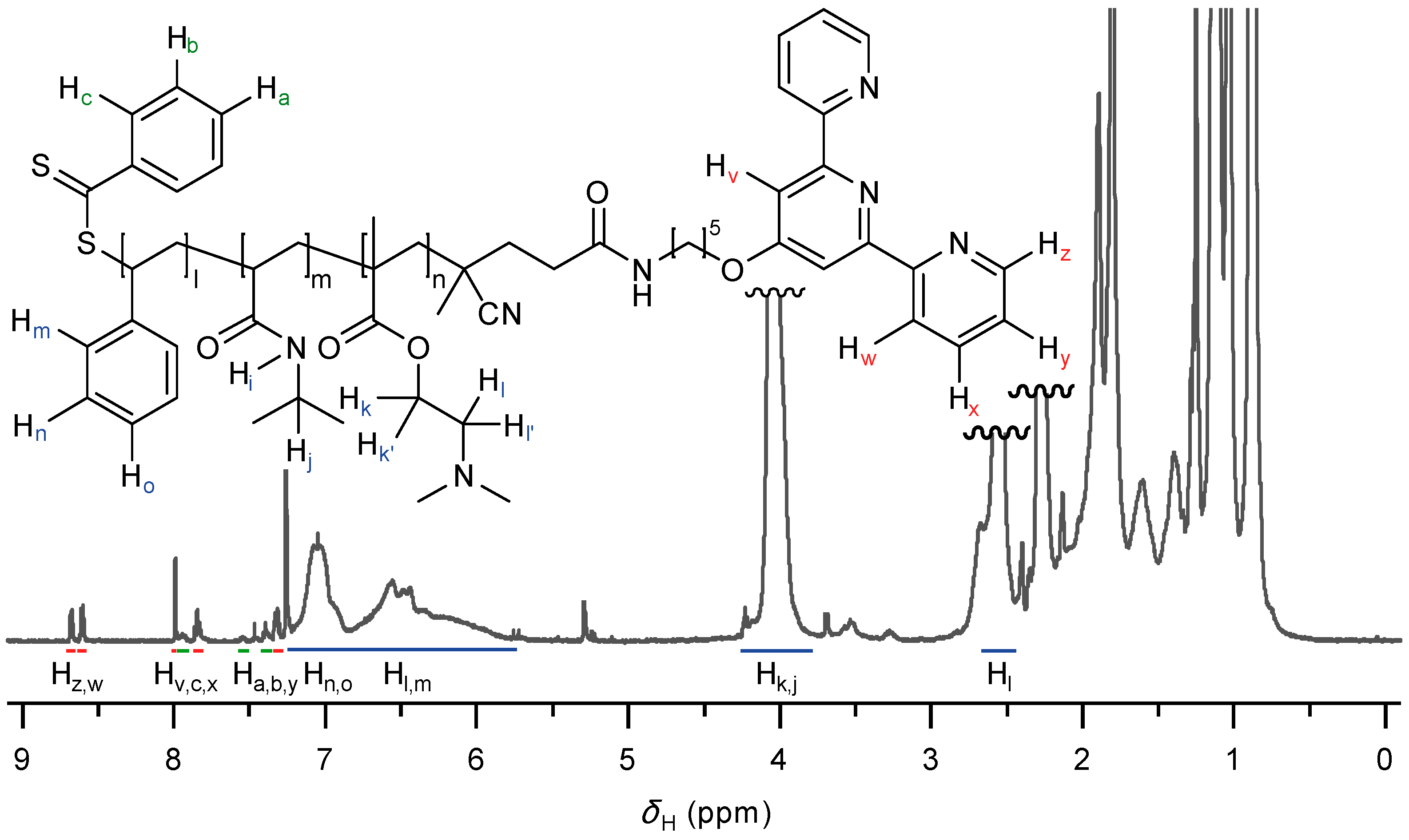
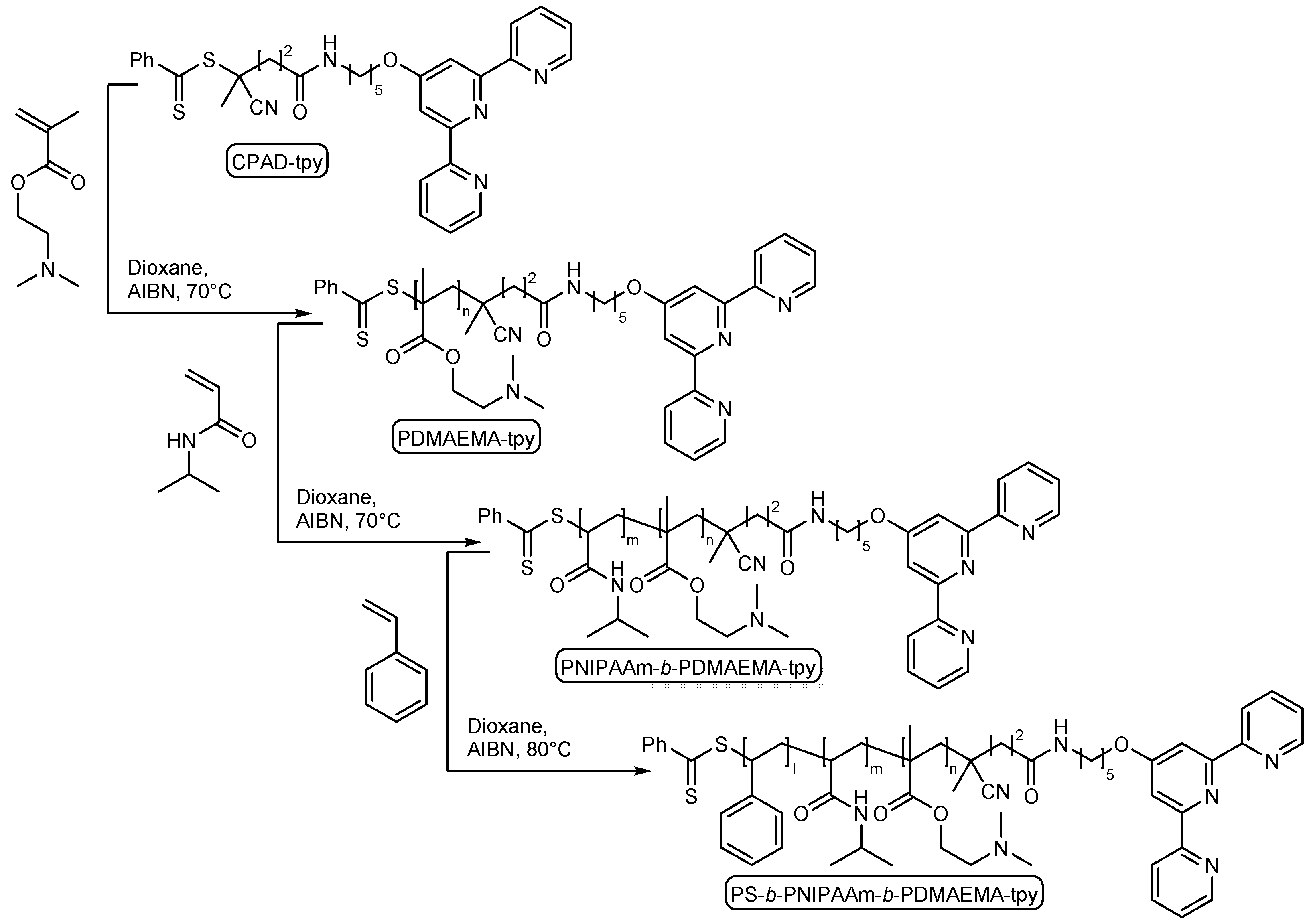
2.1. Synthesis of Functional Building Block
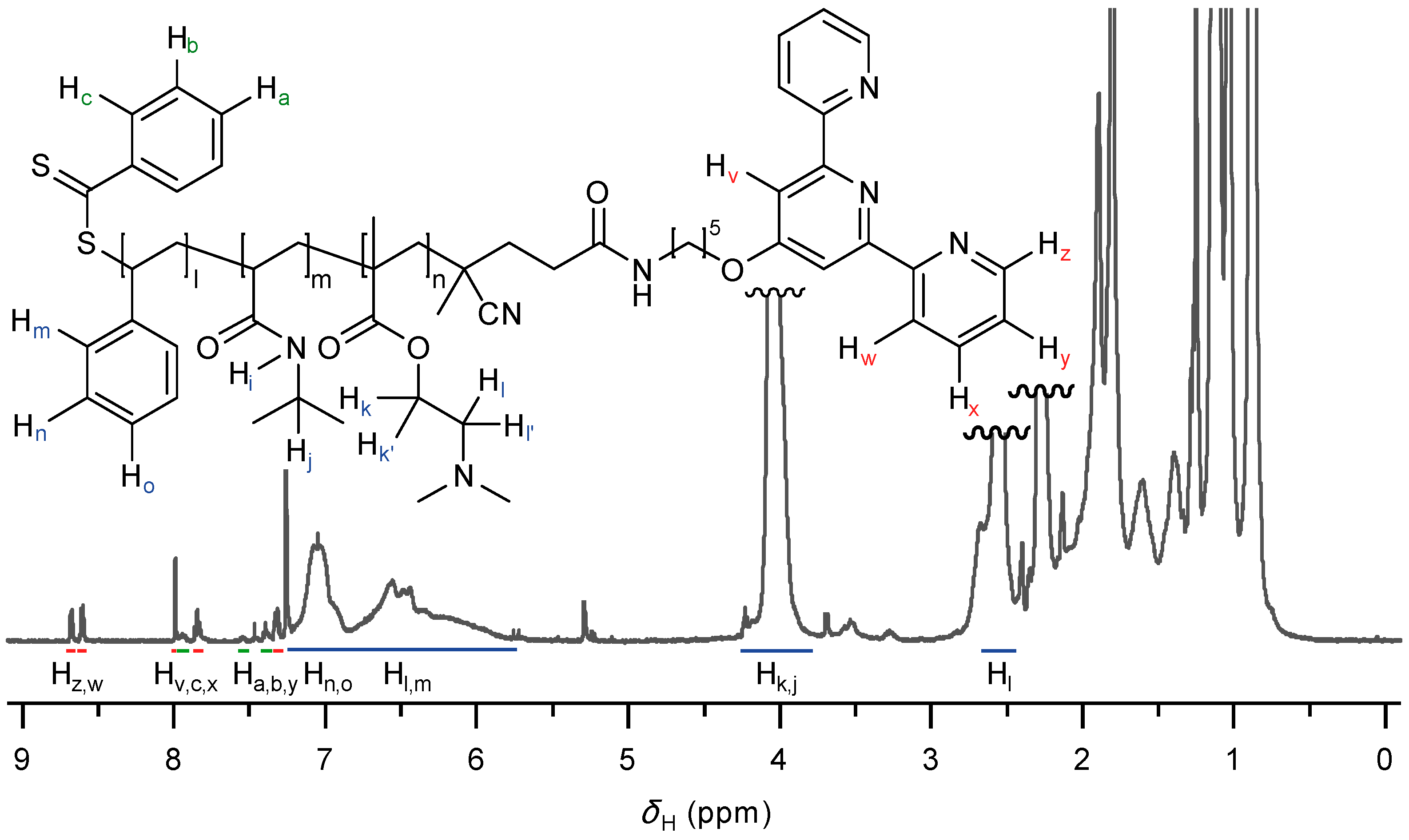

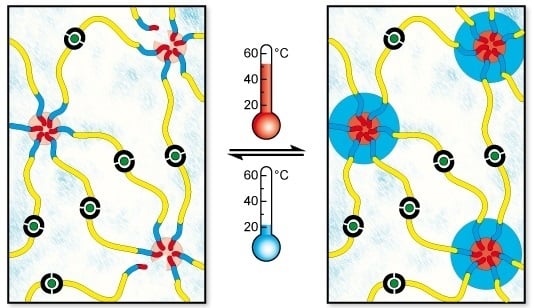
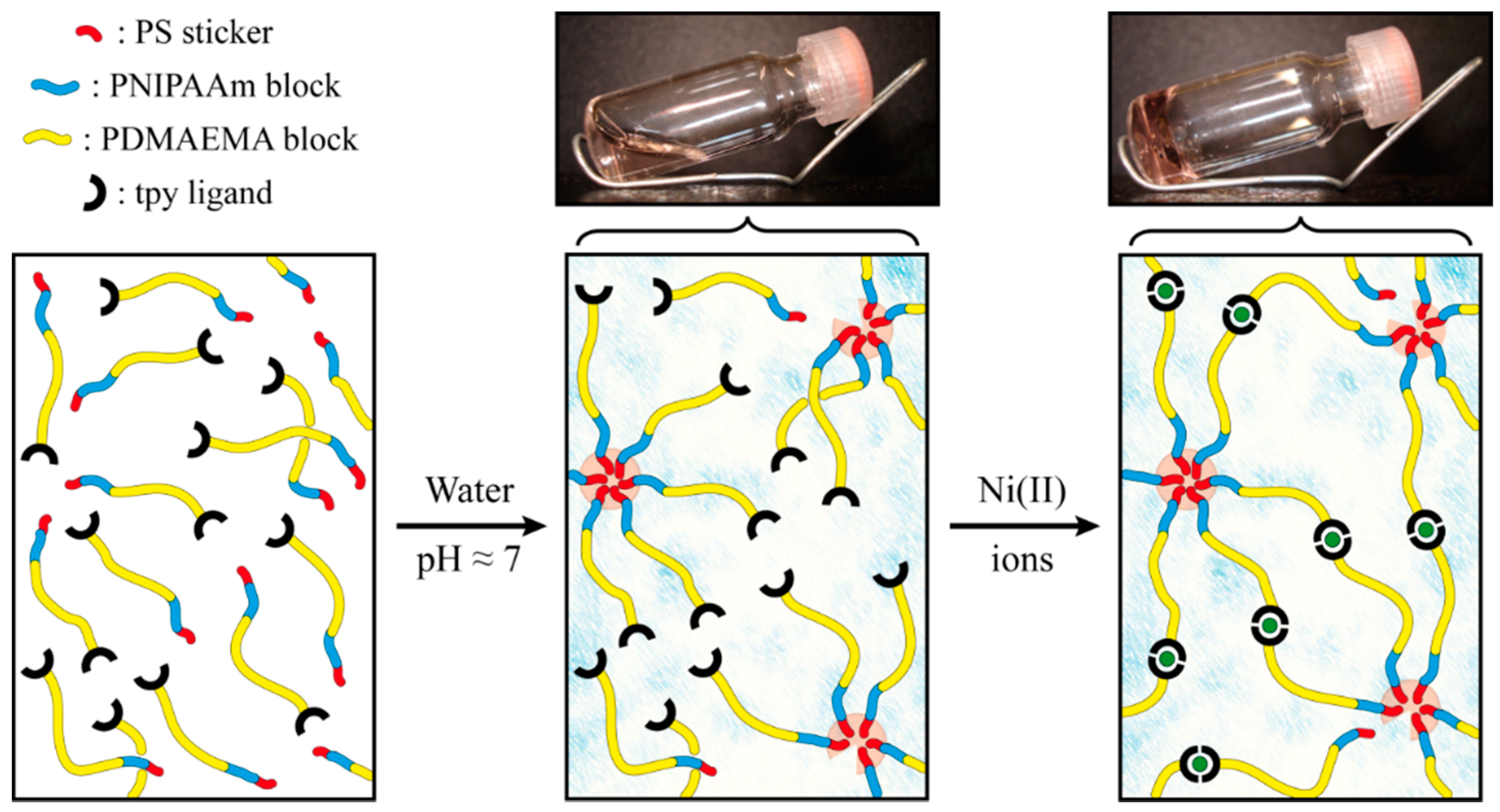
2.2. Self-Assembly into Metallo-Supramolecular Hydrogel
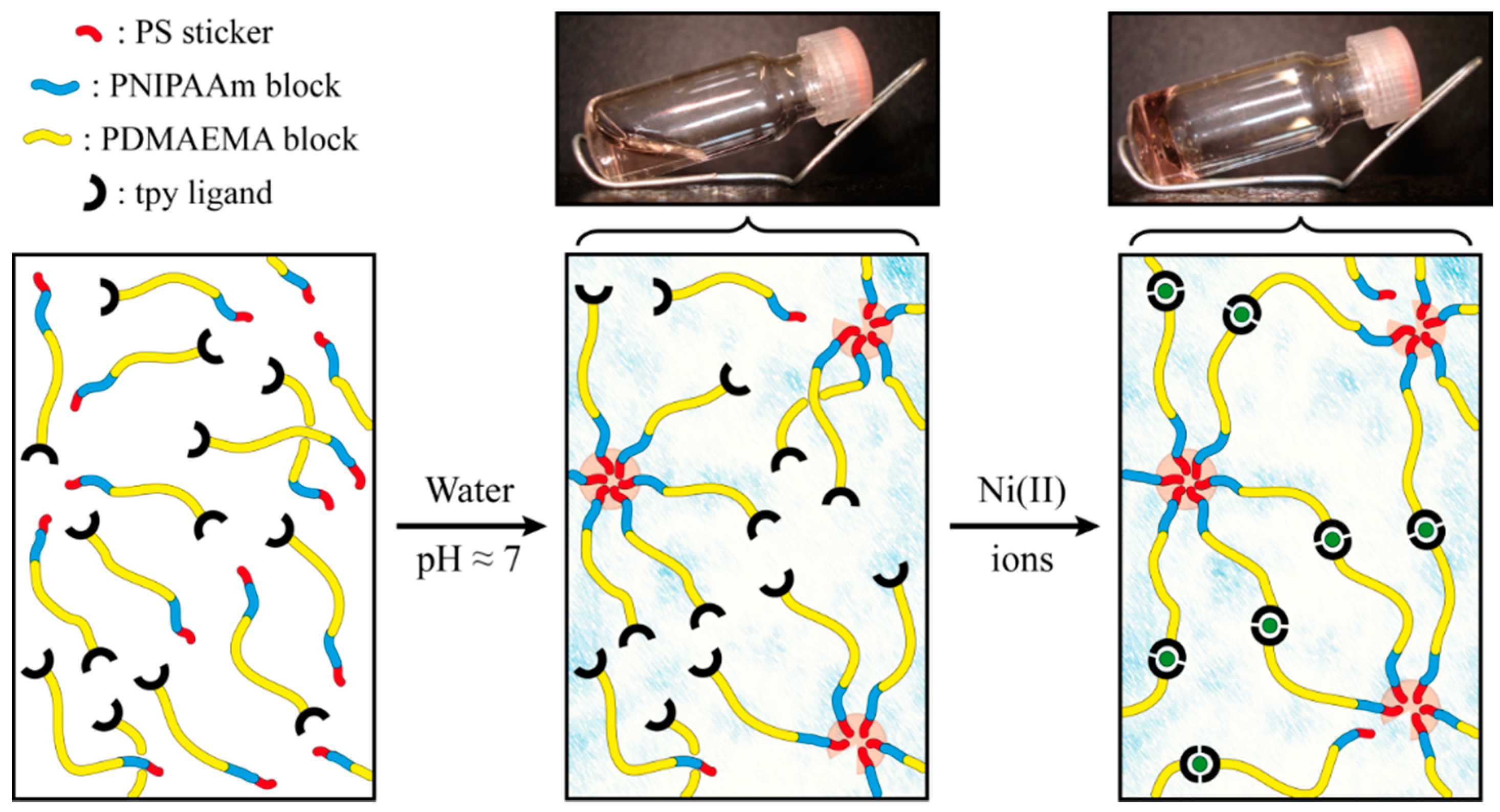

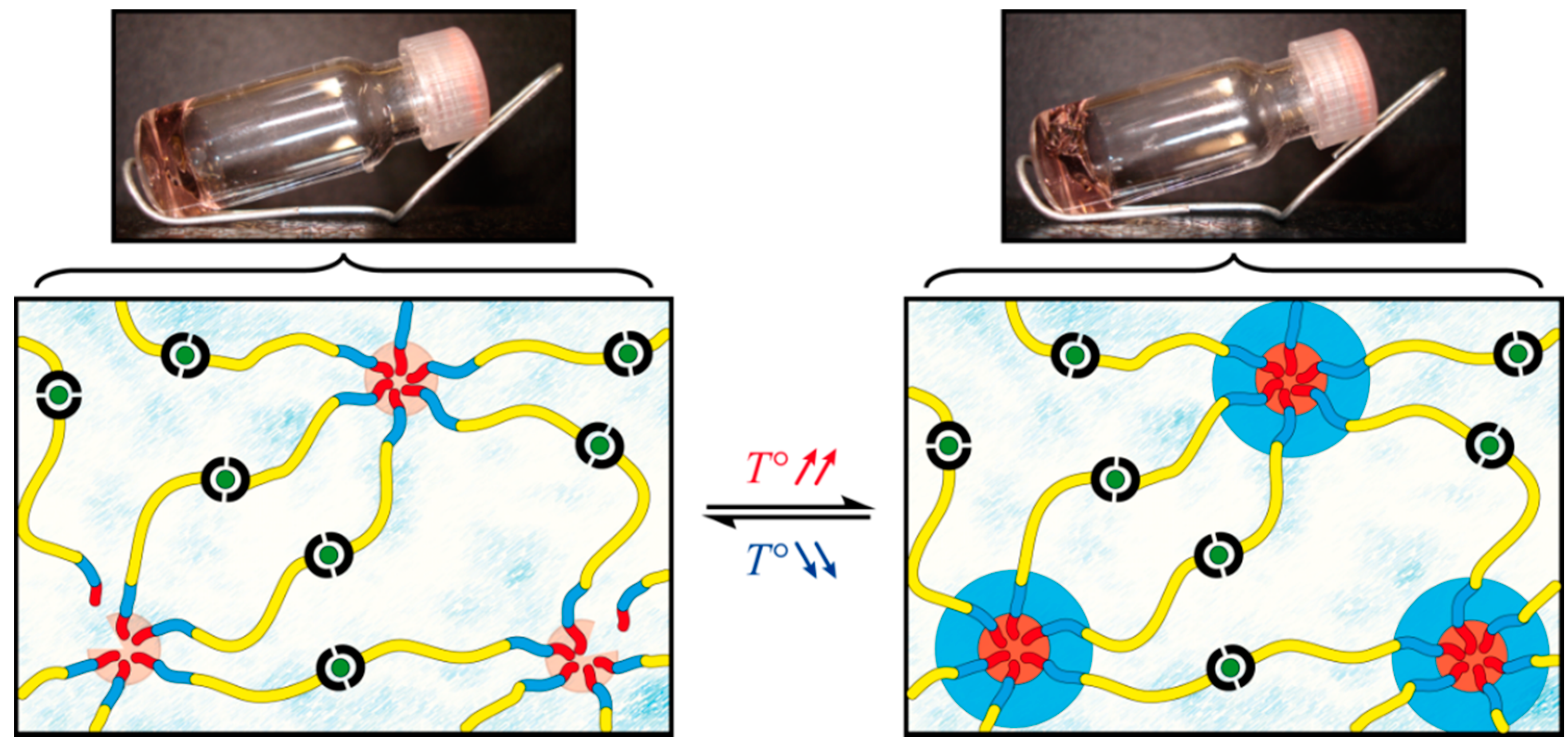
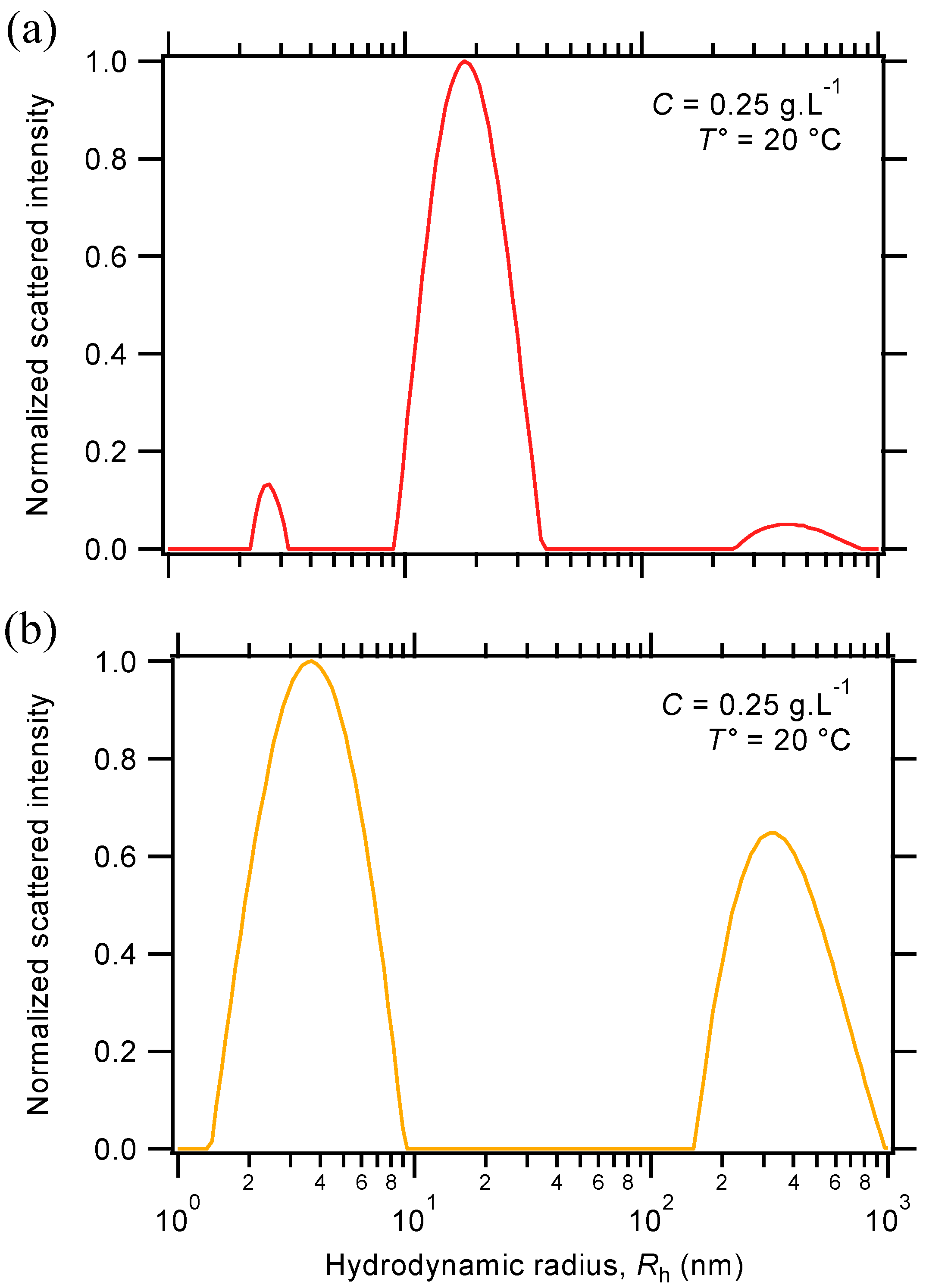
2.3. Characterization of the Rheological Response
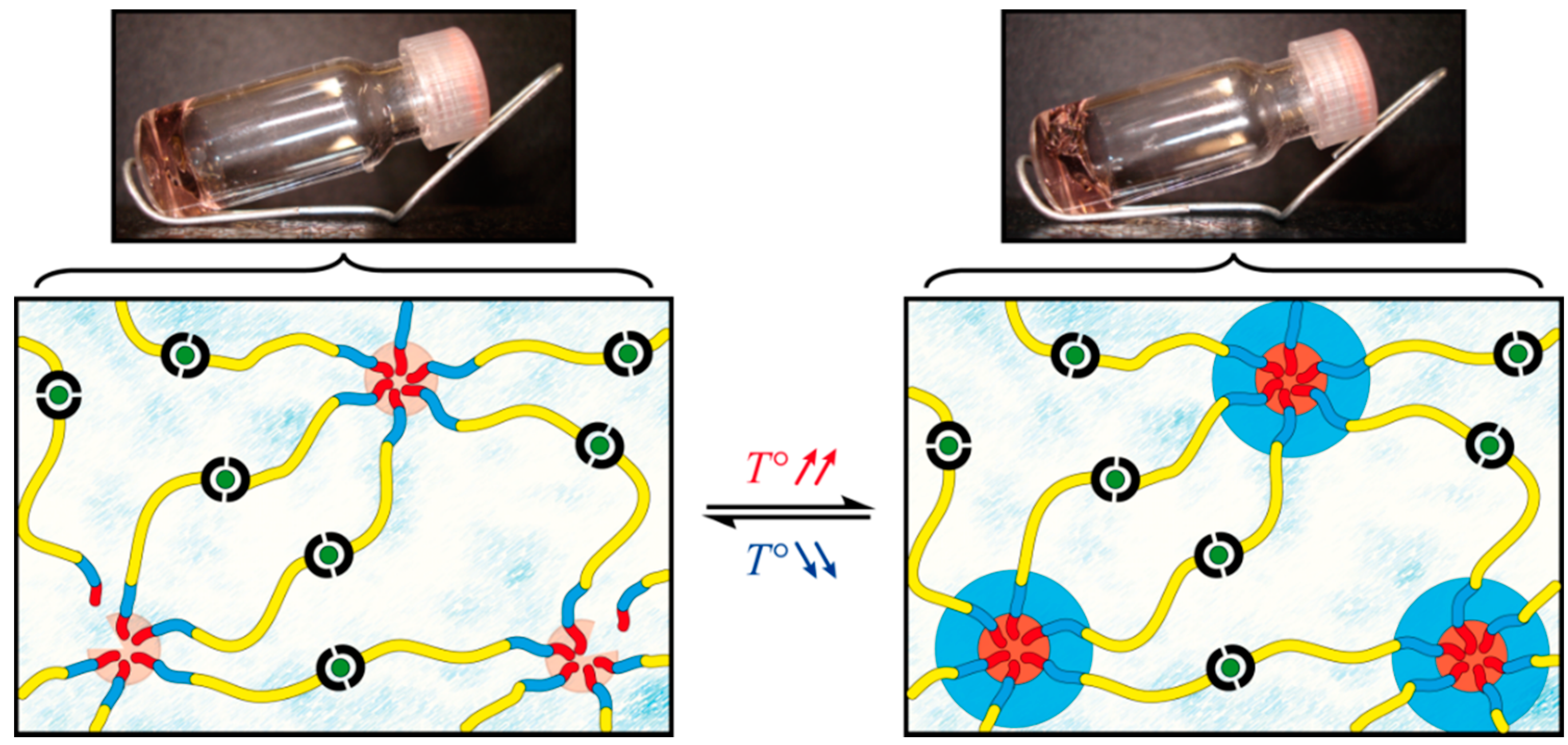
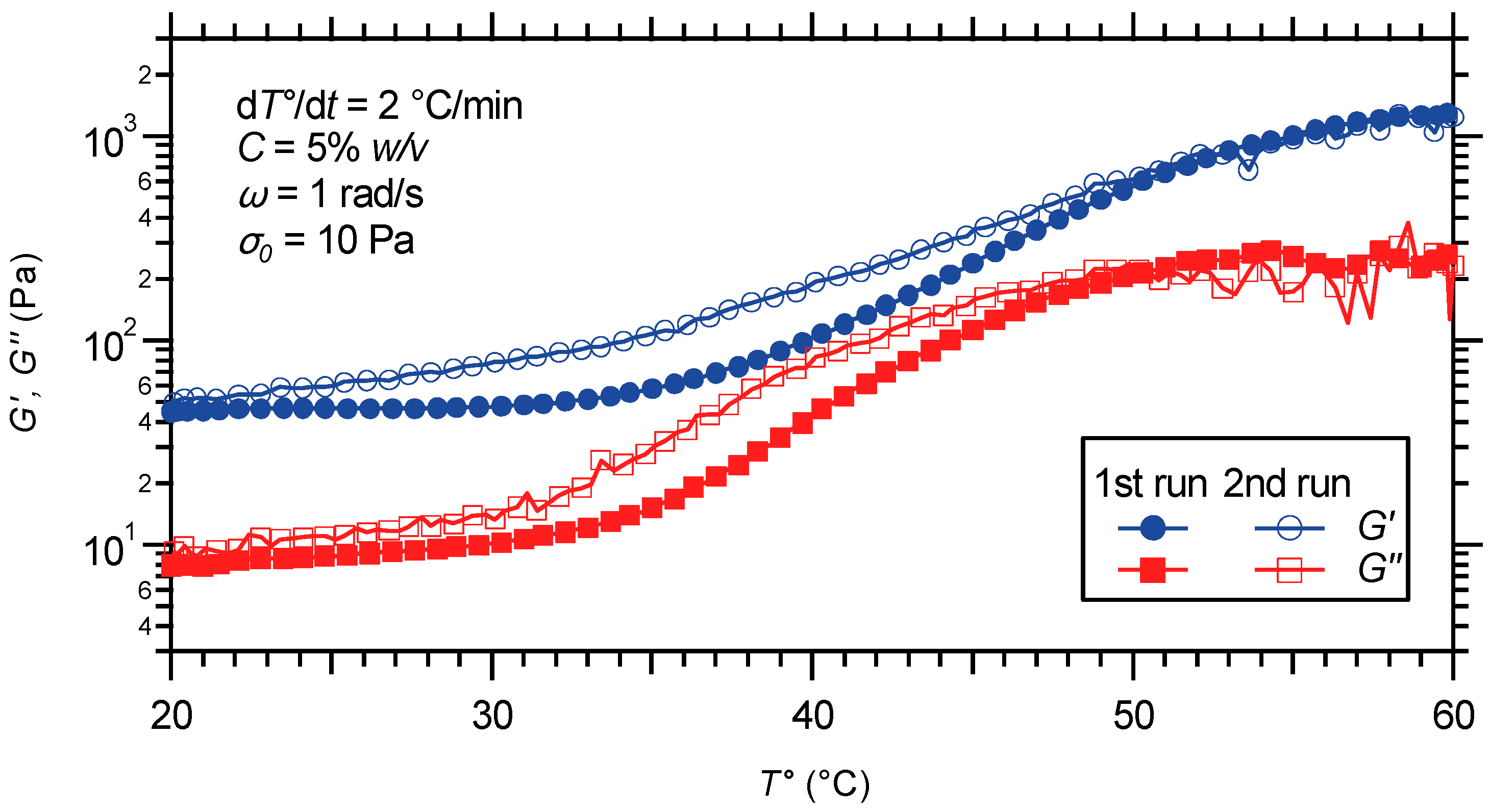
2.3.1. Oscillatory Temperature Sweep

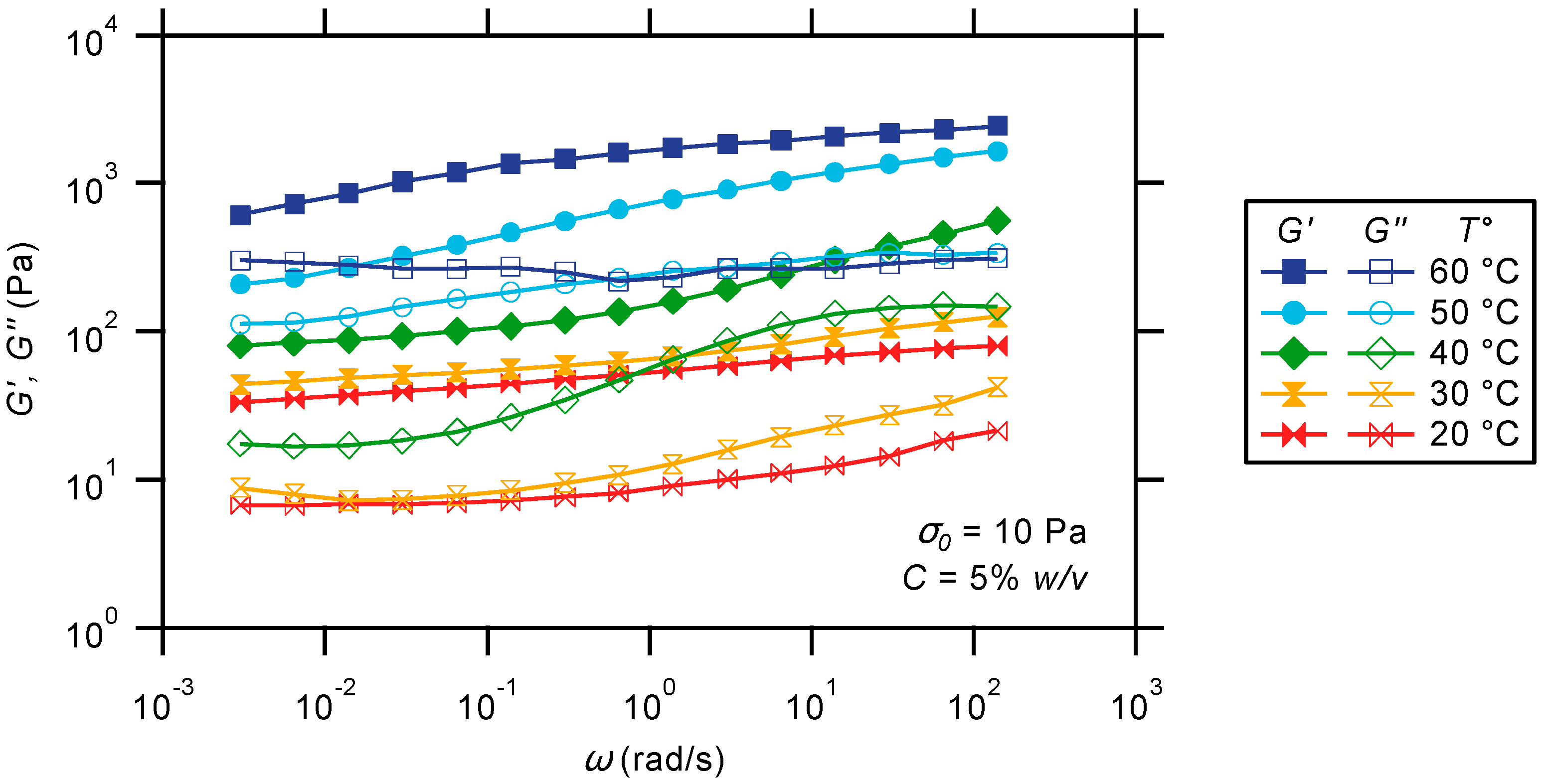
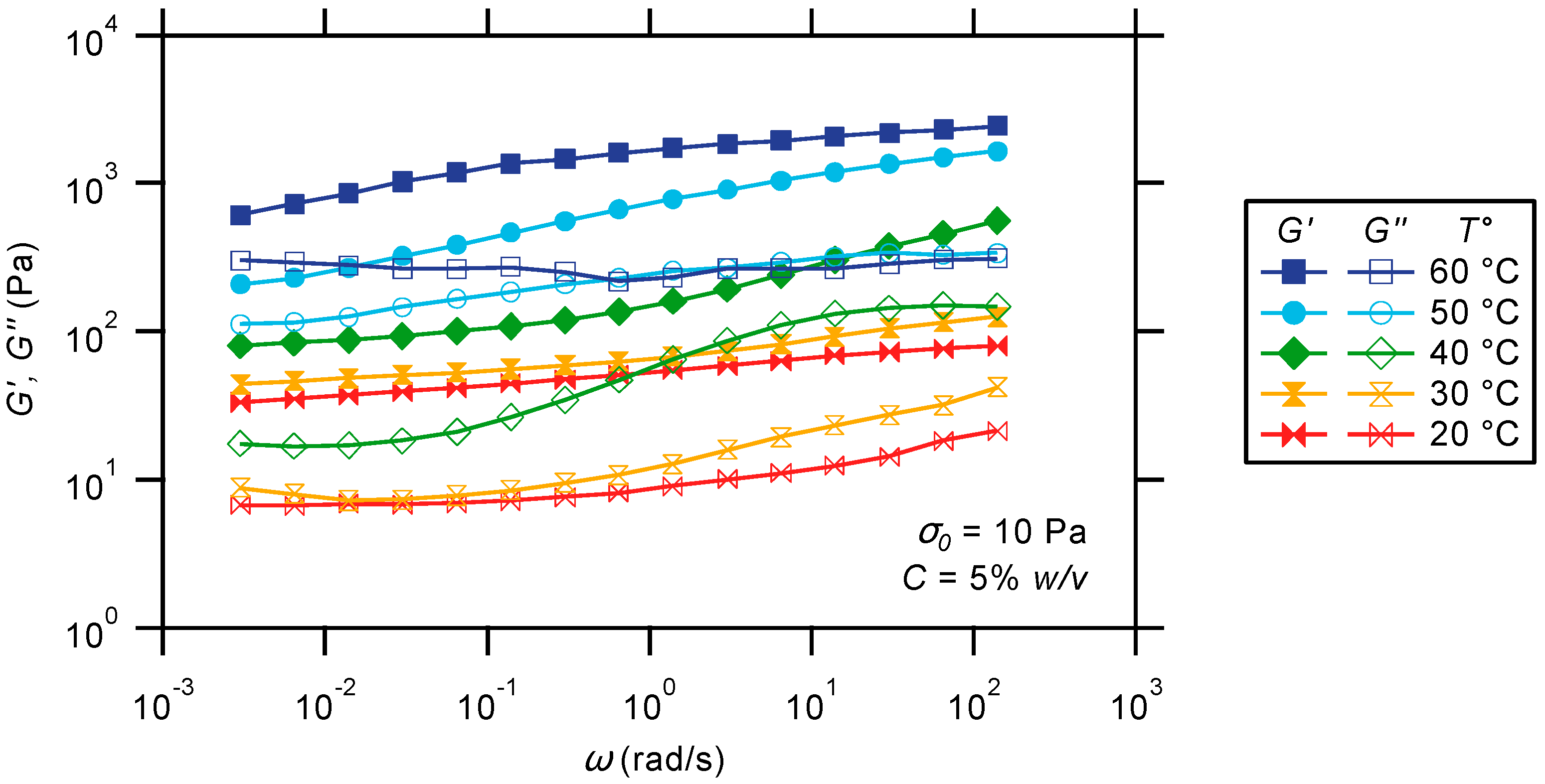
2.3.2. Oscillatory Frequency Sweep
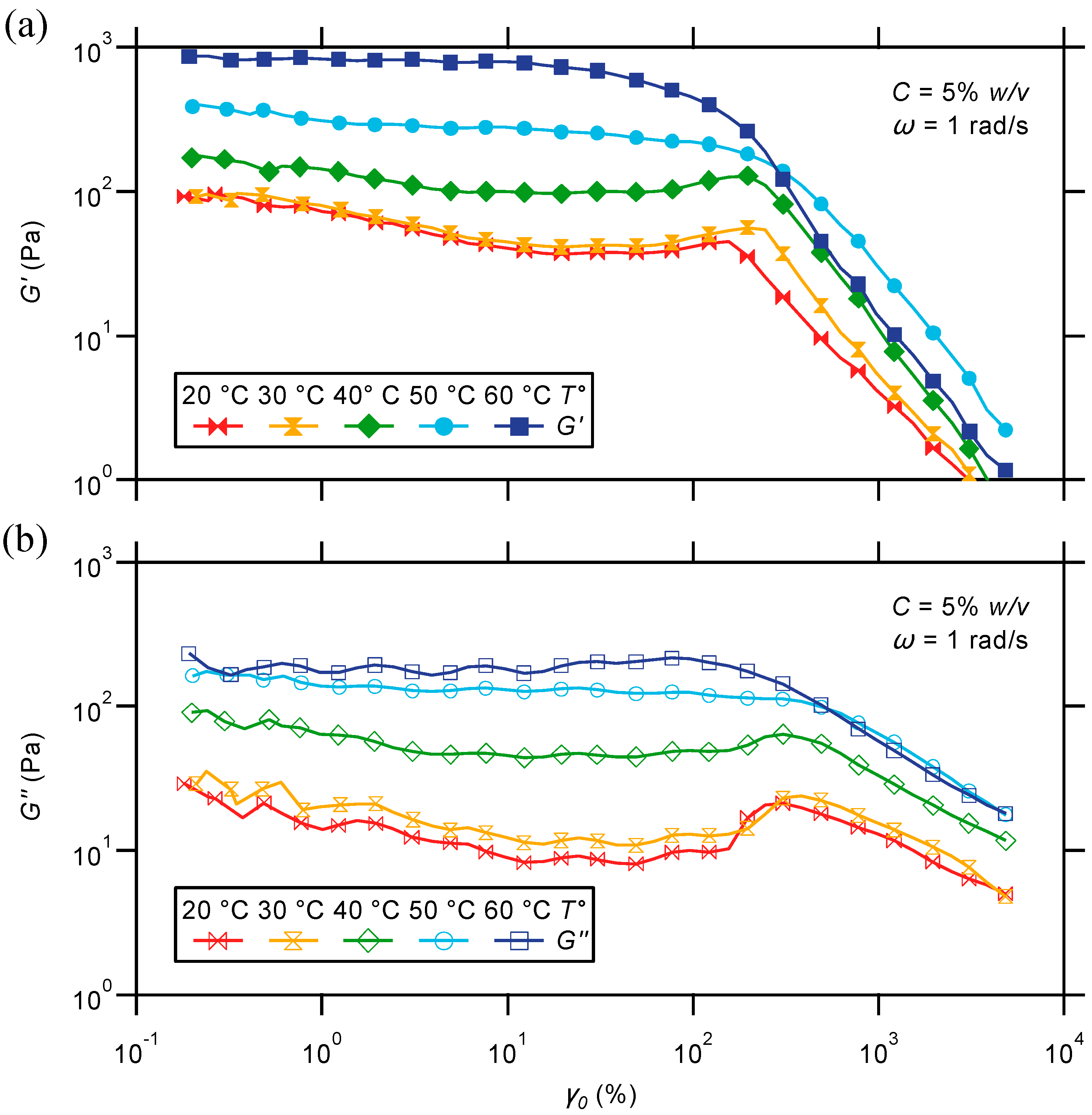
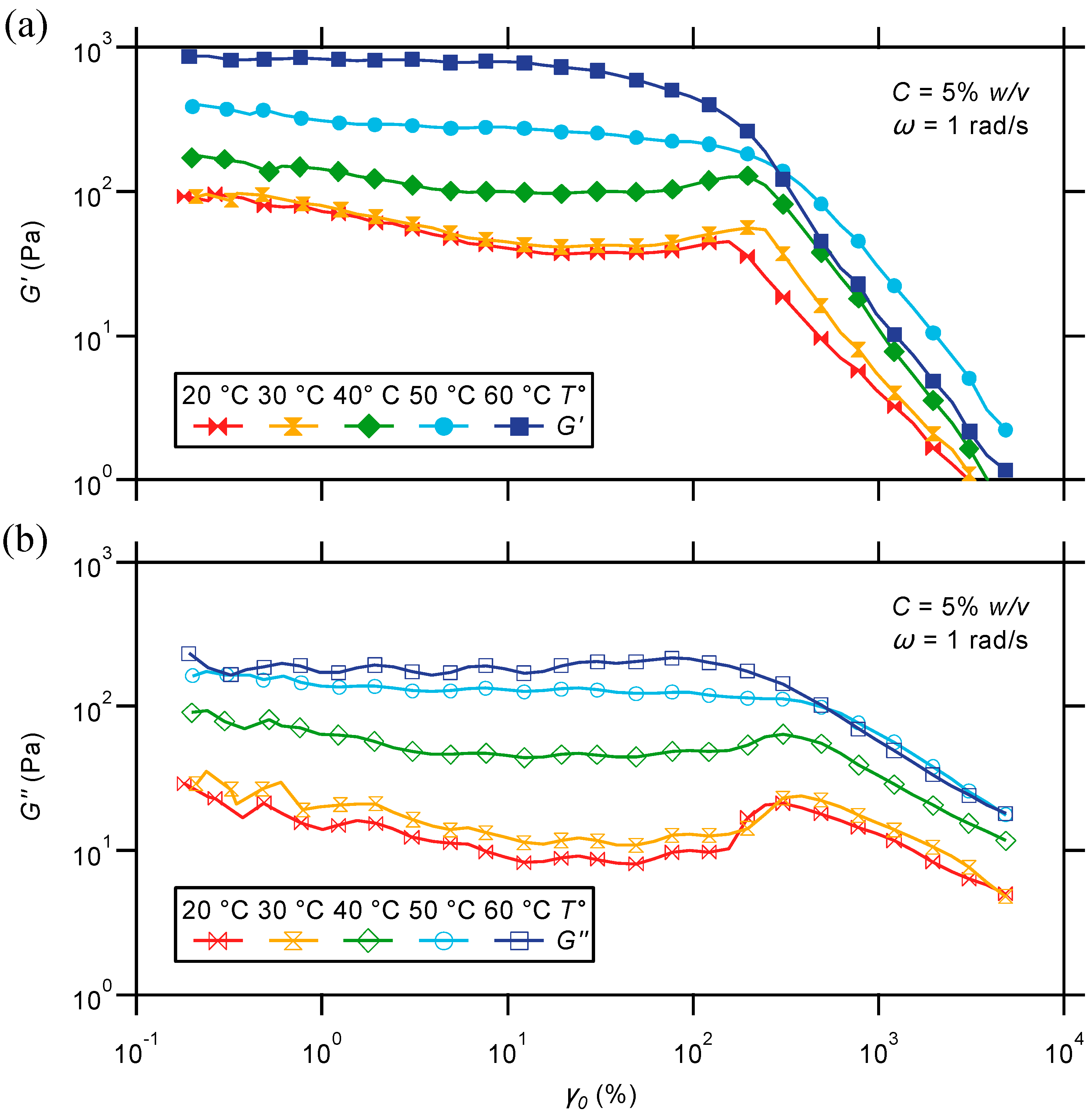
2.2.3. Oscillatory Strain Sweep
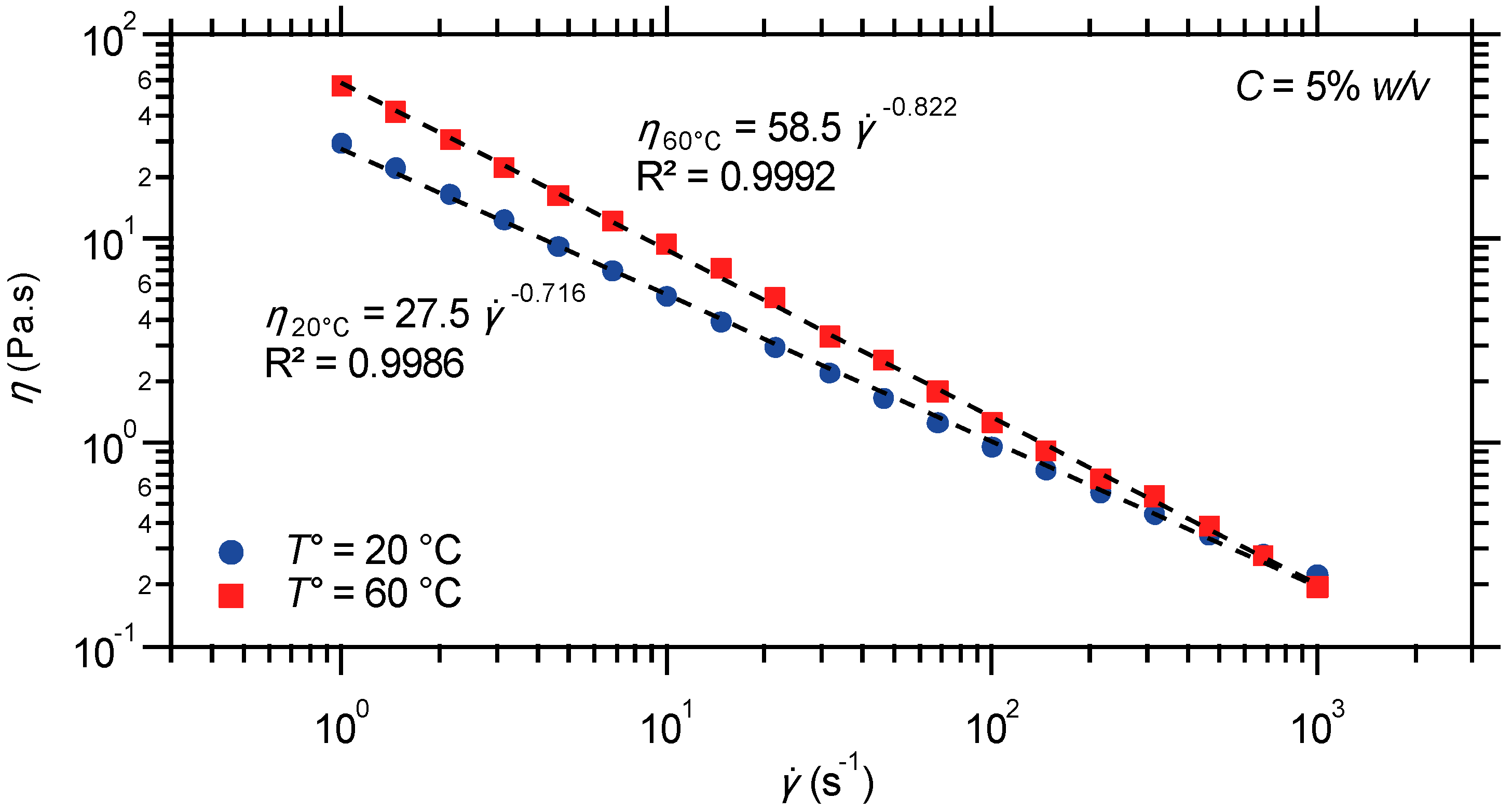
2.2.4. Steady Shear Viscometry

3. Conclusions
4. Experimental Section
4.1. Materials
4.2. Instrumentation
4.3. Synthesis of PDMAEMA103-tpy
4.4. Synthesis of PNIPAAm73-b-PDMAEMA103-tpy
4.5. Synthesis of PS12-b-PNIPAAm73-b-PDMAEMA103-tpy
4.6. Sample Preparation
4.7. Loading and Testing Protocol
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dai, L. Intelligent Macromolecules for Smart Devices; Springer: New York, NY, USA, 2003; p. 496. [Google Scholar]
- Ahn, S.K.; Kasi, R.M.; Kim, S.C.; Sharma, N.; Zhou, Y.X. Stimuli-responsive polymer gels. Soft Matter 2008, 4, 1151–1157. [Google Scholar] [CrossRef]
- Pasparakis, G.; Vamvakaki, M. Multiresponsive polymers: Nano-sized assemblies, stimuli-sensitive gels and smart surfaces. Polym. Chem. 2011, 2, 1234–1248. [Google Scholar] [CrossRef]
- Kopeček, J.; Yang, J.Y. Hydrogels as smart biomaterials. Polym. Int. 2007, 56, 1078–1098. [Google Scholar] [CrossRef]
- Osada, Y.; Gong, J. Stimuli-responsive polymer gels and their application to chemomechanical systems. Prog. Polym. Sci. 1993, 18, 187–226. [Google Scholar] [CrossRef]
- Roy, D.; Brooks, W.L.A.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.X.; Fraylich, M.; Saunders, B.R. Thermoresponsive copolymers: From fundamental studies to applications. Colloid Polym. Sci. 2009, 287, 627–643. [Google Scholar] [CrossRef]
- Beck, J.B.; Rowan, S.J. Multistimuli, multiresponsive metallo-supramolecular polymers. J. Am. Chem. Soc. 2003, 125, 13922–13923. [Google Scholar] [CrossRef] [PubMed]
- Noro, A.; Matsushita, Y.; Lodge, T.P. Thermoreversible supramacromolecular ion gels via hydrogen bonding. Macromolecules 2008, 41, 5839–5844. [Google Scholar] [CrossRef]
- Jochum, F.D.; Theato, P. Temperature- and light-responsive smart polymer materials. Chem. Soc. Rev. 2013, 42, 7468–7483. [Google Scholar] [CrossRef] [PubMed]
- Burnworth, M.; Tang, L.M.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Weder, C. Optically healable supramolecular polymers. Nature 2011, 472, 334–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qu, D.-H.; Wu, J.; Ma, X.; Wang, Q.; Tian, H. A dual-modality photoswitchable supramolecular polymer. Langmuir 2013, 29, 5345–5350. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Ravi, P.; Tam, K.C. pH-Responsive polymers: Synthesis, properties and applications. Soft Matter 2008, 4, 435–449. [Google Scholar] [CrossRef]
- Charbonneau, C.L.; Chassenieux, C.; Colombani, O.; Nicolai, T. Controlling the dynamics of self-assembled triblock copolymer networks via the pH. Macromolecules 2011, 44, 4487–4495. [Google Scholar] [CrossRef]
- Yao, X.; Chen, L.; Chen, X.; He, C.; Zhang, J.; Chen, X. Metallo-supramolecular nanogels for intracellular pH-responsive drug release. Macromol. Rapid. Commun. 2014, 35, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ten Brinke, G.; Ruokolainen, J.; Ikkala, O. Supramolecular materials based on hydrogen-bonded polymers. Adv. Polym. Sci. 2007, 207, 113–177. [Google Scholar]
- Cordier, P.; Tournilhac, F.; Soulie-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.F.; Antonietti, M. Ionic self-assembly: Facile synthesis of supramolecular materials. Adv. Mater. 2003, 15, 673–683. [Google Scholar] [CrossRef]
- Faul, C.F.J. Ionic self-assembly for functional hierarchical nanostructured materials. Acc. Chem. Res. 2014, 47, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P.H.J. About supramolecular assemblies of π-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Brassinne, J.; Fustin, C.-A.; Gohy, J.-F. Polymer gels constructed through metal-ligand coordination. J. Inorg. Organomet. Polym. Mater. 2013, 23, 24–40. [Google Scholar] [CrossRef]
- Goshe, A.J.; Crowley, J.D.; Bosnich, B. Supramolecular recognition: Use of cofacially disposed bis-terpyridyl square-planar complexes in self-assembly and molecular recognition. Helv. Chim. Acta 2001, 84, 2971–2985. [Google Scholar] [CrossRef]
- Goshe, A.J.; Steele, I.M.; Ceccarelli, C.; Rheingold, A.L.; Bosnich, B. Supramolecular recognition: On the kinetic lability of thermodynamically stable host-guest association complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 4823–4829. [Google Scholar] [CrossRef] [PubMed]
- Paulusse, J.M.J.; Huijbers, J.P.J.; Sijbesma, R.P. Quantification of ultrasound-induced chain scission in PdII–phosphine coordination polymers. Chem. Eur. J. 2006, 12, 4928–4934. [Google Scholar] [CrossRef] [PubMed]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. Chem. 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Plamper, F.A.; Ruppel, M.; Schmalz, A.; Borisov, O.; Ballauff, M.; Muller, A.H.E. Tuning the thermoresponsive properties of weak polyelectrolytes: Aqueous solutions of star-shaped and linear poly(N,N-dimethylaminoethyl methacrylate). Macromolecules 2007, 40, 8361–8366. [Google Scholar] [CrossRef]
- Li, F.-M.; Chen, S.-J.; Du, F.-S.; Wu, Z.-Q.; Li, Z.-C. Stimuli-responsive behavior of N,N-dimethylaminoethyl methacrylate polymers and their hydrogels. ACS Symp. Ser. 1999, 726, 266–276. [Google Scholar]
- Liu, Q.; Yu, Z.; Ni, P. Micellization and applications of narrow-distribution poly[2-(dimethylamino)ethyl methacrylate]. Colloid Polym. Sci. 2004, 282, 387–393. [Google Scholar] [CrossRef]
- Butun, V.; Armes, S.; Billingham, N. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. [Google Scholar] [CrossRef]
- He, Y.; Lodge, T.P. Thermoreversible ion gels with tunable melting temperatures from triblock and pentablock copolymers. Macromolecules 2008, 41, 167–174. [Google Scholar] [CrossRef]
- Sugihara, S.; Kanaoka, S.; Aoshima, S. Stimuli-responsive ABC triblock copolymers by sequential living cationic copolymerization: Multistage self-assemblies through micellization to open association. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 2601–2611. [Google Scholar] [CrossRef]
- Dyakonova, M.A.; Stavrouli, N.; Popescu, M.T.; Kyriakos, K.; Grillo, I.; Philipp, M.; Jaksch, S.; Tsitsilianis, C.; Papadakis, C.M. Physical hydrogels via charge driven self-organization of a triblock polyampholyte—Rheological and structural investigations. Macromolecules 2014, 47, 7561–7572. [Google Scholar] [CrossRef]
- Piogé, S.; Fustin, C.-A.; Gohy, J.-F. Temperature-responsive aqueous micelles from terpyridine end-capped poly(N-isopropylacrylamide)-block-polystyrene diblock copolymers. Macromol. Rapid. Commun. 2012, 33, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Brassinne, J.; Poggi, E.; Fustin, C.-A.; Gohy, J.-F. Synthesis and self-assembly of terpyridine end-capped poly(N-isopropylacrylamide)-block-poly(2-(dimethylamino)ethyl methacrylate) diblock copolymers. Macromol. Rapid Commun. 2015, 36, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Mugemana, C.; Guillet, P.; Fustin, C.-A.; Gohy, J.-F. Metallo-supramolecular block copolymer micelles: Recent achievements. Soft Matter 2011, 7, 3673–3678. [Google Scholar] [CrossRef]
- Brassinne, J.; Stevens, A.M.; van Ruymbeke, E.; Gohy, J.-F.; Fustin, C.-A. Hydrogels with dual relaxation and two-step gel–sol transition from heterotelechelic polymers. Macromolecules 2013, 46, 9134–9143. [Google Scholar] [CrossRef]
- Jochum, F.D.; Brassinne, J.; Fustin, C.-A.; Gohy, J.-F. Metallo-supramolecular hydrogels based on copolymers bearing terpyridine side-chain ligands. Soft Matter 2013, 9, 2314–2320. [Google Scholar] [CrossRef]
- Mugemana, C.; Joset, A.; Guillet, P.; Appavou, M.-S.; de Souza, N.; Fustin, C.-A.; Leyh, B.; Gohy, J.-F. Structure of metallo-supramolecular micellar gels. Macromol. Chem. Phys. 2013, 214, 1699–1709. [Google Scholar] [CrossRef]
- Guillet, P.; Mugemana, C.; Stadler, F.J.; Schubert, U.S.; Fustin, C.-A.; Bailly, C.; Gohy, J.-F. Connecting micelles by metallo-supramolecular interactions: Towards stimuli responsive hierarchical materials. Soft Matter 2009, 5, 3409–3411. [Google Scholar] [CrossRef]
- Brassinne, J.; Gohy, J.-F.; Fustin, C.-A. Controlling the cross-linking density of supramolecular hydrogels formed by heterotelechelic associating copolymers. Macromolecules 2014, 47, 4514–4524. [Google Scholar] [CrossRef]
- Brassinne, J.; Mugemana, C.; Guillet, P.; Bertrand, O.; Auhl, D.; Bailly, C.; Fustin, C.-A.; Gohy, J.-F. Tuning micellar morphology and rheological behaviour of metallo-supramolecular micellar gels. Soft Matter 2012, 8, 4499–4506. [Google Scholar] [CrossRef]
- Brassinne, J.; Fustin, C.-A.; Gohy, J.-F. Thermo-responsive metallo-supramolecular gels based on terpyridine end-functionalized amphiphilic diblock copolymers. Mater. Res. Soc. Symp. Proc. 2013, 1499. [Google Scholar] [CrossRef]
- Brassinne, J.; Bourgeois, J.-P.; Fustin, C.-A.; Gohy, J.-F. Thermo-responsive properties of metallo-supramolecular block copolymer micellar hydrogels. Soft Matter 2014, 10, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, F.; Zheng, B.; Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 2012, 41, 6042–6065. [Google Scholar] [CrossRef] [PubMed]
- Holyer, R.H.; Hubbard, C.D.; Kettle, S.F.A.; Wilkins, R.G. The kinetics of replacement reactions of complexes of the transition metals with 2,2',2"-terpyridine. Inorg. Chem. 1966, 5, 622–625. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R. Exchange studies of certain chelate compounds of the transitional metals. Part VIII. 2,2',2"-terpyridine complexes. J. Chem. Soc. 1962, 341–350. [Google Scholar] [CrossRef]
- Schubert, U.; Hofmeier, H.; Newkome, G.R. Modern Terpyridine Chemistry; Wiley-VCH: Weinheim, Germany, 2006; p. 229. [Google Scholar]
- Schubert, U.; Winter, A.; Newkome, G.R. Terpyridine-Based Materials: For Catalytic, Optoelectronic and Life Science Applications; Wiley-VCH: Weinheim, Germany, 2011; p. 522. [Google Scholar]
- Aamer, K.A.; Tew, G.N. Supramolecular polymers containing terpyridine-metal complexes in the side chain. Macromolecules 2007, 40, 2737–2744. [Google Scholar] [CrossRef]
- Ott, C.; Ulbricht, C.; Hoogenboom, R.; Schubert, U.S. Metallo-supramolecular materials based on amine-grafting onto polypentafluorostyrene. Macromol. Rapid. Commun. 2012, 33, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Hackelbusch, S.; Rossow, T.; van Assenbergh, P.; Seiffert, S. Chain dynamics in supramolecular polymer networks. Macromolecules 2013, 46, 6273–6286. [Google Scholar] [CrossRef]
- Calzia, K.J.; Tew, G.N. Methacrylate polymers containing metal binding ligands for use in supramolecular materials: Random copolymers containing terpyridines. Macromolecules 2002, 35, 6090–6093. [Google Scholar] [CrossRef]
- El-ghayoury, A.; Hofmeier, H.; de Ruiter, B.; Schubert, U.S. Combining covalent and noncovalent cross-linking: A novel terpolymer for two-step curing applications. Macromolecules 2003, 36, 3955–3959. [Google Scholar] [CrossRef]
- Hofmeier, H.; Schubert, U.S. Supramolecular branching and crosslinking of terpyridine-modified copolymers: Complexation and decomplexation studies in diluted solution. Macromol. Chem. Phys. 2003, 204, 1391–1397. [Google Scholar] [CrossRef]
- Brassinne, J.; Jochum, F.; Fustin, C.-A.; Gohy, J.-F. Revealing the supramolecular nature of side-chain terpyridine-functionalized polymer networks. Int. J. Mol. Sci. 2015, 16, 990–1007. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Lohmeijer, B.G.G.; Wouters, D.; Schubert, U.S. Terpyridine-terminated homo and diblock copolymer LEGO units by nitroxide-mediated radical polymerization. Macromol. Chem. Phys. 2006, 207, 1439–1449. [Google Scholar] [CrossRef]
- Lohmeijer, B.G.G.; Schubert, U.S. The LEGO toolbox: Supramolecular building blocks by nitroxide-mediated controlled radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6331–6344. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, C.; Zhang, J.; Zhang, M.; Liu, Y.; Wang, X.; Fu, G. Controlled polymerization of 2-(diethylamino)ethyl methacrylate and its block copolymer with N-isopropylacrylamide by RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3294–3305. [Google Scholar] [CrossRef]
- Nuopponen, M.; Ojala, J.; Tenhu, H. Aggregation behaviour of well defined amphiphilic diblock copolymers with poly(N-isopropylacrylamide) and hydrophobic blocks. Polymer 2004, 45, 3643–3650. [Google Scholar] [CrossRef]
- Rossow, T.; Habicht, A.; Seiffert, S. Relaxation and dynamics in transient polymer model networks. Macromolecules 2014, 47, 6473–6482. [Google Scholar] [CrossRef]
- Rossow, T.; Seiffert, S. Supramolecular polymer gels with potential model-network structure. Polym. Chem. 2014, 5, 3018–3029. [Google Scholar] [CrossRef]
- Chiper, M.; Meier, M.A.R.; Kranenburg, J.M.; Schubert, U.S. New insights into nickel(II), iron(II), and cobalt(II) bis-complex-based metallo-supramolecular polymers. Macromol. Chem. Phys. 2007, 208, 679–689. [Google Scholar] [CrossRef]
- Vermonden, T.; van Steenbergen, M.J.; Besseling, N.A.M.; Marcelis, A.T.M.; Hennink, W.E.; Sudhölter, E.J.R.; Stuart, M.A.C. Linear rheology of water-soluble reversible neodymium(III) coordination polymers. J. Am. Chem. Soc. 2004, 126, 15802–15808. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, T.; Colombani, O.; Chassenieux, C. Dynamic polymeric micelles versus frozen nanoparticles formed by block copolymers. Soft Matter 2010, 6, 3111–3118. [Google Scholar] [CrossRef]
- Van Stam, J.; Creutz, S.; de Schryver, F.C.; Jérôme, R. Tuning of the exchange dynamics of unimers between block copolymer micelles with temperature, cosolvents, and cosurfactants. Macromolecules 2000, 33, 6388–6395. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, R.; Quirk, R.P.; Mattice, W.L. Detection of the rate of exchange of chains between micelles formed by diblock copolymers in aqueous solution. Polym. Bull. 1992, 28, 333–338. [Google Scholar] [CrossRef]
- Jacquin, M.; Muller, P.; Talingting-Pabalan, R.; Cottet, H.; Berret, J.; Futterer, T.; Theodoly, O. Chemical analysis and aqueous solution properties of charged amphiphilic block copolymers PBA-b-PAA synthesized by MADIX®. J. Colloid Interface Sci. 2007, 316, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Farina, R.D.; Hogg, R.; Wilkins, R.G. Rate-pH profile for the dissociation of iron(II)-and cobalt(II)-2,2',2"-terpyridine complexes. Inorg. Chem. 1968, 7, 170–172. [Google Scholar] [CrossRef]
- Mugemana, C.; Guillet, P.; Hoeppener, S.; Schubert, U.S.; Fustin, C.-A.; Gohy, J.-F. Metallo-supramolecular diblock copolymers based on heteroleptic cobalt(III) and nickel(II) bis-terpyridine complexes. Chem. Commun. 2010, 46, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Mugemana, C.; Gohy, J.-F.; Fustin, C.-A. Functionalized nanoporous thin films from metallo-supramolecular diblock copolymers. Langmuir 2012, 28, 3018–3023. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L.A.; Lane, J.A.; Derry, M.J.; Mykhaylyk, O.O.; Armes, S.P. Thermo-responsive diblock copolymer worm gels in non-polar solvents. J. Am. Chem. Soc. 2014, 136, 5790–5798. [Google Scholar] [CrossRef] [PubMed]
- Dreiss, C.A. Wormlike micelles: Where do we stand? Recent developments, linear rheology and scattering techniques. Soft Matter 2007, 3, 956–970. [Google Scholar] [CrossRef]
- Clausen, T.; Vinson, P.; Minter, J.; Davis, H.; Talmon, Y.; Miller, W. Viscoelastic micellar solutions: Microscopy and rheology. J. Phys. Chem. 1992, 96, 474–484. [Google Scholar] [CrossRef]
- Tam, K.; Jenkins, R.; Winnik, M.; Bassett, D. A structural model of hydrophobically modified urethane-ethoxylate (HEUR) associative polymers in shear flows. Macromolecules 1998, 31, 4149–4159. [Google Scholar] [CrossRef]
- Howe, A.J.; Howe, A.M.; Routh, A.F. The viscosity of dilute poly(N-isopropylacrylamide) dispersions. J. Colloid Interface Sci. 2011, 357, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.R. Handbook of Surfactants; Springer: New York, NY, USA, 1991; p. 227. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brassinne, J.; Zhuge, F.; Fustin, C.-A.; Gohy, J.-F. Precise Control over the Rheological Behavior of Associating Stimuli-Responsive Block Copolymer Gels. Gels 2015, 1, 235-255. https://doi.org/10.3390/gels1020235
Brassinne J, Zhuge F, Fustin C-A, Gohy J-F. Precise Control over the Rheological Behavior of Associating Stimuli-Responsive Block Copolymer Gels. Gels. 2015; 1(2):235-255. https://doi.org/10.3390/gels1020235
Chicago/Turabian StyleBrassinne, Jérémy, Flanco Zhuge, Charles-André Fustin, and Jean-François Gohy. 2015. "Precise Control over the Rheological Behavior of Associating Stimuli-Responsive Block Copolymer Gels" Gels 1, no. 2: 235-255. https://doi.org/10.3390/gels1020235