Syntheses of Macromolecular Ruthenium Compounds: A New Approach for the Search of Anticancer Drugs


Abstract
:1. Introduction
2. Multinuclear Approaches
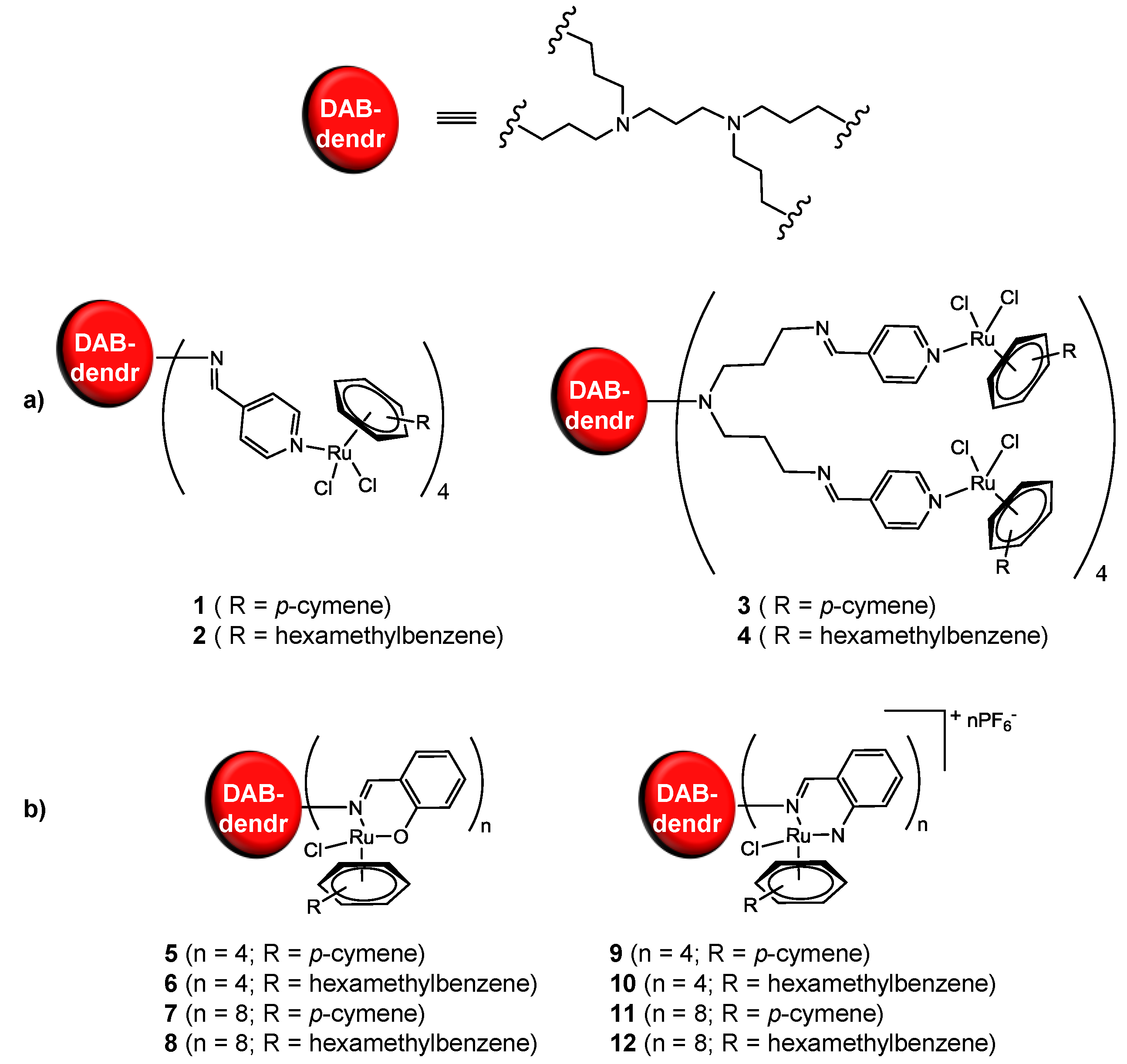
2.1. Ruthenium-Based Dendrimers
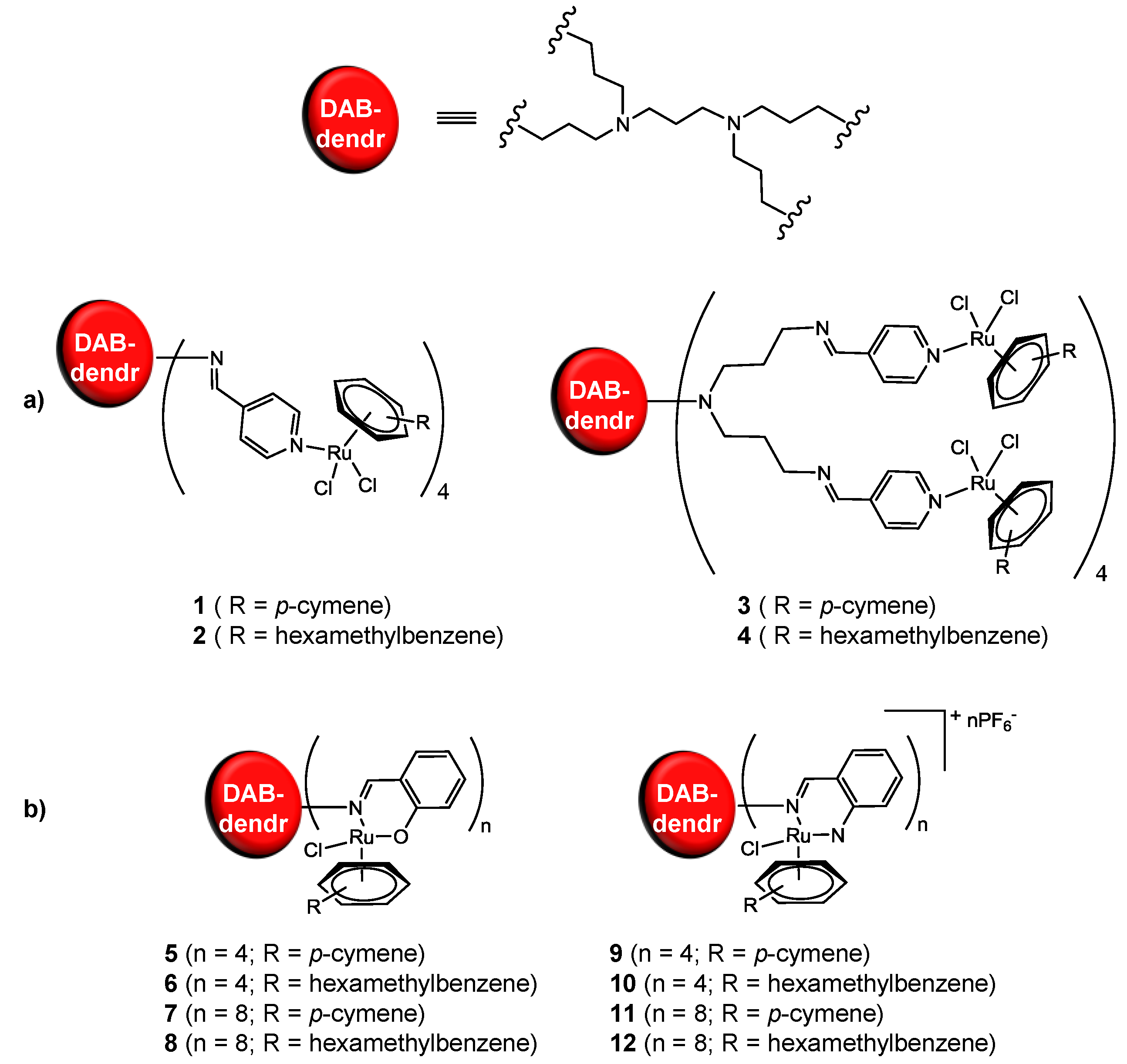

| Compound | IC50 (µM) per metallodendrimer | IC50 (µM) mononuclear Ru-derivative |
|---|---|---|
| 1 a | 43 | ≈100 |
| 2 a | 40 | |
| 3 a | 21 | |
| 4 a | 20 | |
| 5 b | 50 | 20–50 |
| 6 b | 27 | |
| 7 b | 22 | |
| 8 b | 10 | |
| 9 b | >200 | >200 |
| 10 b | 32 | |
| 11 b | 23 | |
| 12 b | 4 |
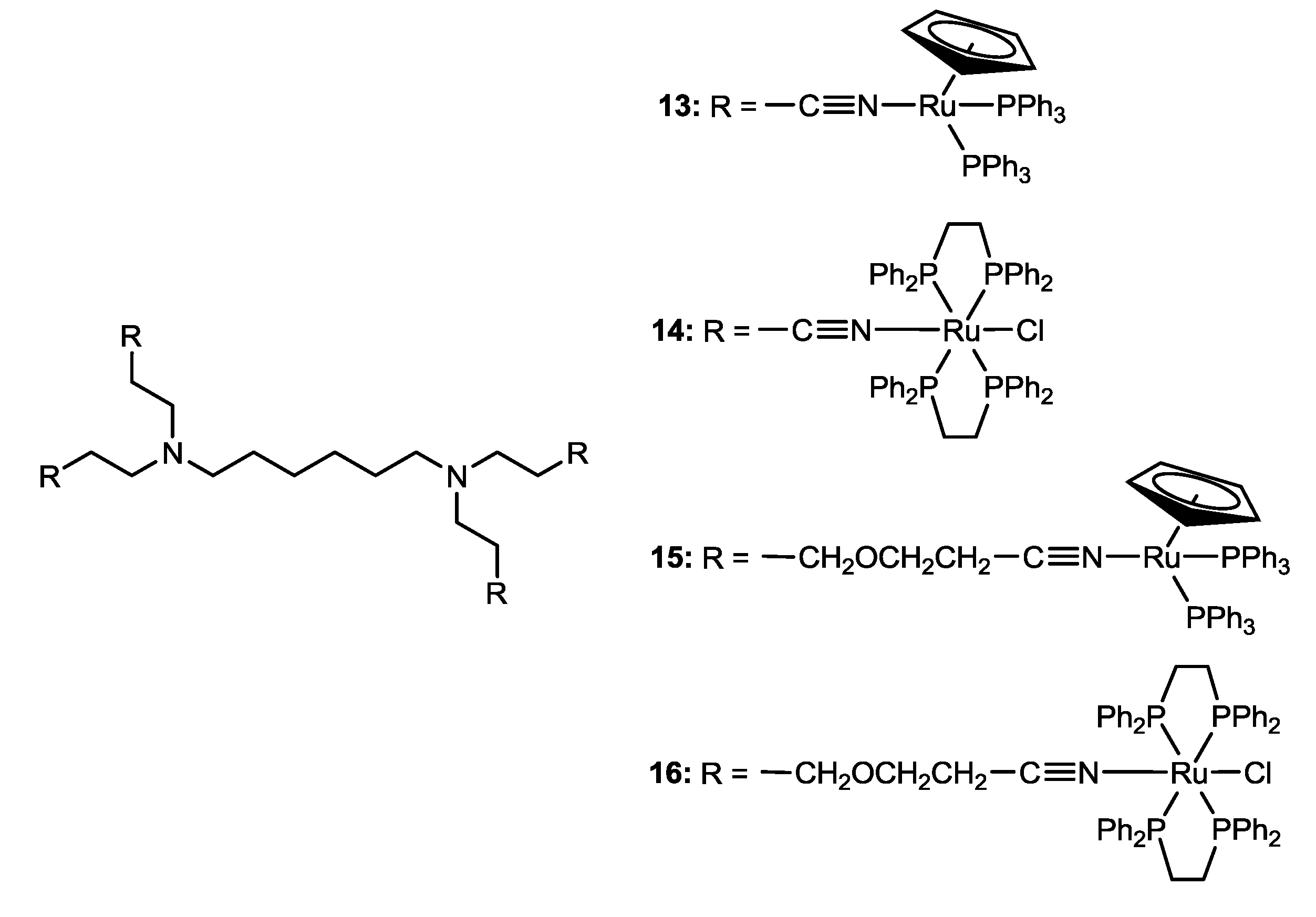
2.2. Ruthenium-Based Coordination-Cage Conjugates
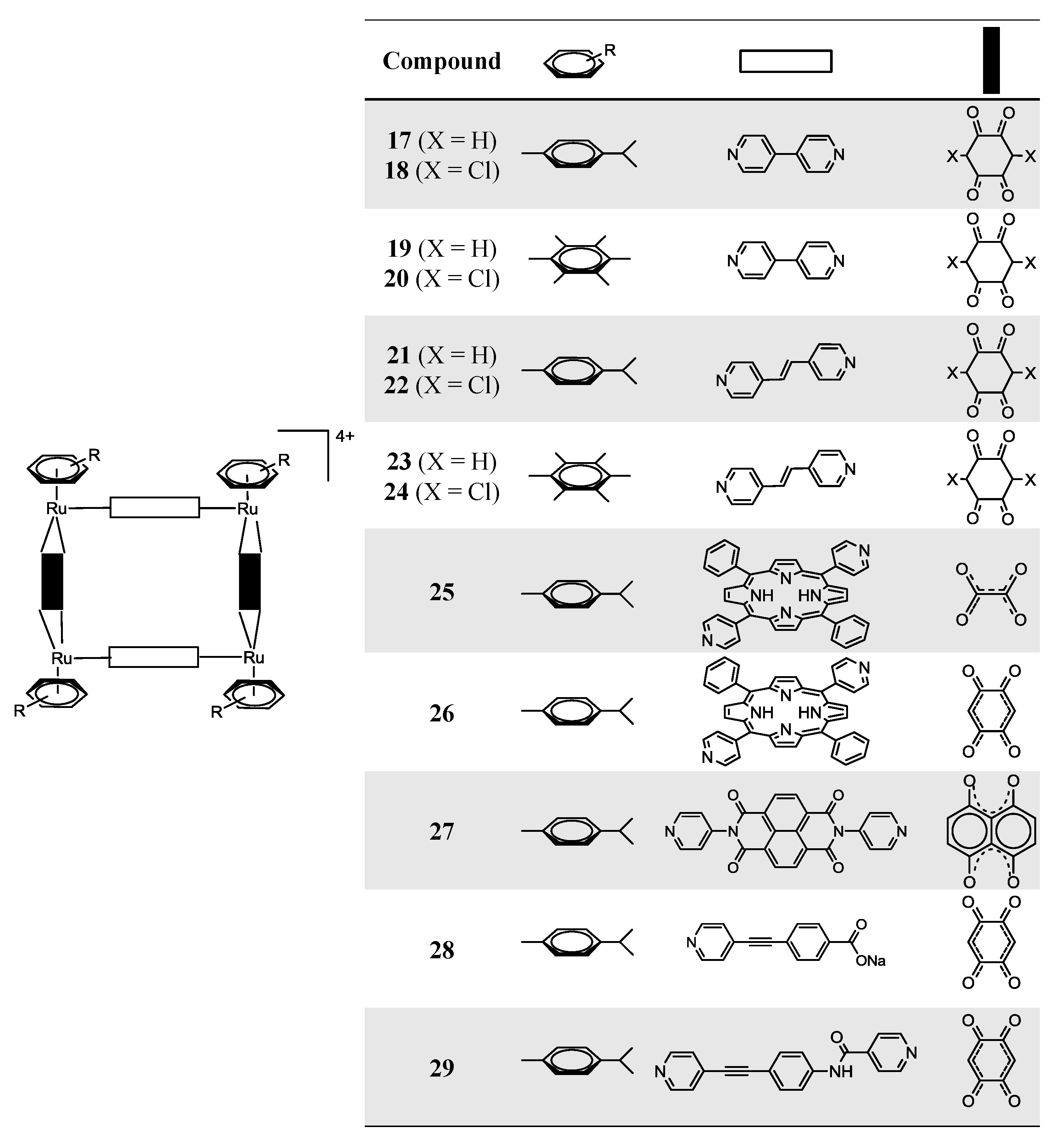
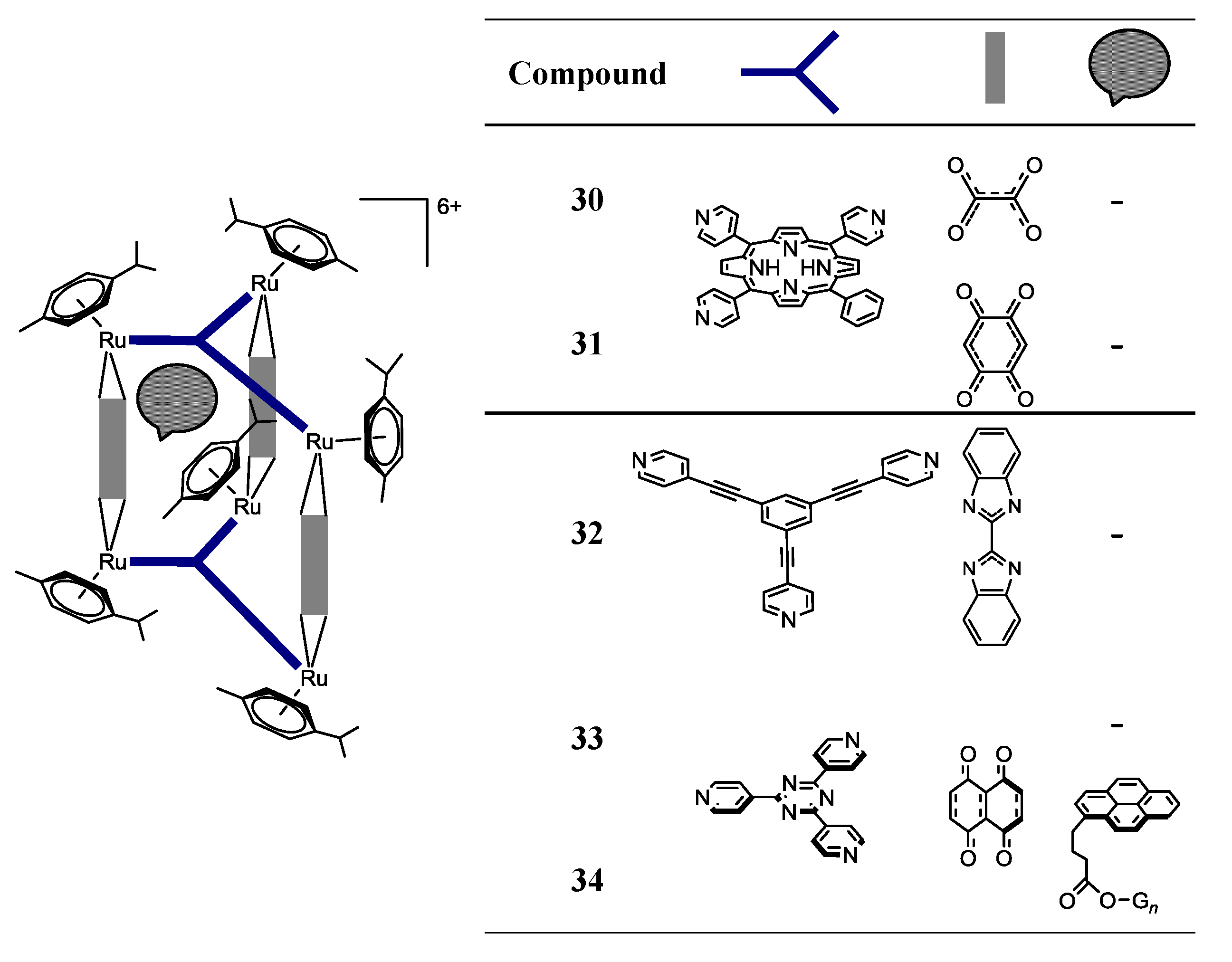
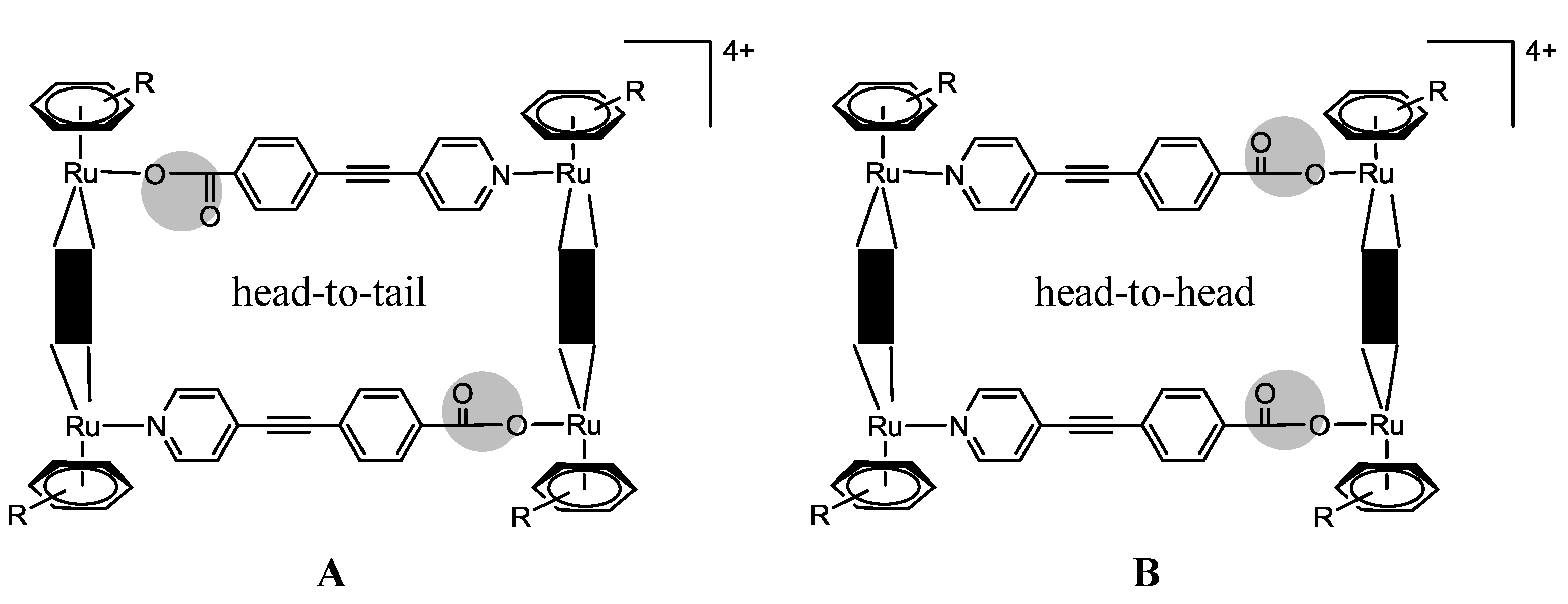
 OO)2Cl2] (arene = p-cymene, hexamethylbenzene; OO OO = 2,5-dihydroxy-1,4-benzoquinonato; 2,5-dichloro-1,4-benzoquinonato) with pyrazine or bipyridine linkers (N N = 4,4'-bipyridine; 1,2-bis(4-pyridyl)ethylene) in methanol, at room temperature, using AgCF3SO3 as a halide scavenger, afford the synthesis of the water soluble tetranuclear metallacyclic cations of general formula [Ru4(arene)4(N N)2(OO OO)2]4+ (Scheme 3, Complexes 17–26) [24,25,39]. The larger rectangles, incorporating the 1,2-bis(4-pyridyl)-ethylene linker, are ca. five times more cytotoxic (IC50 £ 6 µM) than the 4,4'-bipyridine-containg cations (IC50 ³ 30 µM) for the A2780 human ovarian cancer cells (Table 2) [24]. The authors suggested that these variations could result from the different sized cavities, different flexibilities and different packing arrangements (observed from the X-ray diffraction of [Ru4(hexamethylbenzene)4(4,4'-bipyridine)2(2,5-dihydroxy-1,4-benzo-quinonato)2]4+19 and [Ru4(hexamethylbenzene)4(1,2-bis(4-pyridyl)ethylene)2(2,5-di-hydroxy-1,4-benzoquinonato)2]4+23) [24]. In each case, the hexamethylbenzene complexes exhibit lower IC50 than their p-cymene analogues, probably due to the greater lipophilicity of the second [24].OO)2]2+ (OO OO) = oxalate, 2,5-dioxydo-1,4-benzoquinonato, dobq) have been synthesized in the presence of AgCF3SO3 (the synthesis details are ambiguous) [25]. The compounds are sparingly soluble in water and stable in deuterated water at 60 °C for 48 h (NMR studies) [25]. All the complexes are cytotoxic against A2780 human ovarian cancer cells, the complexes with the dobq ligand (26 and 31) being more cytotoxic than the oxalate derivatives (25 and 30); this feature shows the importance of the spacer in the cytotoxic activity [25].
OO)2Cl2] (arene = p-cymene, hexamethylbenzene; OO OO = 2,5-dihydroxy-1,4-benzoquinonato; 2,5-dichloro-1,4-benzoquinonato) with pyrazine or bipyridine linkers (N N = 4,4'-bipyridine; 1,2-bis(4-pyridyl)ethylene) in methanol, at room temperature, using AgCF3SO3 as a halide scavenger, afford the synthesis of the water soluble tetranuclear metallacyclic cations of general formula [Ru4(arene)4(N N)2(OO OO)2]4+ (Scheme 3, Complexes 17–26) [24,25,39]. The larger rectangles, incorporating the 1,2-bis(4-pyridyl)-ethylene linker, are ca. five times more cytotoxic (IC50 £ 6 µM) than the 4,4'-bipyridine-containg cations (IC50 ³ 30 µM) for the A2780 human ovarian cancer cells (Table 2) [24]. The authors suggested that these variations could result from the different sized cavities, different flexibilities and different packing arrangements (observed from the X-ray diffraction of [Ru4(hexamethylbenzene)4(4,4'-bipyridine)2(2,5-dihydroxy-1,4-benzo-quinonato)2]4+19 and [Ru4(hexamethylbenzene)4(1,2-bis(4-pyridyl)ethylene)2(2,5-di-hydroxy-1,4-benzoquinonato)2]4+23) [24]. In each case, the hexamethylbenzene complexes exhibit lower IC50 than their p-cymene analogues, probably due to the greater lipophilicity of the second [24].OO)2]2+ (OO OO) = oxalate, 2,5-dioxydo-1,4-benzoquinonato, dobq) have been synthesized in the presence of AgCF3SO3 (the synthesis details are ambiguous) [25]. The compounds are sparingly soluble in water and stable in deuterated water at 60 °C for 48 h (NMR studies) [25]. All the complexes are cytotoxic against A2780 human ovarian cancer cells, the complexes with the dobq ligand (26 and 31) being more cytotoxic than the oxalate derivatives (25 and 30); this feature shows the importance of the spacer in the cytotoxic activity [25].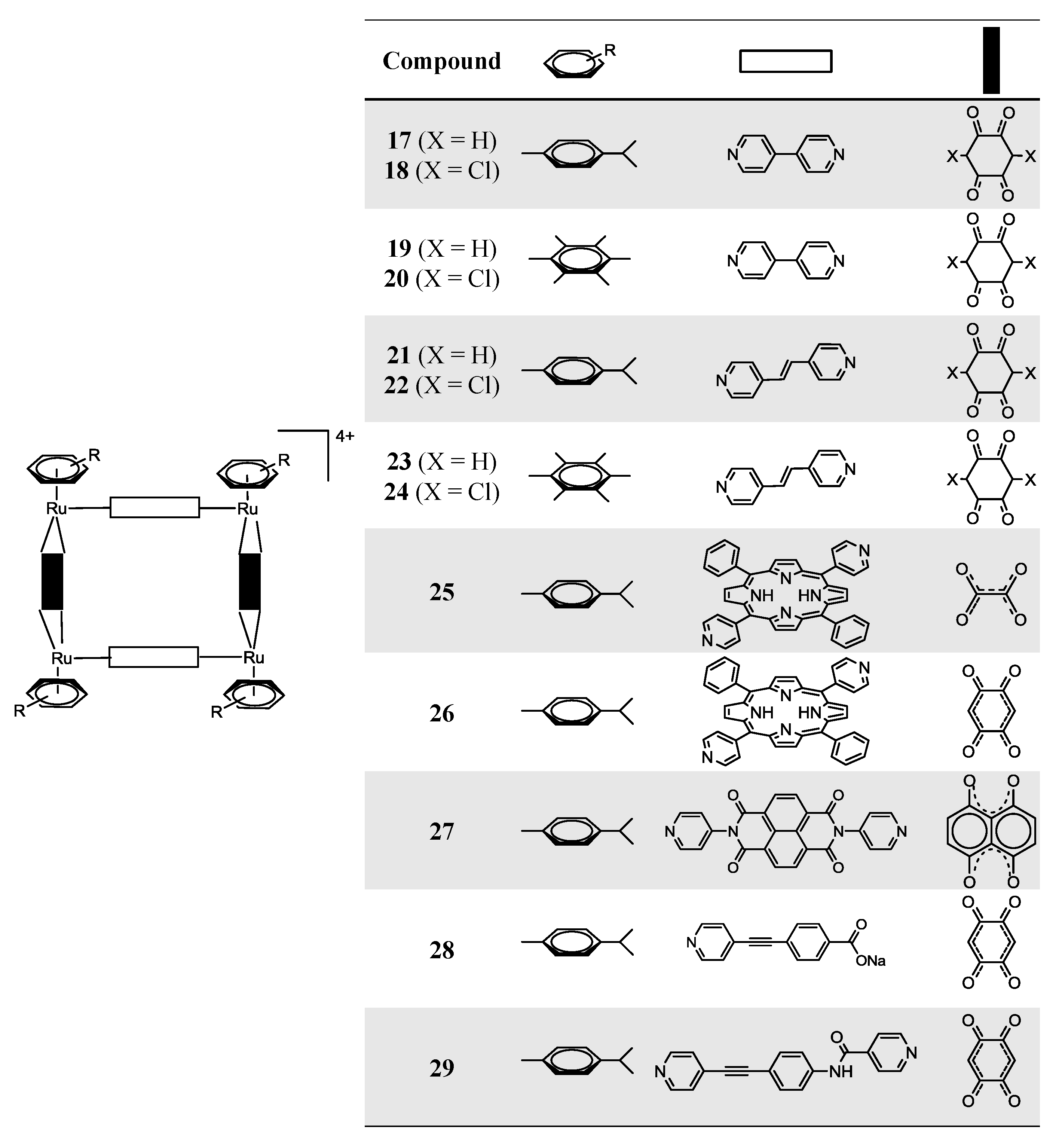
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
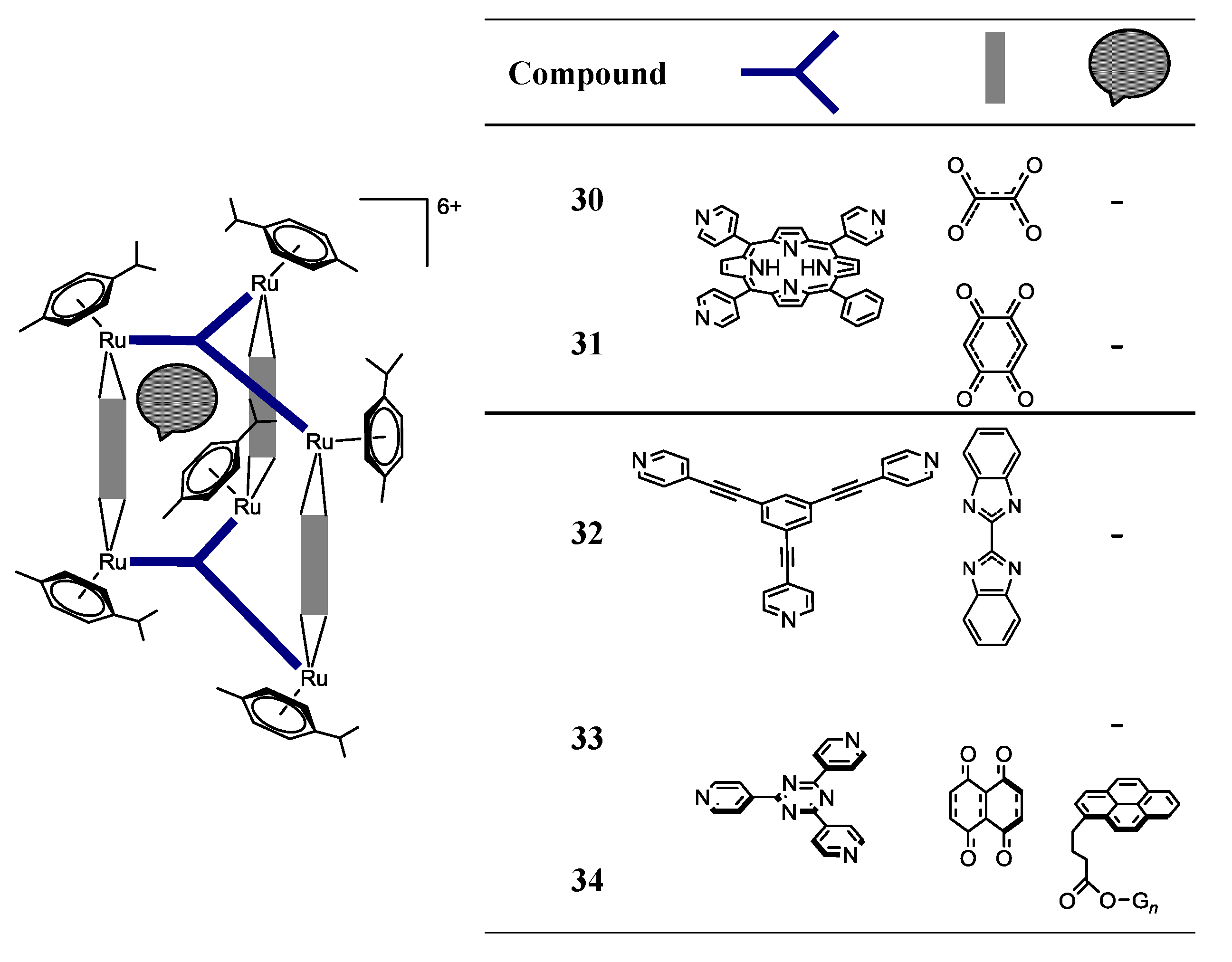
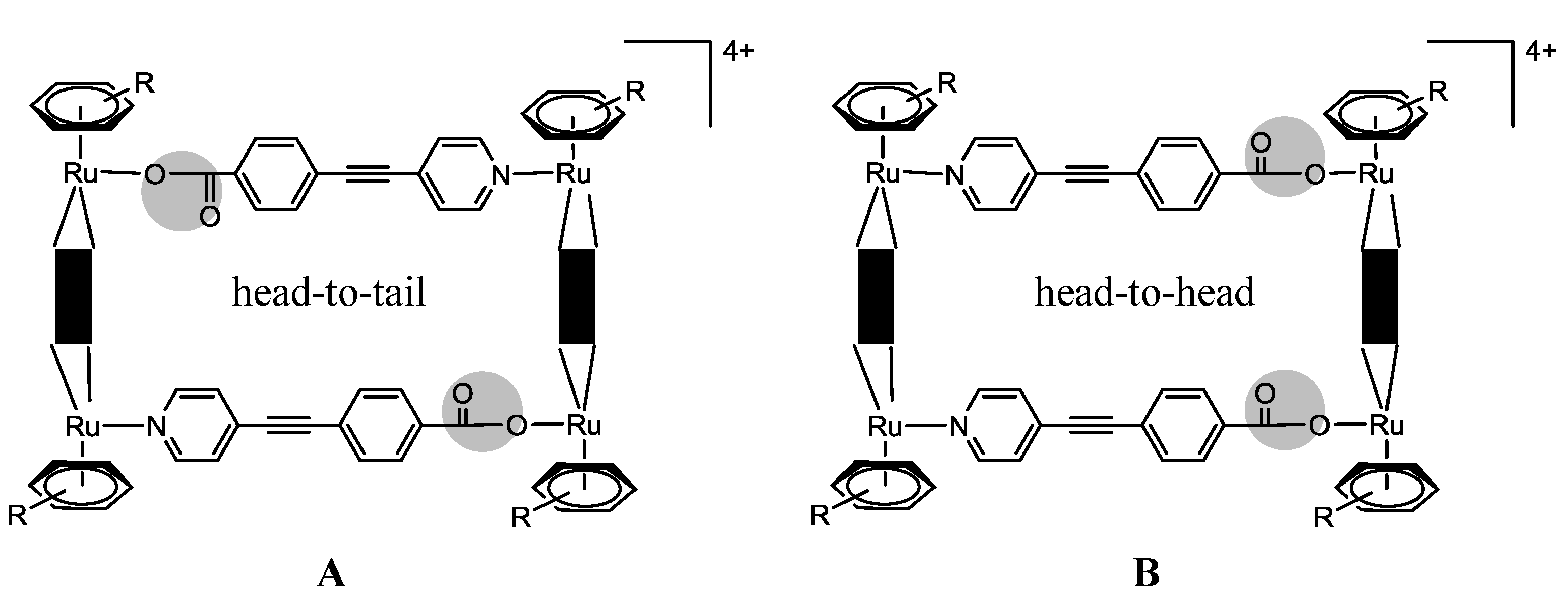 OO)Cl2] (OO OO = oxalato; dobq), affords the cationic organometallic cube, [Ru8(p-cymene)8(tpp-H2)2(OO OO)4]8+ [26]. In addition, the reaction of the dinuclear arene ruthenium dobq clips, [Ru2(indane)2(dobq)Cl2] and [Ru2(nonylbenzene)2(dobq)Cl2], in MeOH for 48 h at reflux temperature, with tpp-H2 in the presence of AgCF3SO3, affords the corresponding cationic cubes, [Ru8(indane)8(tpp-H2)2(dobq)4]8+ and [Ru8(nonylbenzene)8(tpp-H2)2(dobq)4]8+, respectively [26]. However, these octanuclear ruthenium compounds are poorly soluble in H2O and show decreased cytotoxic activity compared with their hexanuclear homologues, showing, in this case, that there is not a direct correlation between the number of ruthenium centers vs. cytotoxicity.
OO)Cl2] (OO OO = oxalato; dobq), affords the cationic organometallic cube, [Ru8(p-cymene)8(tpp-H2)2(OO OO)4]8+ [26]. In addition, the reaction of the dinuclear arene ruthenium dobq clips, [Ru2(indane)2(dobq)Cl2] and [Ru2(nonylbenzene)2(dobq)Cl2], in MeOH for 48 h at reflux temperature, with tpp-H2 in the presence of AgCF3SO3, affords the corresponding cationic cubes, [Ru8(indane)8(tpp-H2)2(dobq)4]8+ and [Ru8(nonylbenzene)8(tpp-H2)2(dobq)4]8+, respectively [26]. However, these octanuclear ruthenium compounds are poorly soluble in H2O and show decreased cytotoxic activity compared with their hexanuclear homologues, showing, in this case, that there is not a direct correlation between the number of ruthenium centers vs. cytotoxicity.| Compound | IC50 (µM) per coordination-cage |
|---|---|
| 17 a | 66 |
| 18 a | 43 |
| 19 a | 27 |
| 20 a | 33 |
| 21 a | 6 |
| 22 a | 29 |
| 23 a | 4 |
| 24 a | 23 |
| 25 b | 11 |
| 26 b | 5.6 |
| 30 b | 3.1 |
| 31 b | 2.1 |
| 33 | 3.1 |
| 34 | 2.4 |
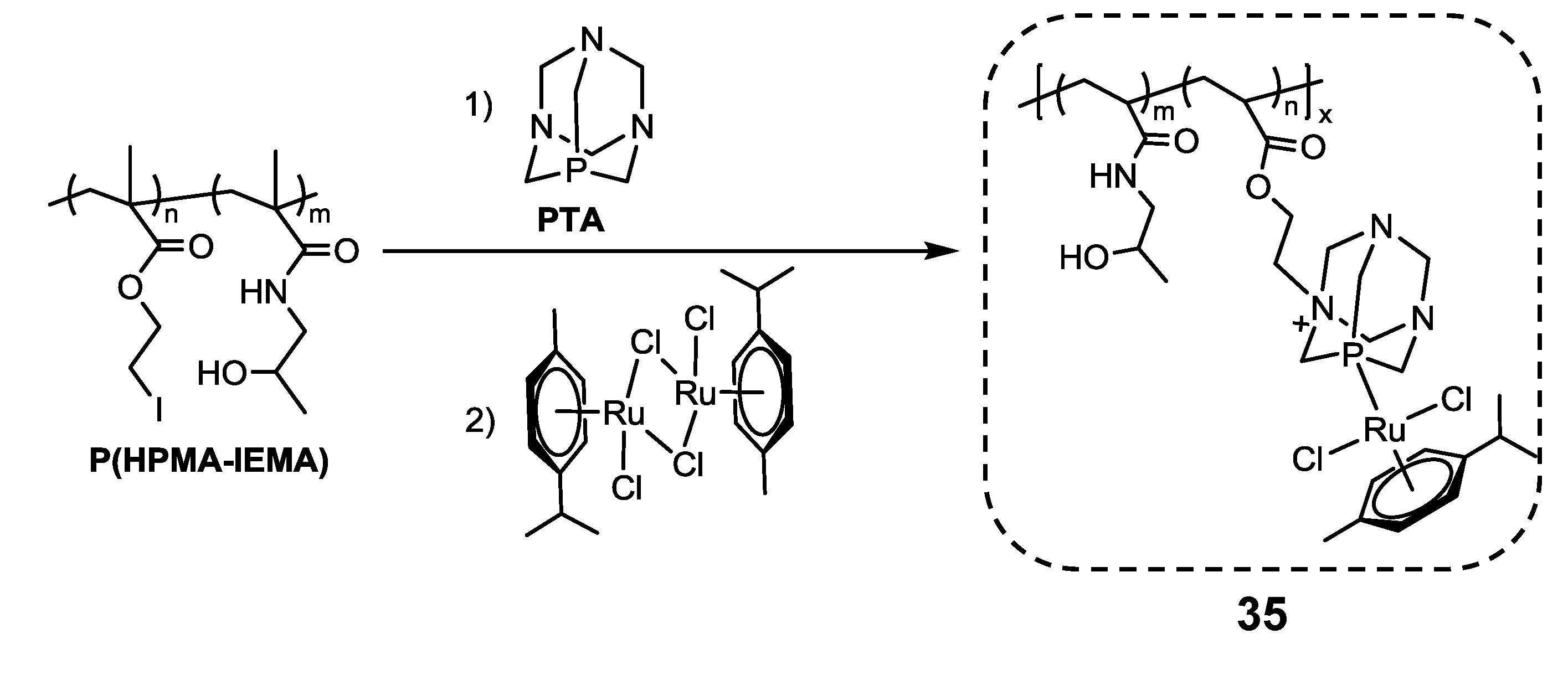
2.3. Ruthenium(II)-Coordinate Polymers

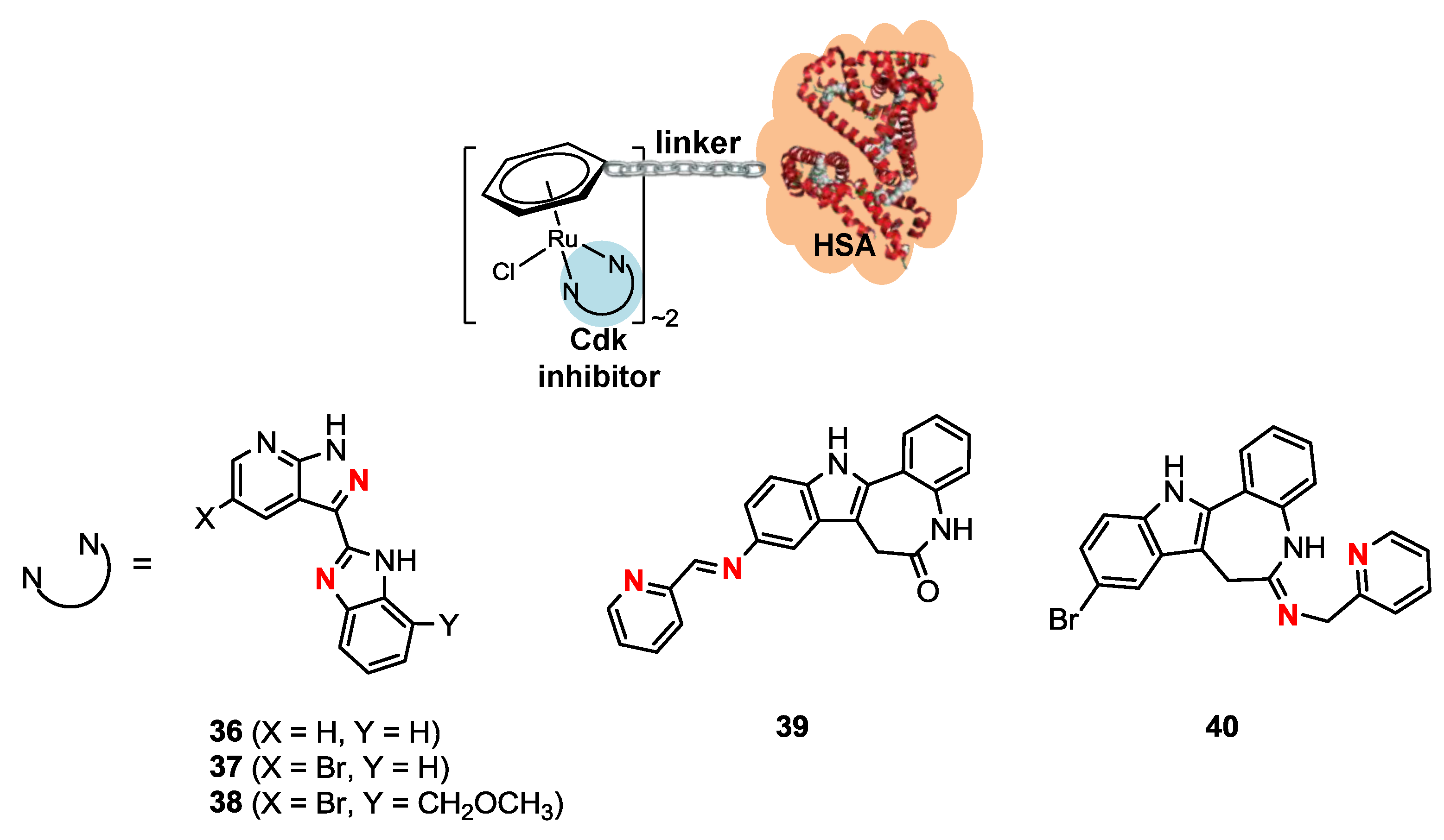
2.4. Ruthenium(II)-HSA Conjugates
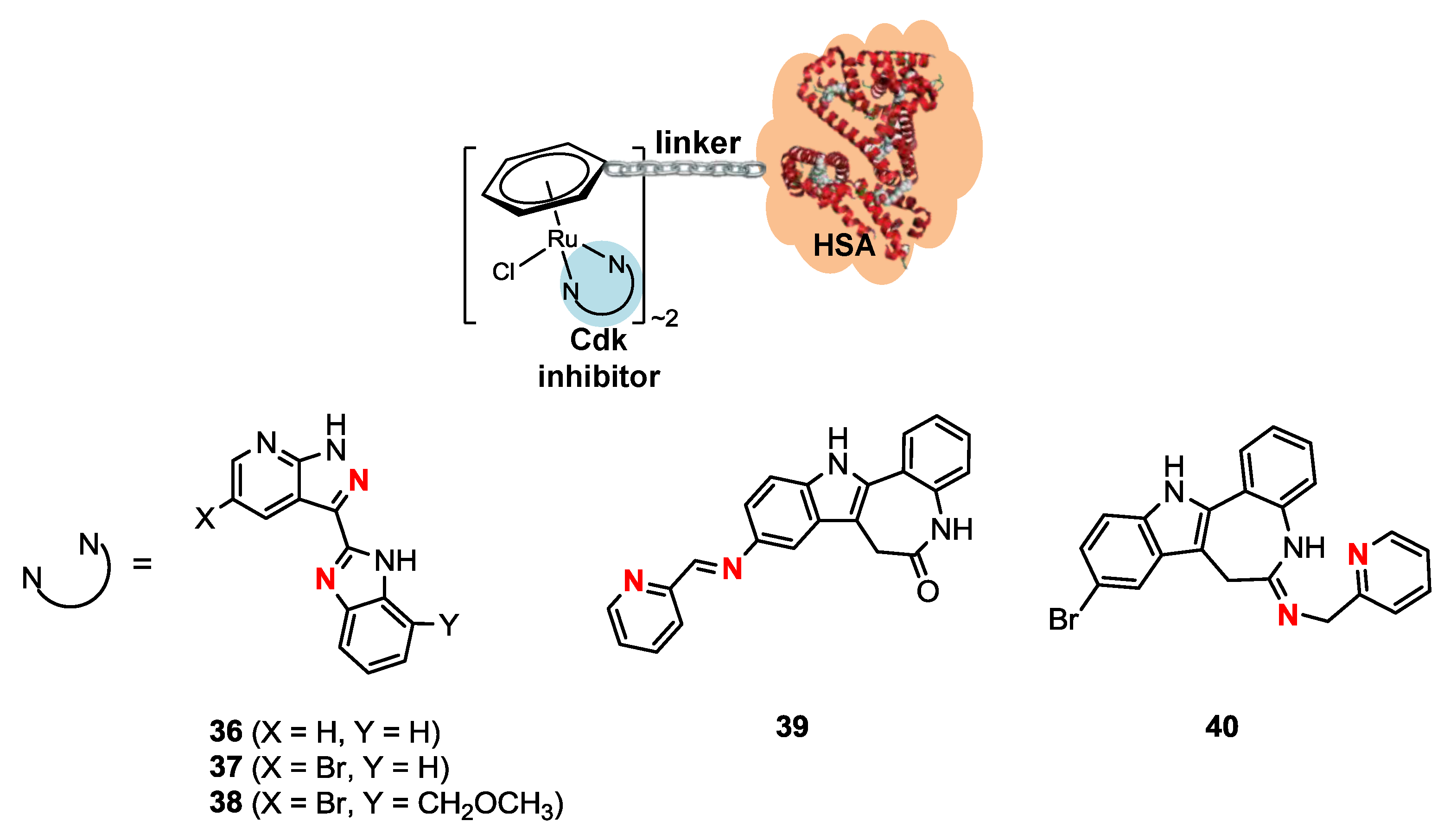
| Compound | IC50 (µM) per compound |
|---|---|
| 36 | >200 |
| 36-rHSA | 45 |
| 37 | >200 |
| 37-rHSA | 43 |
| 38 | >200 |
| 38-rHSA | 46 |
| 39 | >100 |
| 39-rHSA | 49 |
| 40 | 85 |
| 40-rHSA | 26 |
3. Mononuclear Approaches
3.1. Ruthenium Nanoparticles
| Compound | Ru nanoparticles | Mean size (nm) | IC50 (µM) |
|---|---|---|---|
| 41 |  | 8.5 | 29 |
| 42 | 2.8 | 34 | |
| 43 | 2.3 | >200 | |
| 44 | 2.2 | 39 |
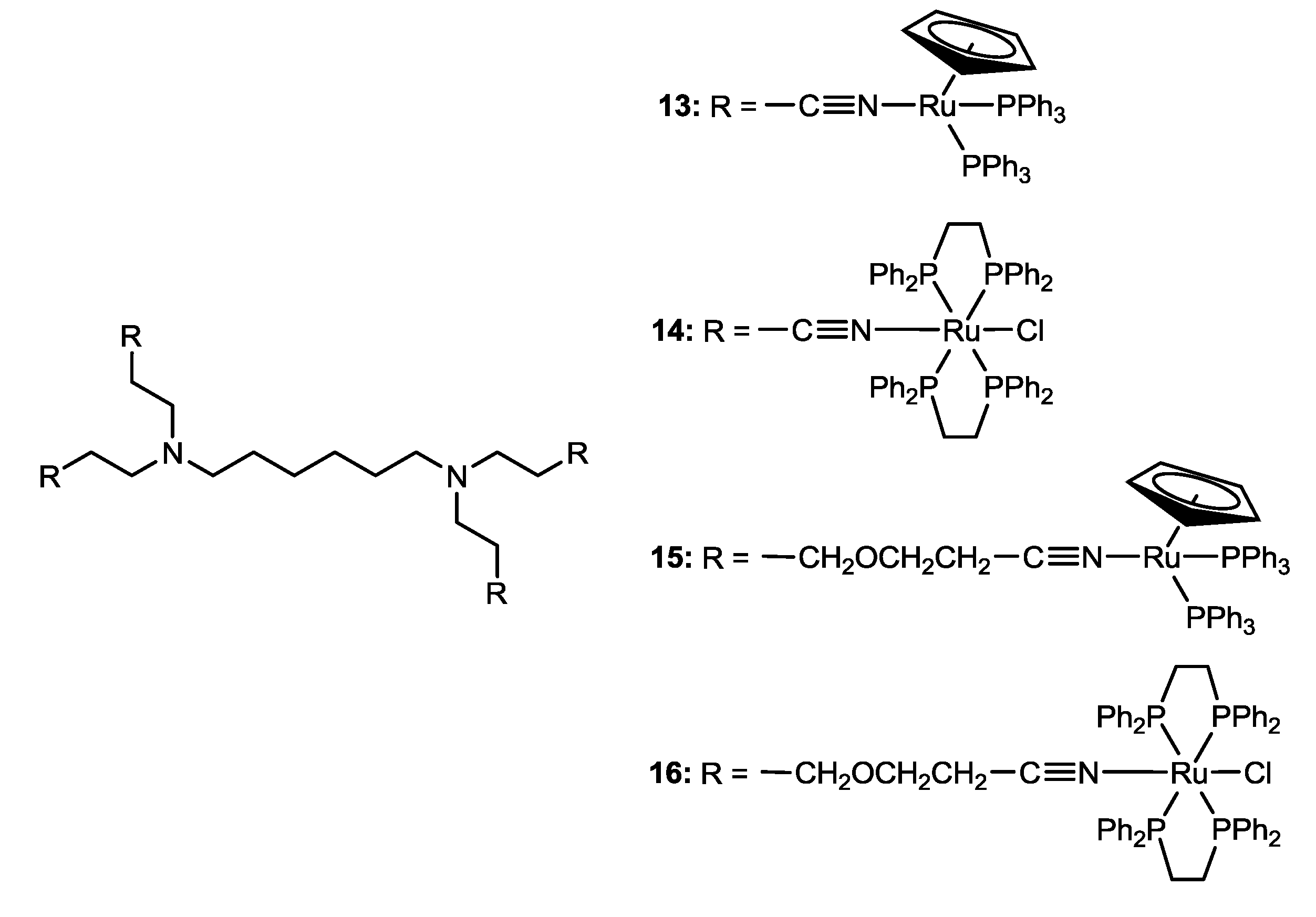
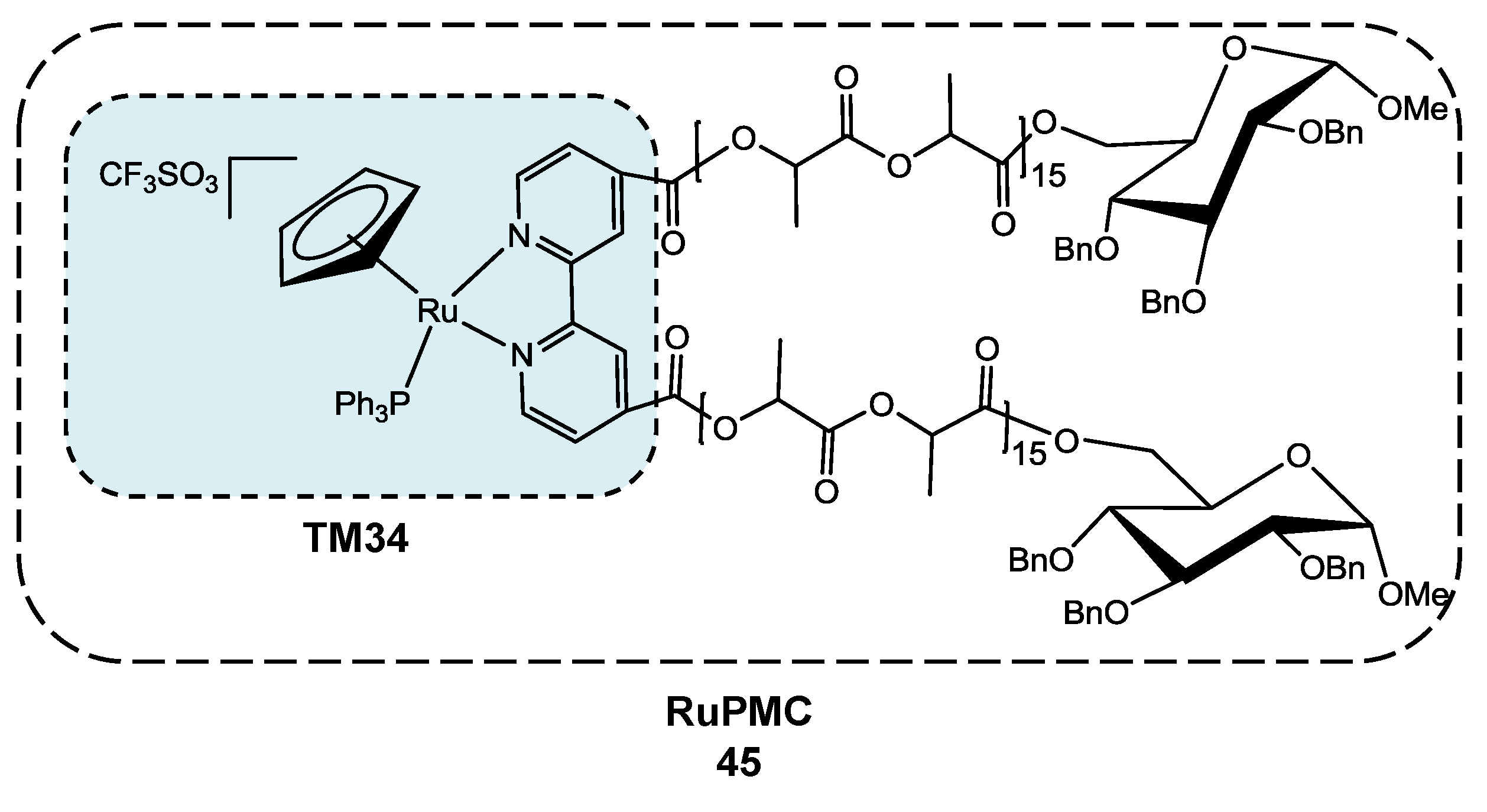
3.2. Polymer-“Ruthenium-Cyclopentadienyl” Conjugate

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chau, I.; Cunningham, D. Oxaliplatin for colorectal cancer in the united states: better late than never. J. Clin. Oncol. 2003, 21, 2049–2051. [Google Scholar] [CrossRef]
- Vicent, M.J.; Duncan, R. Polymer conjugates: Nanosized medicines for treating cancer. Trends Biotechnol. 2006, 24, 39–47. [Google Scholar] [CrossRef]
- Vinditto, V.J.; Szoka, F.C., Jr. Cancer nanomedicines: So many papers and so few drugs! Adv. Drug Deliv. Rev. 2013, 65, 80–88. [Google Scholar] [CrossRef]
- Wang, R.; Billone, P.S.; Mullett, W.M.J. Nanomedicine in action: An overview of cancer nanomedicine on the market and in clinical trials. Nanomaterials 2013, 2013, 629681. [Google Scholar]
- Zhao, X.; Loo, A.C.J.; Lee, P.P.-F.; Tan, T.T.Y.; Chu, C.K. Synthesis and cytotoxic activities of chloropyridylimineplatinum(II) and chloropyridyliminecopper(II) surface-functionalized poly(amido-amine) dendrimers. J. Inorg. Biochem. 2010, 104, 105–110. [Google Scholar] [CrossRef]
- Ahamad, T.; Mapolie, S.F.; Alshehri, S.M. Synthesis and characterization of polyamide metallodendrimers and their anti-bacterial and anti-tumor activities. Med. Chem. Res. 2012, 21, 2023–2031. [Google Scholar] [CrossRef]
- Robilotto, T.J.; Alt, D.S.; von Recum, H.A.; Gray, T.G. Cytotoxic gold(I)-bearing dendrimers from alkyne precursors. Dalton Trans. 2011, 40, 8083–8085. [Google Scholar] [CrossRef]
- Hurley, A.L.; Mohler, D.L. Organometallic photonucleases: Synthesis and DNA-cleavage studies of cyclopentadienyl metal-substituted dendrimers designed to increase double-strand scission. Org. Lett. 2000, 2, 2745–2748. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y. A new concept for macromoecular therapeutics in cancer chemotherapy: mechanisms of tumoritropic accumulation of proteins and the antitumor agent smancs. Crit. Rev. Ther. Drug. Carrier Syst. 1986, 46, 6387–6392. [Google Scholar]
- Kopecek, J.; Kopeckova, P.; Minko, T.; Lu, Z.R. HPMA copolymer-anticancer drug conjugates: Design, activity, and mechanism of action. Eur. J. Pharm. Biopharm. 2000, 50, 61–81. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar]
- Duncan, R.; Dimitrijevic, S.; Evagorou, E.G. The role of polymer conjugates in the diagnosis and treatment of cancer. STP Pharma Sci. 1996, 6, 237–263. [Google Scholar]
- Ang, W.H.; Dyson, P.J. Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur. J. Inorg. Chem. 2006, 20, 4003–4018. [Google Scholar]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Emadi, A. Ruthenium-based chemotherapeutics: Are they ready for prime time? Cancer Chemother. Pharmacol. 2010, 66, 1–9. [Google Scholar] [CrossRef]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G. Approaching tumour therapy beyond platinum drugs: Status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifrieda, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent—Preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Alessio, E.; Mestroni, G.; Bergamo, A.; Sava, G.; Alessio, E.; Mestroni, G.; Bergamo, A.; Sava, G. Ruthenium antimetastatic agents. Curr. Topics Med. Chem. 2004, 4, 1525–1535. [Google Scholar] [CrossRef]
- Govender, P.; Antonels, N.C.; Mattsson, J.; Renfrew, A.K.; Dyson, P.J.; Moss, J.R.; Therrien, B.; Smith, G.S. Anticancer activity of multinuclear arene ruthenium complexes coordinated to dendritic polypyridyl scaffolds. J. Organomet. Chem. 2009, 694, 3470–3476. [Google Scholar] [CrossRef]
- Govender, P.; Renfrew, A.K.; Clavel, C.M.; Dyson, P.J.; Therrien, B.; Smith, G.S. Antiproliferative activity of chelating N,O- and N,N-ruthenium(II) arene functionalised poly(propyleneimine) dendrimer scaffolds. Dalton Trans. 2011, 40, 1158–1167. [Google Scholar] [CrossRef]
- Rodrigues, J.; Jardim, M.G.; Figueira, J.; Gouveia, M.; Tomás, H.; Rissanen, K. Poly(alkylidenamines) dendrimers as scaffolds for the preparation of low-generation ruthenium based metallodendrimers. New J. Chem. 2011, 35, 1938–1943. [Google Scholar] [CrossRef]
- Mattsson, J.; Govindaswamy, P.; Renfrew, A.K.; Dyson, P.J.; Štěpnička, P.; Süss-Fink, G.; Therrien, B. Synthesis, molecular structure, and anticancer activity of cationic arene ruthenium metallarectangles. Organometallics 2009, 28, 4350–4357. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Zava, O.; Furrer, J.; Dyson, P.J.; Therrien, B. Anticancer activity of opened arene ruthenium metalla-assemblies. Dalton Trans. 2010, 39, 5272–5277. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Zava, O.; Dyson, P.J.; Therrien, B. Synthesis, characterization and anticancer activity of porphyrin-containing organometallic cubes. Aust. J. Chem. 2010, 63, 1529–1537. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Barry, N.P.E.; Zava, O.; Deschenaux, R.; Therrien, B. Encapsulation of pyrene-functionalized poly(benzyl ether) dendrons into a water-soluble organometallic cage. Chem. Asian J. 2011, 6, 1595–1603. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Barry, N.P.E.; Zava, O.; Deschenaux, R.; Dyson, P.J.; Therrien, B. Double targeting of tumours with pyrenyl-modified dendrimers encapsulated in an arene-ruthenium metallaprism. Chem. Eur. J. 2011, 17, 1966–1971. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Edafe, F.; Therrien, B. Anticancer activity of tetracationic arene ruthenium metalla-cycles. Dalton Trans. 2011, 40, 7172–7180. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Zava, O.; Dyson, P.J.; Therrien, B. Excellent correlation between drug release and portal size in metalla-cage drug-delivery systems. Chem. Eur. J. 2011, 17, 9669–9667. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Zava, O.; Dyson, P.J.; Deschenaux, R.; Therrien, B. Enhancement of cytotoxicity by combining pyrenyl-dendrimers and arene ruthenium metallacages. Inorg. Chem. 2012, 51, 7119–7124. [Google Scholar] [CrossRef]
- Furrer, M.A.; Garci, A.; Denoyelle-Di-Muro, E.; Trouillas, P.; Giannini, F.; Furrer, J.; Clavel, C.M.; Dyson, P.J.; Süss-Fink, G.; Therrien, B. Synthesis, characterisation and in vitro anticancer activity of hexanuclear thiolato-bridged arene ruthenium metalla-prisms. Chem. Eur. J. 2013, 19, 3198–3203. [Google Scholar] [CrossRef]
- Dubey, A.; Min, J.W.; Koo, H.J.; Kim, H.; Cook, T.R.; Kang, S.C.; Stang, P.J.; Chi, K.-W. Anticancer potency and multi-resistant studies of self-assembled arene-ruthenium metallarectangles. Chem. Eur. J. 2013, 19, 11622–11628. [Google Scholar] [CrossRef]
- Jung, H.; Dubey, A.; Koo, H.J.; Vajpayee, V.; Cook, T.R.; Kim, H.; Kang, S.C.; Stang, P.J.; Chi, K.-W. Self-assembly of ambidentate pyridyl-carboxylate ligands with octahedral ruthenium metal centers: Self-selection for a single-linkage isomer and anticancer-potency studies. Chem. Eur. J. 2013, 19, 6709–6717. [Google Scholar] [CrossRef]
- Mishra, A.; Jung, H.; Park, J.W.; Kim, H.K.; Kim, H.; Stang, P.J.; Chi, K.-W. Anticancer activity of self-assembled molecular rectangles via arene-ruthenium acceptors and a new unsymmetrical amide ligand. Organometallics 2012, 31, 3519–3526. [Google Scholar] [CrossRef]
- Vajpayee, V.; Lee, S.; Kim, S.-H.; Kang, S.C.; Cook, T.R.; Kim, H.; Kim, D.W.; Verma, S.; Lah, M.S.; Kim, I.S.; et al. Self-assembled metala-rectangles bearing azodipyridyl ligands: Synthesis, characterization and antitumor activity. Dalton Trans. 2013, 42, 466–475. [Google Scholar] [CrossRef]
- Vajpayee, V.; mi Lee, S.; Park, J.W.; Dubey, A.; Kim, H.; Cook, T.R.; Stang, P.J.; Chi, K.-W. Growth inhibitory activity of a bis-benzimidazole-bridged arene ruthenium metalla-rectangle and-prism. Organometallics 2013, 32, 1563–1566. [Google Scholar] [CrossRef]
- Cook, T.R.; Vajpayee, V.; Lee, M.H.; Stang, P.J.; Chi, K.-W. Biomedical and biochemical applications of self-assembled metallacycles and metallacages. Acc. Chem. Res. 2013, 46, 2464–2474. [Google Scholar] [CrossRef]
- Han, Y.-F.; Jia, W.-G.; Lin, Y.-J.; Jin, G.-X. Stepwise formation of molecular rectangles of half-sandwich rhodium and ruthenium complexes containing bridging chloranilate ligands. Organometallics 2008, 27, 5002–5008. [Google Scholar] [CrossRef]
- Blunden, B.M.; Thomas, D.S.; Stenzel, M.H. Macromolecular ruthenium complexes as anti-cancer agents. Polym. Chem. 2012, 3, 2964. [Google Scholar] [CrossRef]
- Stepanenko, I.N.; Casini, A.; Edafe, F.; Novak, M.S.; Arion, V.B.; Dyson, P.J.; Jakupec, M.A.; Keppler, B.K. Conjugation of organoruthenium(II) 3-(1H-benzimidazol-2-yl)pyrazolo[3,4-b]pyridines and indolo[3,2-d]benzazepines to recombinant human serum albumin: A strategy to enhance cytotoxicity in cancer cells. Inorg. Chem. 2011, 50, 12669–12679. [Google Scholar] [CrossRef]
- Warnecke, A.; Fichtner, I.; Garmann, D.; Jaehde, U.; Kratz, F. Synthesis and biological activity of water-soluble maleimide derivatives of the anticancer drug carboplatin designed as albumin-binding prodrugs. Bioconjugate Chem. 2004, 15, 1349–1359. [Google Scholar] [CrossRef]
- Süss-Fink, G.; Khan, F.-A.; Juillerat-Jeanneret, L.; Dyson, P.J.; Renfrew, A.K. Synthesis and anticancer activity of long-chain isonicotinic ester ligand-containing arene ruthenium complexes and nanoparticles. J. Clus. Sci. 2010, 21, 313–324. [Google Scholar] [CrossRef]
- Garcia, M.H.; Morais, T.S.; Florindo, P.; Piedade, M.F.M.; Moreno, V.; Ciudad, C.; Noe, V. Inhibition of cancer cell growth by ruthenium(II) cycplopentadienyl derivative complexes with heteroaromatic ligands, journal of inorganic biochemistry. J. Inorg. Biochem. 2009, 103(3), 354–361. [Google Scholar]
- Garcia, M.H.; Valente, A.; Florindo, P.; Morais, T.S.; Piedade, M.F.M.; Duarte, M.T.; Moreno, V. New ruthenium(II) mixed metallocene derived complexes: Synthesis, characterization by X-ray diffraction studies and evaluation on DNA interaction by atomic force microcopy. Inorg. Chim. Acta 2010, 363, 3765–3775. [Google Scholar] [CrossRef]
- Moreno, V.; Lorenzo, J.; Avilés, F.X.; Garcia, M.H.; Ribeiro, J.; Florindo, P.; Robalo, M.P. Studies of the antiproliferative activity of ruthenium(II) cyclopentadienyl derived complexes with nitrogen coordinated ligands. Bioinorg. Chem. Appl. 2010. [Google Scholar] [CrossRef]
- Moreno, V.; Font-Bardia, M.; Calvet, T.; Lorenzo, J.; Avilés, F.X.; Garcia, M.H.; Morais, T.S.; Valente, A.; Robalo, M.P. DNA interaction and cytotocixity studies of new ruthenium(II) cyclopentadienyl derivative complexes containing heteroaromatic ligands. J. Inorg. Biochem. 2011, 105(2), 241–249. [Google Scholar]
- Tomaz, A.I.; Jakusch, T.; Morais, T.S.; Marques, F.; Almeida, R.F.M.; de Mendes, F.; Enyedy, E.A.; Santos, I.; Pessoa, J.C.; Kiss, T.; et al. [RuII(η5-C5H5)(bipy)(PPh3)]+ a promising large spectrum antitumor agent: cytotoxic activity and interaction with human serum albumin. J. Inorg. Biochem. 2012, 117, 261–269. [Google Scholar]
- Morais, T.S.; Silva, T.J.L.; Marques, F.; Robalo, M.P.; Avecilla, F.; Madeira, P.J.A.; Mendes, P.J.G.; Santos, I.; Garcia, M.H. Synthesis of organometallic ruthenium(II) complexes with strong activity against several human cancer cell lines. J. Inorg. Biochem. 2012, 114, 65–74. [Google Scholar] [CrossRef]
- Morais, T.S.; Santos, F.; Côrte-Real, L.; Marques, F.; Robalo, M.P.; Madeira, P.J.A.; Garcia, M.H. Biological activity and cellular uptake of [RuII(η5-C5H5)(PPh3)(Me2bpy)][CF3SO3]. J. Inorg. Biochem. 2013, 122, 8–17. [Google Scholar] [CrossRef]
- Morais, T.S.; Santos, F.C.; Jorge, T.F.; Côrte-Real, L.; Madeira, P.J.A.; Marques, F.; Robalo, M.P.; Matos, A.; Santos, I.; Garcia, M.H. New water-soluble ruthenium(II) cytotoxic complex: Biological activity and cellular distribution. J. Inorg. Biochem. 2013, 130, 1–14. [Google Scholar]
- Côrte-Real, L.; Matos, A.P.; Alho, I.; Morais, T.S.; Tomaz, A.I.; Garcia, M.H.; Santos, I.; Bicho, M.P.; Marques, F. Cellular uptake mechanisms of an antitumor ruthenium compound: The endossomal/lysosomal system as a target for anticancer metal-based drugs. Microsc.Microanal. 2013, 19, 1122–1130. [Google Scholar] [CrossRef]
- Duncan, R. Designing polymer conjugates as lysosomotropic nanomedicines. Biochem. Soc. Trans. 2007, 35, 56–60. [Google Scholar] [CrossRef]
- Valente, A.; Garcia, M.H.; Marques, F.; Miao, Y.; Rousseau, C.; Zinck, P. First polymer “ruthenium-cyclopentadienyl” complex as potential anticancer agent. J. Inorg. Biochem. 2013, 127, 79–81. [Google Scholar] [CrossRef]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Cabral, H.; Nishiyama, N.; Okazaki, S.; Koyama, H.; Kataoka, K. Preparation and biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-loaded polymeric micelles. J. Control. Rel. 2005, 101, 223–232. [Google Scholar] [CrossRef]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Hayashi, F.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef]
- Nishiyama, N.; Kataoka, K. Preparation and characterization of size-controlled polymeric micelle containing cis-dichlorodiammineplatinum(II) in the core. J. Control. Rel. 2001, 74, 83–94. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, Y.-S.; Park, K.; Lee, S.; Nam, H.Y.; Min, K.H.; Lo, H.G.; Park, J.H.; Choi, K.; Jeong, S.Y.; et al. Antitumor efficacy of cisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J. Control. Rel. 2008, 127, 41–49. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valente, A.; Garcia, M.H. Syntheses of Macromolecular Ruthenium Compounds: A New Approach for the Search of Anticancer Drugs. Inorganics 2014, 2, 96-114. https://doi.org/10.3390/inorganics2010096
Valente A, Garcia MH. Syntheses of Macromolecular Ruthenium Compounds: A New Approach for the Search of Anticancer Drugs. Inorganics. 2014; 2(1):96-114. https://doi.org/10.3390/inorganics2010096
Chicago/Turabian StyleValente, Andreia, and M. Helena Garcia. 2014. "Syntheses of Macromolecular Ruthenium Compounds: A New Approach for the Search of Anticancer Drugs" Inorganics 2, no. 1: 96-114. https://doi.org/10.3390/inorganics2010096
APA StyleValente, A., & Garcia, M. H. (2014). Syntheses of Macromolecular Ruthenium Compounds: A New Approach for the Search of Anticancer Drugs. Inorganics, 2(1), 96-114. https://doi.org/10.3390/inorganics2010096