
Development and Function of the Cardiac Conduction System in Health and Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
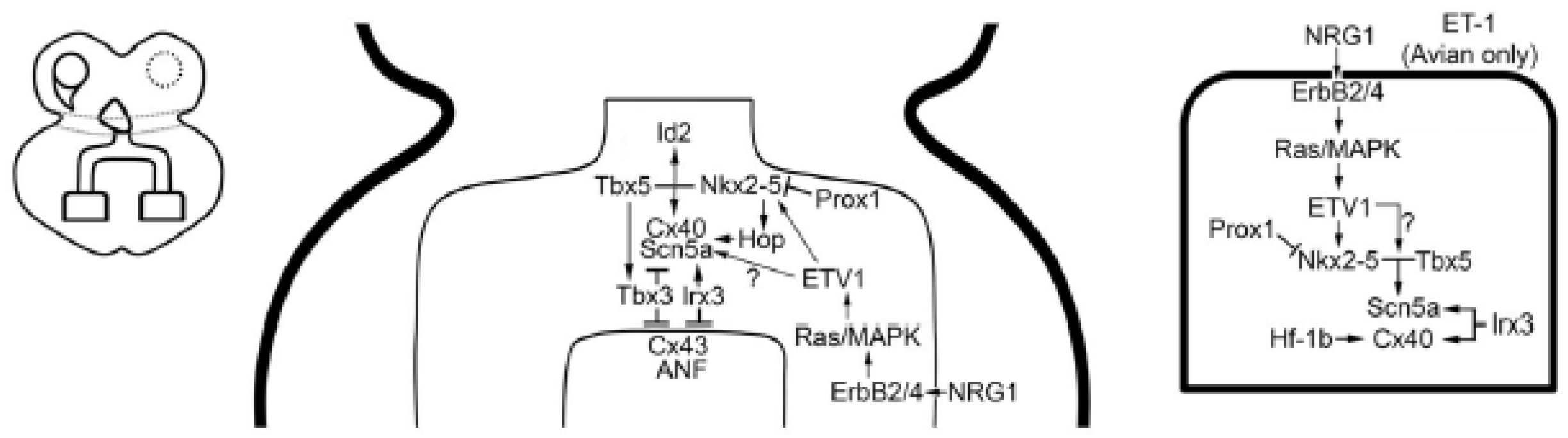
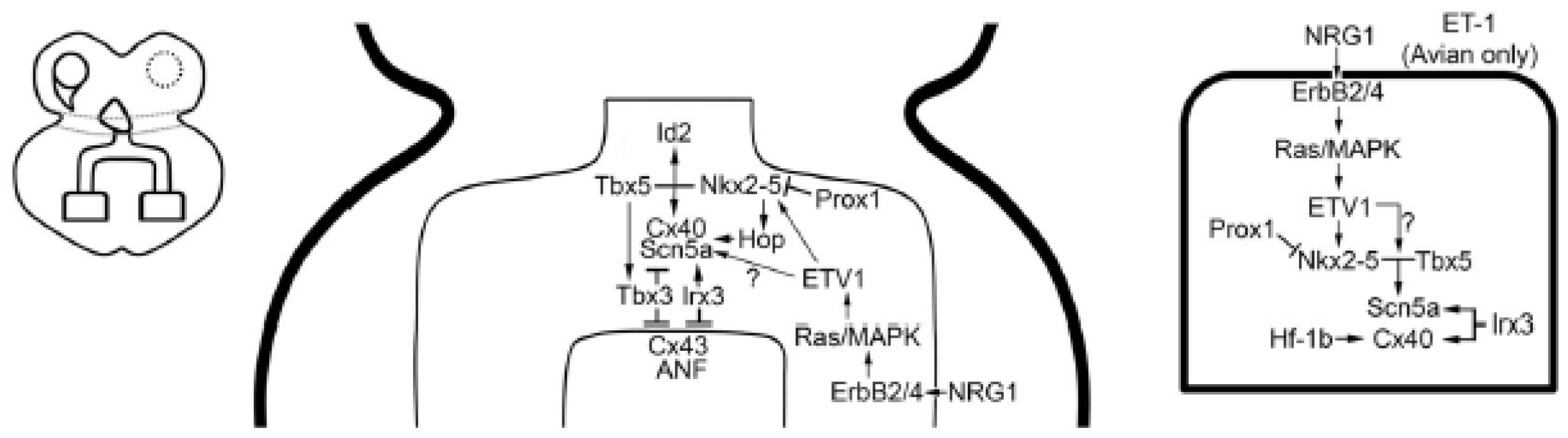{kind=link}
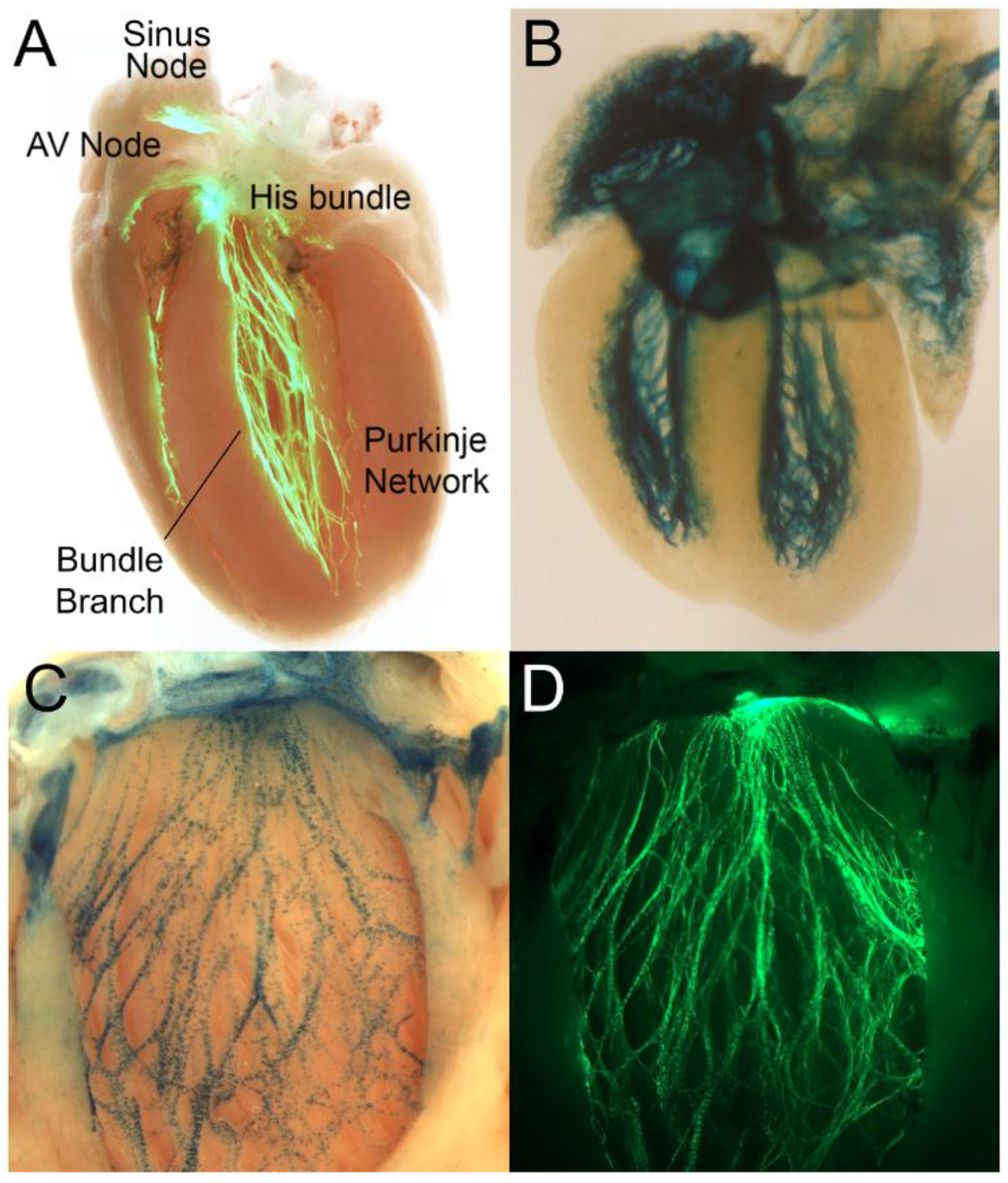
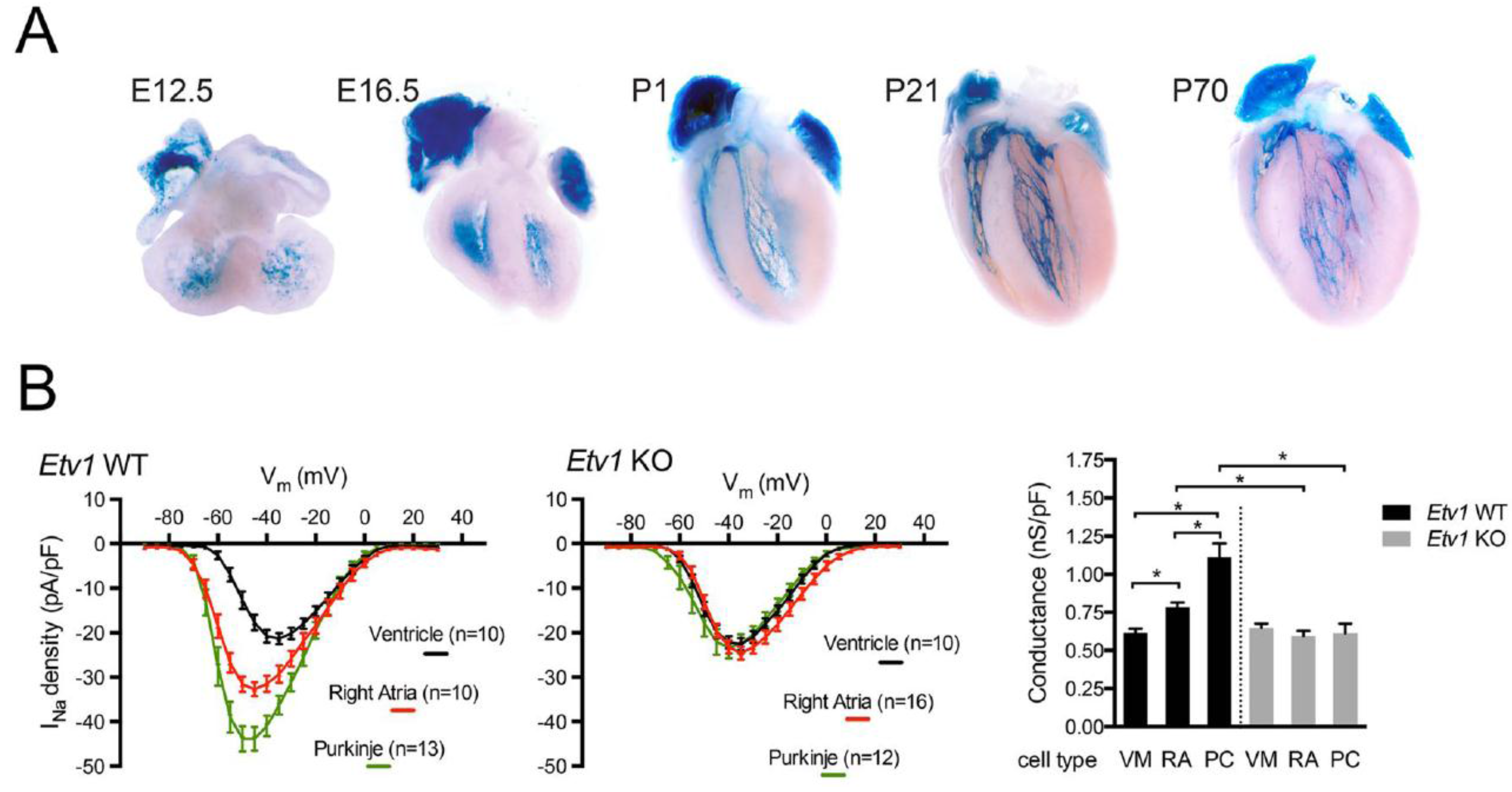{kind=link}
Abstract
:1. Introduction
2. The Sinus Node
3. The Atrioventricular Canal and Node
4. The Ventricular Conduction System
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shekhar, A.; Lin, X.; Liu, F.Y.; Zhang, J.; Mo, H.; Bastarache, L.; Denny, J.C.; Cox, N.J.; Delmar, M.; Roden, D.M.; et al. Transcription factor ETV1 is essential for rapid conduction in the heart. J. Clin. Investig. 2016, 126, 4444–4459. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Tompkins, R.O.; Liu, F.; Zhang, J.; Phoon, C.K.; Zavadil, J.; Fishman, G.I. Pocket proteins critically regulate cell cycle exit of the trabecular myocardium and the ventricular conduction system. Biol. Open 2013, 2, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Paff, G.H.; Boucek, R.J.; Harrell, T.C. Observations on the development of the electrocardiogram. Anat. Rec. 1968, 160, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Aanhaanen, W.T.; Mommersteeg, M.T.; Norden, J.; Wakker, V.; de Gier-de Vries, C.; Anderson, R.H.; Kispert, A.; Moorman, A.F.M.; Christoffels, V.M. Developmental origin, growth, and three-dimensional architecture of the atrioventricular conduction axis of the mouse heart. Circ. Res. 2010, 107, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Difrancesco, D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Wang, J.; Song, Y.; Yu, D.; Sun, C.; Liu, C.; Chen, F.; Zhang, Y.; Wang, F.; Harvey, R.P.; et al. A common Shox2-Nkx2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinus node. Development 2015, 142, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Lewis, R.A.; Liu, H.; Sun, C.; Chen, C.; Jiao, K.; Chen, Y. Ectopic expression of Nkx2.5 suppresses the formation of the sinus node in mice. Dev. Biol. 2011, 356, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Wiese, C.; Grieskamp, T.; Airik, R.; Mommersteeg, M.T.; Gardiwal, A.; de Gier-de Vries, C.; Schuster-Gossler, k.; Moorman, A.F.M.; Kispert, A.; Christoffels, V.M. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 2009, 104, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.P.; Vicente-Steijn, R.; Harryvan, T.J.; Kosmidis, G.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Schalij, M.J.; DeRuiter, M.C.; Jongbloed, M.R.M. The sinus venosus myocardium contributes to the atrioventricular canal: Potential role during atrioventricular node development? J. Cell. Mol. Med. 2015, 19, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell. 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Ammirabile, G.; Tessari, A.; Pignataro, V.; Szumska, D.; Sutera Sardo, F.; Benes, J., Jr.; Balistreri, M.; Bhattacharya, S.; Sedmera, D.; Campione, M. Pitx2 confers left morphological, molecular, and functional identity to the sinus venosus myocardium. Cardiovasc. Res. 2012, 93, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Mommersteeg, M.T.; Hoogaars, W.M.; Prall, O.W.; de Gier-de Vries, C.; Wiese, C.; Clout, D.E.; Papaioannou, Y.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.M.; et al. Molecular pathway for the localized formation of the sinus node. Circ. Res. 2007, 100, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Fishman, G.I. Cardiac Electrophysiology: From Cell to Bedside; Zipes, D.P., Jalife, J., Eds.; Elsevier: Philadelphia, PA, USA, 2014; pp. 287–296. [Google Scholar]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E; et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef]
- Moskowitz, I.P.; Pizard, A.; Patel, V.V.; Bruneau, B.G.; Kim, J.B.; Kupershmidt, S.; Roden, D.; Berul, C.I.; Seidman, C.E.; Seidman, J.G. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 2004, 131, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.D.; Zhu, Y.; Vahora, I.; Nieman, B.; Koshiba-Takeuchi, K.; Davidson, L.; Pizard, A.; Seidmanf, J.G.; Seidman, C.E.; Chend, X.J.; et al. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev. Biol. 2006, 297, 566–586. [Google Scholar] [CrossRef] [PubMed]
- Puskaric, S.; Schmitteckert, S.; Mori, A.D.; Glaser, A.; Schneider, K.U.; Bruneau, B.G.; Blaschke, R.J.; Steinbeisser, H.; Rappold, G. Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum. Mol. Genet. 2010, 19, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.M.; Tessari, A.; Moorman, A.F.; de Boer, P.A.; Hagoort, J.; Soufan, A.T.; Campione, M.; Christoffels, V.M. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc. Res. 2004, 62, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.U.; Carter, K.L.; Thomas, K.R.; Burr, R.M.; Bakker, M.L.; Coetzee, W.A.; Tristani-Firouzi, M.; Bamshad, M.J.; Christoffels, V.M.; Moona, A.M. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, E154–E163. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.M.; Engel, A.; Brons, J.F.; Verkerk, A.O.; de Lange, F.J.; Wong, L.Y.; Bakker, M.L.; Clout, D.E.; Wakker, V.; Barnett, P.; et al. Tbx3 controls the sinus node gene program and imposes pacemaker function on the atria. Genes Dev. 2007, 21, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Peng, S.; Yang, J.; Tu, Z.; Cai, X.; Cai, C.L.; Wang, Z.; Zhao, Y. Baf250a orchestrates an epigenetic pathway to repress the Nkx2.5-directed contractile cardiomyocyte program in the sinus node. Cell. Res. 2014, 24, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.; Liang, W.; Marban, E.; Cho, H.C. Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat. Biotechnol. 2013, 31, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, R.J.; Hahurij, N.D.; Kuijper, S.; Just, S.; Wisse, L.J.; Deissler, K.; Maxelon, T.; Anastassiadis, K.; Spitzer, J.; Hardt, S.E.; et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinus and pacemaking development. Circulation 2007, 115, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Lewis, R.A.; Yu, L.; He, F.; Liu, H.; Tang, R.; Shi, J.; Sund, X.; Martind, J.F.; Wang, D.; Yang, J.; et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 2009, 327, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Berger, I.M.; Glaser, A.; Bacon, C.; Li, L.; Gretz, N.; Steinbeisser, H.; Rottbauer, W.; Just, S.; Rappold, G. Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 2013, 108, 339. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhang, Q.; Cattaneo, P.; Zhuang, S.; Gong, X.; Spann, N.J.; Jiang, C.; Cao, X.; Zhao, X.; Zhang, X.; et al. Transcription factor ISL1 is essential for pacemaker development and function. J. Clin. Investig. 2015, 125, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Vedantham, V.; Galang, G.; Evangelista, M.; Deo, R.C.; Srivastava, D. RNA sequencing of mouse sinus node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circ. Res. 2015, 116, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Tessari, A.; Pietrobon, M.; Notte, A.; Cifelli, G.; Gage, P.J.; Schneider, M.D.; Lembo, G.; Campione, M. Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ. Res. 2008, 102, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Campione, M.; Ros, M.A.; Icardo, J.M.; Piedra, E.; Christoffels, V.M.; Schweickert, A.; Blumc, M.; Francoa, D.; Moormana, A.F.M. Pitx2 expression defines a left cardiac lineage of cells: Evidence for atrial and ventricular molecular isomerism in the iv/iv mice. Dev. Biol. 2001, 231, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Klysik, E.; Sood, S.; Johnson, R.L.; Wehrens, X.H.; Martin, J.F. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. USA 2010, 107, 9753–9758. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bai, Y.; Li, N.; Ye, W.; Zhang, M.; Greene, S.B.; Tao, Y.; Chen, Y.; Wehrensa, X.H.T.; Martin, J.F. Pitx2-microRNA pathway that delimits sinus node development and inhibits predisposition to atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 9181–9186. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Arnar, D.O.; Helgadottir, A.; Gretarsdottir, S.; Holm, H.; Sigurdsson, A.; Jonasdottir, A.; Baker, A.; Thorleifsson, G.; Kristjansson, K. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007, 448, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; van den Boogaard, M.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8, 354ra115. [Google Scholar] [CrossRef] [PubMed]
- Aanhaanen, W.T.; Brons, J.F.; Dominguez, J.N.; Rana, M.S.; Norden, J.; Airik, R.; Wakker, V.; de Gier-de Vries, C.; Brown, N.A.; Kisper, A.; et al. The Tbx2+ primary myocardium of the atrioventricular canal forms the atrioventricular node and the base of the left ventricle. Circ. Res. 2009, 104, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Horsthuis, T.; Buermans, H.P.; Brons, J.F.; Verkerk, A.O.; Bakker, M.L.; Wakker, V.; Clout, D.E.W.; Moorman, A.F.M.; Hoen, P.A.C.; Christoffels, V.M. Gene expression profiling of the forming atrioventricular node using a novel tbx3-based node-specific transgenic reporter. Circ. Res. 2009, 105, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quintana, D.; Ho, S.Y. Anatomy of cardiac nodes and atrioventricular specialized conduction system. Rev. Esp. Cardiol. 2003, 56, 1085–1092. [Google Scholar] [CrossRef]
- Tawara, S. Das Reizleitungssystem Des Säugetierherzens: Eine Anatomisch-Histologische Studie Über Das Atrioventrikularbündel Und Die Purkinjeschen Fäden; Fischer: Jena, Germany, 1906. [Google Scholar]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Hoogaars, W.M.; Barnett, P.; Grieskamp, T.; Rana, M.S.; Buermans, H.; Farin, H.F.; Petry, M.; Heallen, T.; Martin, J.F.; et al. Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell. Mol. Life Sci. 2012, 69, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Bressan, M.; Yang, P.B.; Louie, J.D.; Navetta, A.M.; Garriock, R.J.; Mikawa, T. Reciprocal myocardial-endocardial interactions pattern the delay in atrioventricular junction conduction. Development 2014, 141, 4149–4157. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.M.; Boukens, B.J.; Phelps, A.L.; Brown, C.L.; Toomer, K.A.; Burns, T.A.; Burnsa, T.A.; Mukherjeec, R.D.; Norrisa, R.A.; Trusk, T.C. Alk3 mediated Bmp signaling controls the contribution of epicardially derived cells to the tissues of the atrioventricular junction. Dev. Biol. 2014, 396, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Gaussin, V.; Morley, G.E.; Cox, L.; Zwijsen, A.; Vance, K.M.; Emile, L.; Tian, Y.; Liu, J.; Hong, C.; Myers, D.; et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ. Res. 2005, 97, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.M.; Gaussin, V.; Burch, J.B.; Yu, C.; Mishina, Y.; Schneider, M.D.; Fishman, G.J.; Morley, G.E.; et al. Abnormal conduction and morphology in the atrioventricular node of mice with atrioventricular canal targeted deletion of Alk3/Bmpr1a receptor. Circulation 2007, 116, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Thakuria, J.V.; Cox, G.F.; Wang, X.; Bi, W.; Bray, M.S.; Shaw, C.; Cheung, S.W.; Chinault, A.C.; Boggs, B.A.; et al. 20p12.3 microdeletion predisposes to Wolff-Parkinson-White syndrome with variable neurocognitive deficits. J. Med. Genet. 2009, 46, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Linden, H.; Williams, R.; King, J.; Blair, E.; Kini, U. Ulnar Mammary syndrome and TBX3: Expanding the phenotype. Am. J. Med. Genet. Part A 2009, 149, 2809–2812. [Google Scholar] [CrossRef] [PubMed]
- Boogerd, K.J.; Wong, L.Y.; Christoffels, V.M.; Klarenbeek, M.; Ruijter, J.M.; Moorman, A.F.; Barnett, P. Msx1 and Msx2 are functional interacting partners of T-box factors in the regulation of Connexin43. Cardiovasc. Res. 2008, 78, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Hoogaars, W.M.; Tessari, A.; Clout, D.E.; Moorman, A.F.; Campione, M. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev. Dyn. 2004, 229, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.L.; Boukens, B.J.; Mommersteeg, M.T.; Brons, J.F.; Wakker, V.; Moorman, A.F.; Christoffels, V.M. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ. Res. 2008, 102, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Harrelson, Z.; Kelly, R.G.; Goldin, S.N.; Gibson-Brown, J.J.; Bollag, R.J.; Silver, L.M.; Papaioannou, V.E. Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development 2004, 131, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Aanhaanen, W.T.; Boukens, B.J.; Sizarov, A.; Wakker, V.; de Gier-de Vries, C.; van Ginneken, A.C.; Moorman, A.F.M; Coronel, R.; Christoffels, V.M. Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J. Clin. Investig. 2011, 121, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Rutenberg, J.B.; Fischer, A.; Jia, H.; Gessler, M.; Zhong, T.P.; Mercola, M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development 2006, 133, 4381–4390. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Horsthuis, T.; Farin, H.F.; Grieskamp, T.; Norden, J.; Petry, M.; Wakker, V.; Moorman, A.F.M.; Christoffels, V.M.; Kispert, A. Tbx20 interacts with smads to confine tbx2 expression to the atrioventricular canal. Circ. Res. 2009, 105, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Barnett, P.; van Duijvenboden, K.; Weber, D.; Gessler, M.; Christoffels, V.M. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun. 2014, 5, 3680. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.V.; McAnally, J.; Bezprozvannaya, S.; Berry, J.M.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 2009, 136, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.P.; Bhakta, M.; Bezprozvannaya, S.; Wang, L.; Lubczyk, C.; Olson, E.N.; Munshi, N.V. MyoR modulates cardiac conduction by repressing Gata4. Mol. Cell. Biol. 2015, 35, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.C.; Shaw, R.M.; De Val, S.; Kang, G.; Jan, L.Y.; Black, B.L.; Stainier, D.Y.R. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008, 22, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, M.M.; Patel, N.N.; Schillinger, K.J.; Wang, T.; Patel, V.V. GATA-Binding Factor 6 Contributes to Atrioventricular Node Development and Function. Circ. Cardiovasc. Genet. 2015, 8, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ranganayakulu, G.; Elliott, D.A.; Harvey, R.P.; Olson, E.N. Divergent roles for NK-2 class homeobox genes in cardiogenesis in flies and mice. Development 1998, 125, 3037–3048. [Google Scholar] [PubMed]
- Tanaka, M.; Chen, Z.; Bartunkova, S.; Yamasaki, N.; Izumo, S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999, 126, 1269–1280. [Google Scholar] [PubMed]
- Jay, P.Y.; Harris, B.S.; Maguire, C.T.; Buerger, A.; Wakimoto, H.; Tanaka, M.; Kupershmidt, S.; Roden, D.M.; Schultheiss, T.M.; O’Brien, T.X; et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Investig. 2004, 113, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Pashmforoush, M.; Lu, J.T.; Chen, H.; Amand, T.S.; Kondo, R.; Pradervand, S.; Evans, S.M.; Clark, B.; Feramisco, J.R.; Giles, W.; et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 2004, 117, 373–386. [Google Scholar] [CrossRef]
- Rentschler, S.; Harris, B.S.; Kuznekoff, L.; Jain, R.; Manderfield, L.; Lu, M.M.; Morley, G.E.; Patel, V.V.; Epstein, J.A. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J. Clin. Investig. 2011, 121, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Gillers, B.S.; Chiplunkar, A.; Aly, H.; Valenta, T.; Basler, K.; Christoffels, V.M.; Efimov, I.R.; Boukens, B.J.; Rentschler, S. Canonical wnt signaling regulates atrioventricular junction programming and electrophysiological properties. Circ. Res. 2015, 116, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, M.C.; Haase, C.; Christoffels, V.M.; Weidinger, G.; Bakkers, J. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Haissaguerre, M.; Shoda, M.; Jais, P.; Nogami, A.; Shah, D.C.; Kautzner, J.; Arentz, T.; Kalushe, D.; Lamaison, D.; Griffith, M.; et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation 2002, 106, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Noujaim, S.F.; Tolkacheva, E.G.; Talkachou, A.; O’Connell, R.; Berenfeld, O.; Anumonwo, J.; Pandit, S.V.; Vikstrom, K.; Napolitano, C.; et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2007, 101, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Ben Caref, E.; Boutjdir, M.; Himel, H.D.; El-Sherif, N. Role of subendocardial Purkinje network in triggering torsade de pointes arrhythmia in experimental long QT syndrome. Europace 2008, 10, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Giovannone, S.F.; Liu, N.; Liu, F.Y.; Zhang, J.; Priori, S.G.; Fishman, G.J. Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ. Res. 2010, 107, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; Daubert, J.C.; Erdmann, E.; Freemantle, N.; Gras, D.; Kappenberger, L.; Luigi Tavazzi, M.D. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N. Engl. J. Med. 2005, 352, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Vermeulen, J.L.M.; Verbeek, F.Z.; Viragh, S.Z.; Kalman, F.; Lamers, W.H.; Moorman, A.F.M. Spatial distribution of “tissue-specific” antigens in the developing human heart and skeletal muscle III. An immunohistochemical analysis of the distribution of the neural tissue antigen G1N2 in the embryonic heart; implications for the development of the atrioventricular conduction system. Anat. Rec. 1992, 232, 97–111. [Google Scholar] [PubMed]
- Remme, C.A.; Verkerk, A.O.; Hoogaars, W.M.; Aanhaanen, W.T.; Scicluna, B.P.; Annink, C.; Wilde, A.A.M.; van Veen, T.A.B.; Veldkamp, M.W.; de Bakker, J.M.T.; et al. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res. Cardiol. 2009, 104, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Miquerol, L.; Meysen, S.; Mangoni, M.; Bois, P.; van Rijen, H.V.; Abran, P.; Jongsma, H.; Nargeot, J.; Gros, D. Architectural and functional asymmetry of the His-Purkinje system of the murine heart. Cardiovasc. Res. 2004, 63, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.J.; Alshinawi, C.; Kyndt, F.; Probst, V.; Hoorntje, T.M.; Hulsbeek, M.; Wilde, A.A.M.; Escande, D.; Mannens, M.M.A.M.; Marec, H.L. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999, 23, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Seki, A.; Sumitomo, N.; Chkourko, H.; Fukuhara, S.; Watanabe, H.; Shimizu, W.; Bezzina, C.R.; Hasdemir, C.; Mugishima, H; et al. A connexin40 mutation associated with a malignant variant of progressive familial heart block type I. Circ. Arrhythm. Electrophysiol. 2012, 5, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Lev, M. Anatomic Basis for Atrioventricular Block. Am. J. Med. 1964, 37, 742–748. [Google Scholar] [CrossRef]
- Lenegre, J.; Moreau, P.H. Le bloc auriculo-ventriculaire chronique. Etude anatomique, clinique et histologique. Arch. Mal. Coeur. 1963, 56, 867–888. [Google Scholar] [PubMed]
- Moskowitz, I.P.; Kim, J.B.; Moore, M.L.; Wolf, C.M.; Peterson, M.A.; Shendure, J.; Nobrega, M.A; Yokota, Y.; Berul, C.; Izumo, S.; et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 2007, 129, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.S.; Spruill, L.; Edmonson, A.M.; Rackley, M.S.; Benson, D.W.; O’Brien, T.X.; Gourdie, R.G. Differentiation of cardiac Purkinje fibers requires precise spatiotemporal regulation of Nkx2-5 expression. Dev. Dyn. 2006, 235, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Meysen, S.; Marger, L.; Hewett, K.W.; Jarry-Guichard, T.; Agarkova, I.; Chauvin, J.P.; Perriard, J.C.; Izumo, C.; Gourdiec, R.G.; Mangoni, M.E.; et al. Nkx2.5 cell-autonomous gene function is required for the postnatal formation of the peripheral ventricular conduction system. Dev. Biol. 2007, 303, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, H.; Wakimoto, H.; Liu, M.; Maguire, C.T.; Converso, K.L.; Shioi, T.; Huang, W-Y.; Manning, W.J.; Paul, D.; Lawitts, J.; et al. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J. Clin. Investig. 2001, 108, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Risebro, C.A.; Petchey, L.K.; Smart, N.; Gomes, J.; Clark, J.; Vieira, J.M.; Yanni, J.; Dobrzynski, H.; Davidson, S.; Zuberi, Z.; et al. Epistatic rescue of Nkx2.5 adult cardiac conduction disease phenotypes by prospero-related homeobox protein 1 and HDAC3. Circ. Res. 2012, 111, e19–e31. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, S.; Zander, J.; Meyers, K.; France, D.; Levine, R.; Porter, G.; Rivkees, S.A.; Morley, G.E.; Fishman, G.I. Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc. Natl. Acad. Sci. USA 2002, 99, 10464–10469. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, D.E.; Moskowitz, I.P. Inducible recombination in the cardiac conduction system of minK: CreERT(2) BAC transgenic mice. Genesis 2011, 49, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, D.E.; Liu, F.; Fahrenbach, J.P.; Kim, G.H.; Schillinger, K.J.; Smemo, S.; McNally, E.M.; Nobrega, M.A.; Patel, V.V.; Moskowitz, I.P. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Investig. 2012, 122, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Holm, H.; Gudbjartsson, D.F.; Arnar, D.O.; Thorleifsson, G.; Thorgeirsson, G.; Stefansdottir, H.; Gudjonsson, S.A; Jonasdottir, A.; Mathiesen, E.B.; Njølstad, I.; et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 2010, 42, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Pfeufer, A.; van Noord, C.; Marciante, K.D.; Arking, D.E.; Larson, M.G.; Smith, A.V.; Tarasov, K.V.; Müller, M.; Sotoodehnia, N.; Sinner, M.F.; et al. Genome-wide association study of PR interval. Nat. Genet. 2010, 42, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Magnani, J.W.; Palmer, C.; Meng, Y.A.; Soliman, E.Z.; MuSNi, S.K.; Kerr, K.F.; Schnabel, R.B.; Lubitz, S.A.; Sotoodehnia, N.; et al. Genome-wide association studies of the PR interval in African Americans. PLoS Genet. 2011, 7, e1001304. [Google Scholar] [CrossRef] [PubMed]
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.; Dorr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Müller, M.; Eijgelsheim, M.; Alonso, A.; et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; van de Werken, H.J.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.; Wong, L.Y.; Tessadori, F.; Bakker, M.L.; Dreizehnter, L.K.; Wakker, V.; Bezzina, C.R.; Hoen, P.A.C.; Bakkers, J.; Barnett, P.; et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Investig. 2012, 122, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Tamaddon, H.S.; Vaidya, D.; Simon, A.M.; Paul, D.L.; Jalife, J.; Morley, G.E. High-resolution optical mapping of the right bundle branch in connexin40 knockout mice reveals slow conduction in the specialized conduction system. Circ. Res. 2000, 87, 929–936. [Google Scholar] [CrossRef] [PubMed]
- van Rijen, H.V.; van Veen, T.A.; van Kempen, M.J.; Wilms-Schopman, F.J.; Potse, M.; Krueger, O.; Willecke, K.; Opthof, T.; Jongsma, H.J.; de Bakker, J.M.T. Impaired conduction in the bundle branches of mouse hearts lacking the gap junction protein connexin40. Circulation 2001, 103, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Rosen, A.; Hussein, S.M.; Puviindran, V.; Korogyi, A.S.; Chiarello, C.; Nagy, A.; Hui, C.; Backxb, P.H. Irx3 is required for postnatal maturation of the mouse ventricular conduction system. Sci. Rep. 2016, 6, 19197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Kim, K.H.; Rosen, A.; Smyth, J.W.; Sakuma, R.; Delgado-Olguin, P.; Davise, M.; Chih, N.C.; Puviindranc, V.; Gaborit, N.; et al. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc. Natl. Acad. Sci. USA 2011, 108, 13576–13581. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; SaSNo, T.; Kimura, W.; Miyamoto, Y.; Aiba, T.; Ishikawa, T.; Nogami, A.; Fukamizu, S.; Sakurada, H.; Takahashi, Y.; et al. Genetic defects in a His-Purkinje system transcription factor, IRX3, cause lethal cardiac arrhythmias. Eur. Heart J. 2015, 37, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Tran, V.T.; Kubalak, S.W.; Minamisawa, S.; Fiset, C.; Wollert, K.C.; Brown, A.B.; Ruiz-Lozano1, P.; Barrere-Lemaire, S.; Kondo, R.; Norman, L.W.; et al. A novel genetic pathway for sudden cardiac death via defects in the transition between ventricular and conduction system cell lineages. Cell 2000, 102, 671–682. [Google Scholar] [CrossRef]
- Hewett, K.W.; Norman, L.W.; Sedmera, D.; Barker, R.J.; Justus, C.; Zhang, J.; Kubalak, S.W.; Gourdie, R.G. Knockout of the neural and heart expressed gene HF-1b results in apical deficits of ventricular structure and activation. Cardiovasc. Res. 2005, 67, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Ismat, F.A.; Zhang, M.; Kook, H.; Huang, B.; Zhou, R.; Ferrari, V.A.; Epstein, J.A.; Patel, V.V. Homeobox protein Hop functions in the adult cardiac conduction system. Circ. Res. 2005, 96, 898–903. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.S.; Fishman, G.I. Development and Function of the Cardiac Conduction System in Health and Disease. J. Cardiovasc. Dev. Dis. 2017, 4, 7. https://doi.org/10.3390/jcdd4020007
Park DS, Fishman GI. Development and Function of the Cardiac Conduction System in Health and Disease. Journal of Cardiovascular Development and Disease. 2017; 4(2):7. https://doi.org/10.3390/jcdd4020007
Chicago/Turabian StylePark, David S., and Glenn I. Fishman. 2017. "Development and Function of the Cardiac Conduction System in Health and Disease" Journal of Cardiovascular Development and Disease 4, no. 2: 7. https://doi.org/10.3390/jcdd4020007
APA StylePark, D. S., & Fishman, G. I. (2017). Development and Function of the Cardiac Conduction System in Health and Disease. Journal of Cardiovascular Development and Disease, 4(2), 7. https://doi.org/10.3390/jcdd4020007