Anterior Hox Genes in Cardiac Development and Great Artery Patterning


Abstract
:1. Introduction
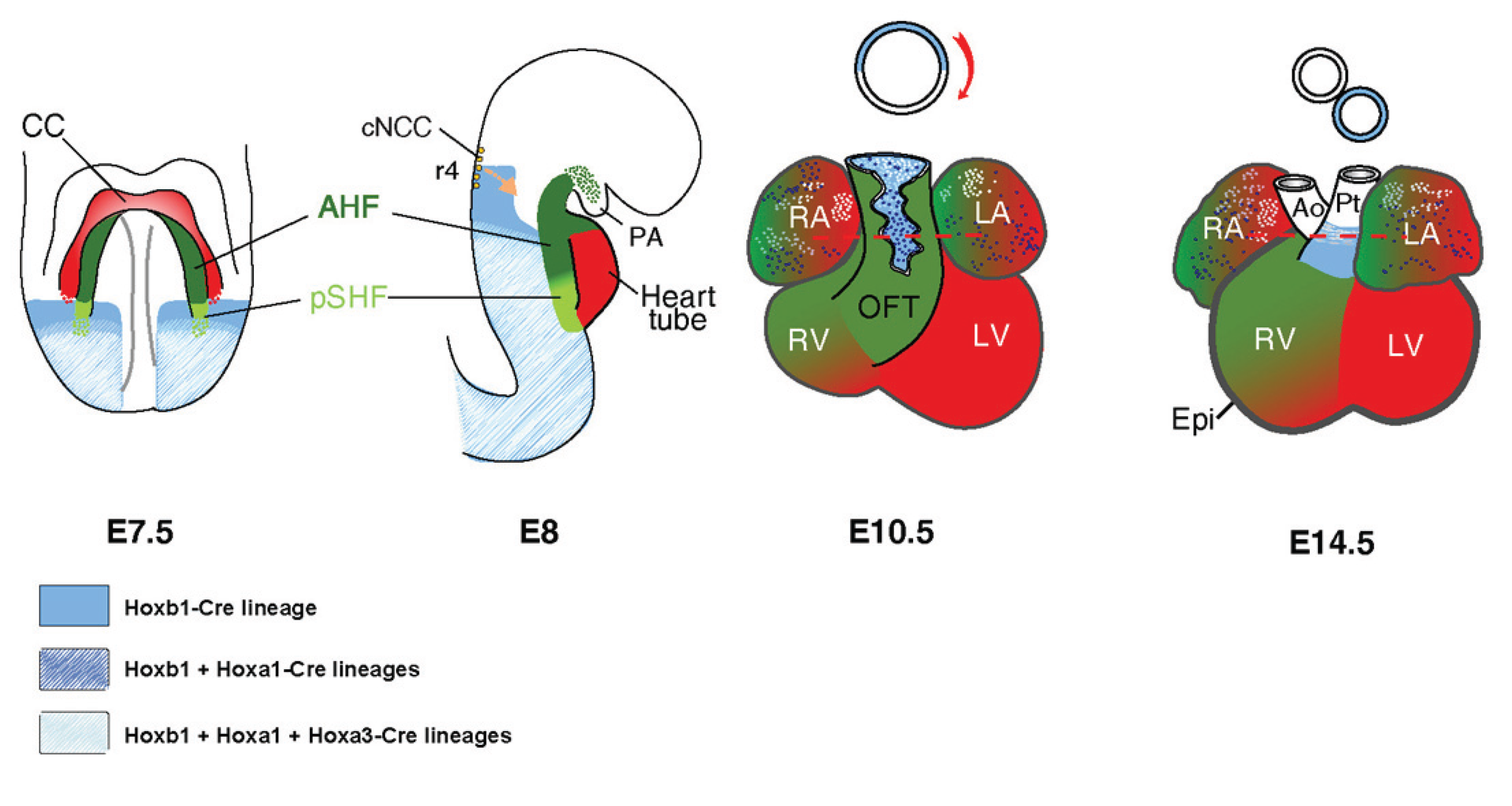

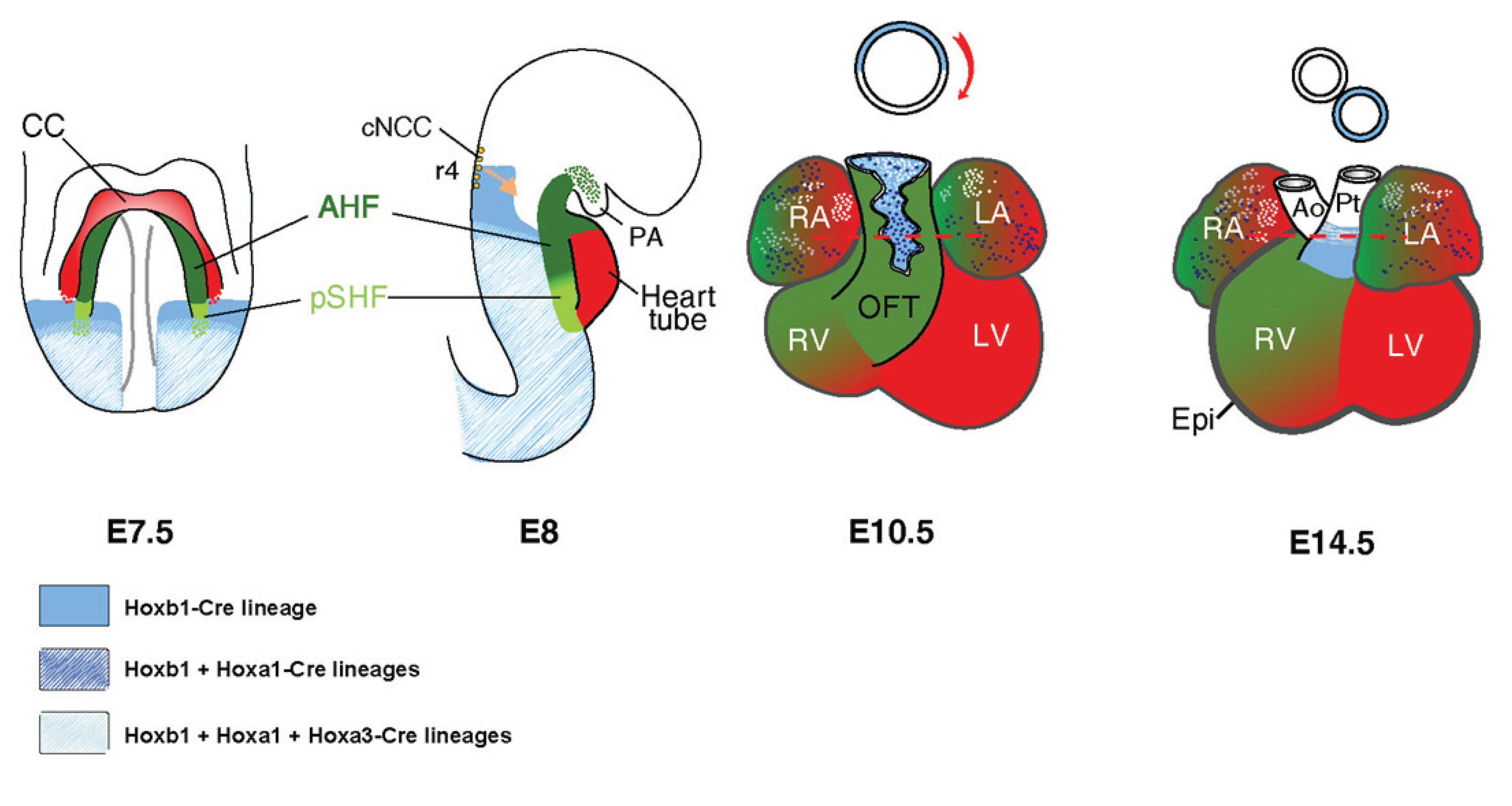{kind=link}
| Gene | Mutants | Phenotypes | References |
|---|---|---|---|
| Hoxa1 | Hox-1.6−/− | No reported cardiac phenotype | [42,43] |
| Hoxa1GFPneo/GFPneo | No reported cardiac phenotype | [44] | |
| Hoxa1−/− | IAAB, ASC, RAA, BAV, VSD, TOF | [34,45] | |
| Hoxa3 | Hox-1.5−/− | PDA, RV hypoplasia, hypertrophy of RA and LV, stenosis of AV, bicuspid pulmonary valve | [35] |
| Hoxa3−/− | Degeneration of the 3rd arch artery Malformation of the carotid artery system | [36] | |
| HoxA/HoxB | Hoxa−/−;Hoxb−/− | Heart looping defects | [37] |
| Pbx1 | Pbx1−/− | Lethality by E15-E16. PTA and VSD | [41] |
| Pbx1+/−;Pbx2+/−;Pbx3+/− | BAV | [41] | |
| Pbx1+/−;Pbx2−/− | Overriding aorta, VSD, BAV, bicuspid pulmonary valve | [41] | |
| Pbx1+/−;Pbx2−/−;Pbx3+/− | TOF | [41] | |
| Pbx2 | Pbx2−/− | No cardiac phenotype | [41] |
| Pbx3 | Pbx3−/− | No cardiac phenotype | [41] |
| Meis1 | Meis1−/− | Overriding aorta, VSD | [41] |
| α-MHC-Cre;Meis1f/f | Increased postnatal cardiomyocyte proliferation | [41] | |
| Meis2 | Zebrafish Meis2-MO | Heart looping defects | [46] |
2. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Zaffran, S.; Kelly, R.G. New developments in the second heart field. Differ. Res. Biol. Divers. 2012, 84, 17–24. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An nkx2-5/bmp2/smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef]
- Baldini, A. Dissecting contiguous gene defects: Tbx1. Curr. Opin. Genet. Dev. 2005, 15, 279–284. [Google Scholar] [CrossRef]
- Galli, D.; Dominguez, J.N.; Zaffran, S.; Munk, A.; Brown, N.A.; Buckingham, M.E. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as pitx2c is expressed. Development 2008, 135, 1157–1167. [Google Scholar] [CrossRef]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar]
- Bertrand, N.; Roux, M.; Ryckebusch, L.; Niederreither, K.; Dolle, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef]
- Dominguez, J.N.; Meilhac, S.M.; Bland, Y.S.; Buckingham, M.E.; Brown, N.A. Asymmetric fate of the posterior part of the second heart field results in unexpected left/right contributions to both poles of the heart. Circ. Res. 2012, 111, 1323–1335. [Google Scholar] [CrossRef]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar] [CrossRef]
- Ward, C.; Stadt, H.; Hutson, M.; Kirby, M.L. Ablation of the secondary heart field leads to tetralogy of fallot and pulmonary atresia. Dev. Biol. 2005, 284, 72–83. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Rhee, J.T.; Bader, D. Expression of the atrial-specific myosin heavy chain amhc1 and the establishment of anteroposterior polarity in the developing chicken heart. Development 1994, 120, 871–883. [Google Scholar]
- Xavier-Neto, J.; Neville, C.M.; Shapiro, M.D.; Houghton, L.; Wang, G.F.; Nikovits, W., Jr.; Stockdale, F.E.; Rosenthal, N. A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 1999, 126, 2677–2687. [Google Scholar]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.; Cardoso, W.V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of Raldh2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating isl1 through the fgf8 pathway. Dev. Dynam. 2008, 237, 1627–1635. [Google Scholar] [CrossRef]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 2008, 15, 923–934. [Google Scholar] [CrossRef]
- Diman, N.Y.; Remacle, S.; Bertrand, N.; Picard, J.J.; Zaffran, S.; Rezsohazy, R. A retinoic acid responsive hoxa3 transgene expressed in embryonic pharyngeal endoderm, cardiac neural crest and a subdomain of the second heart field. PloS One 2011, 6. [Google Scholar] [CrossRef]
- Nolte, C.; Jinks, T.; Wang, X.; Martinez Pastor, M.T.; Krumlauf, R. Shadow enhancers flanking the hoxb cluster direct dynamic hox expression in early heart and endoderm development. Dev. Biol. 2013, 383, 158–173. [Google Scholar] [CrossRef]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar]
- Hutson, M.R.; Kirby, M.L. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res. C Embryo Today 2003, 69, 2–13. [Google Scholar] [CrossRef]
- Nishibatake, M.; Kirby, M.L.; Van Mierop, L.H. Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation 1987, 75, 255–264. [Google Scholar] [CrossRef]
- Arima, Y.; Miyagawa-Tomita, S.; Maeda, K.; Asai, R.; Seya, D.; Minoux, M.; Rijli, F.M.; Nishiyama, K.; Kim, K.S.; Uchijima, Y.; et al. Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Scholl, A.M.; Kirby, M.L. Signals controlling neural crest contributions to the heart. Wiley Interdisc. Rev. Syst. Biol. Med. 2009, 1, 220–227. [Google Scholar] [CrossRef]
- Hutson, M.R.; Zhang, P.; Stadt, H.A.; Sato, A.K.; Li, Y.X.; Burch, J.; Creazzo, T.L.; Kirby, M.L. Cardiac arterial pole alignment is sensitive to fgf8 signaling in the pharynx. Dev. Biol. 2006, 295, 486–497. [Google Scholar] [CrossRef]
- Alexander, T.; Nolte, C.; Krumlauf, R. Hox genes and segmentation of the hindbrain and axial skeleton. Ann. Rev. Cell Dev. Biol. 2009, 25, 431–456. [Google Scholar] [CrossRef]
- Wellik, D.M. Hox genes and vertebrate axial pattern. Curr. Top. Dev. Biol. 2009, 88, 257–278. [Google Scholar] [CrossRef]
- Duboule, D.; Dolle, P. The structural and functional organization of the murine hox gene family resembles that of drosophila homeotic genes. EMBO J. 1989, 8, 1497–1505. [Google Scholar]
- Searcy, R.D.; Yutzey, K.E. Analysis of hox gene expression during early avian heart development. Dev. Dyn. 1998, 213, 82–91. [Google Scholar] [CrossRef]
- Holve, S.; Friedman, B.; Hoyme, H.E.; Tarby, T.J.; Johnstone, S.J.; Erickson, R.P.; Clericuzio, C.L.; Cunniff, C. Athabascan brainstem dysgenesis syndrome. Am. J. Med. Genet. A 2003, 120A, 169–173. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Bosley, T.M.; Salih, M.A.; Alorainy, I.A.; Sener, E.C.; Nester, M.J.; Oystreck, D.T.; Chan, W.M.; Andrews, C.; Erickson, R.P.; et al. Homozygous hoxa1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 2005, 37, 1035–1037. [Google Scholar] [CrossRef]
- Makki, N.; Capecchi, M.R. Cardiovascular defects in a mouse model of hoxa1 syndrome. Human Mol. Genet. 2012, 21, 26–31. [Google Scholar] [CrossRef]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef]
- Kameda, Y.; Watari-Goshima, N.; Nishimaki, T.; Chisaka, O. Disruption of the hoxa3 homeobox gene results in anomalies of the carotid artery system and the arterial baroreceptors. Cell Tissue Res. 2003, 311, 343–352. [Google Scholar]
- Soshnikova, N.; Dewaele, R.; Janvier, P.; Krumlauf, R.; Duboule, D. Duplications of hox gene clusters and the emergence of vertebrates. Dev. Biol. 2013, 378, 194–199. [Google Scholar] [CrossRef]
- Ladam, F.; Sagerstrom, C.G. Hox regulation of transcription: More complex(es). Dev. Dynam. 2014, 243, 4–15. [Google Scholar] [CrossRef]
- Moens, C.B.; Selleri, L. Hox cofactors in vertebrate development. Dev. Biol. 2006, 291, 193–206. [Google Scholar] [CrossRef]
- Chang, C.P.; Stankunas, K.; Shang, C.; Kao, S.C.; Twu, K.Y.; Cleary, M.L. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development 2008, 135, 3577–3586. [Google Scholar] [CrossRef]
- Stankunas, K.; Shang, C.; Twu, K.Y.; Kao, S.C.; Jenkins, N.A.; Copeland, N.G.; Sanyal, M.; Selleri, L.; Cleary, M.L.; Chang, C.P. Pbx/meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res. 2008, 103, 702–709. [Google Scholar] [CrossRef]
- Chisaka, O.; Musci, T.S.; Capecchi, M.R. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene hox-1.6. Nature 1992, 355, 516–520. [Google Scholar] [CrossRef]
- Lufkin, T.; Dierich, A.; LeMeur, M.; Mark, M.; Chambon, P. Disruption of the hox-1.6 homeobox gene results in defects in a region corresponding to its rostral domain of expression. Cell 1991, 66, 1105–1119. [Google Scholar] [CrossRef]
- Godwin, A.R.; Stadler, H.S.; Nakamura, K.; Capecchi, M.R. Detection of targeted gfp-hox gene fusions during mouse embryogenesis. Proc. Nat. Acad. Sci. USA 1998, 95, 13042–13047. [Google Scholar] [CrossRef]
- Makki, N.; Capecchi, M.R. Identification of novel hoxa1 downstream targets regulating hindbrain, neural crest and inner ear development. Dev. Biol. 2011, 357, 295–304. [Google Scholar] [CrossRef]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Christodoulou, D.C.; Robinson, J.Y.; Seidman, C.E.; Baldwin, H.S.; Seidman, J.G.; Barnett, J.V. Spatial transcriptional profile of the chick and mouse endocardial cushions identify novel regulators of endocardial emt in vitro. J. Mol. Cell. Cardiol. 2013, 59, 196–204. [Google Scholar] [CrossRef]
- Gavalas, A.; Studer, M.; Lumsden, A.; Rijli, F.M.; Krumlauf, R.; Chambon, P. Hoxa1 and hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development 1998, 125, 1123–1136. [Google Scholar]
- Rossel, M.; Capecchi, M.R. Mice mutant for both hoxa1 and hoxb1 show extensive remodeling of the hindbrain and defects in craniofacial development. Development 1999, 126, 5027–5040. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laforest, B.; Bertrand, N.; Zaffran, S. Anterior Hox Genes in Cardiac Development and Great Artery Patterning. J. Cardiovasc. Dev. Dis. 2014, 1, 3-13. https://doi.org/10.3390/jcdd1010003
Laforest B, Bertrand N, Zaffran S. Anterior Hox Genes in Cardiac Development and Great Artery Patterning. Journal of Cardiovascular Development and Disease. 2014; 1(1):3-13. https://doi.org/10.3390/jcdd1010003
Chicago/Turabian StyleLaforest, Brigitte, Nicolas Bertrand, and Stéphane Zaffran. 2014. "Anterior Hox Genes in Cardiac Development and Great Artery Patterning" Journal of Cardiovascular Development and Disease 1, no. 1: 3-13. https://doi.org/10.3390/jcdd1010003