Microflow Photochemistry—Photodecarboxylations in Microformats



Abstract
:1. Introduction
2. Experimental Section

2.1. General Procedure for Photodecarboxylative Cyclizations
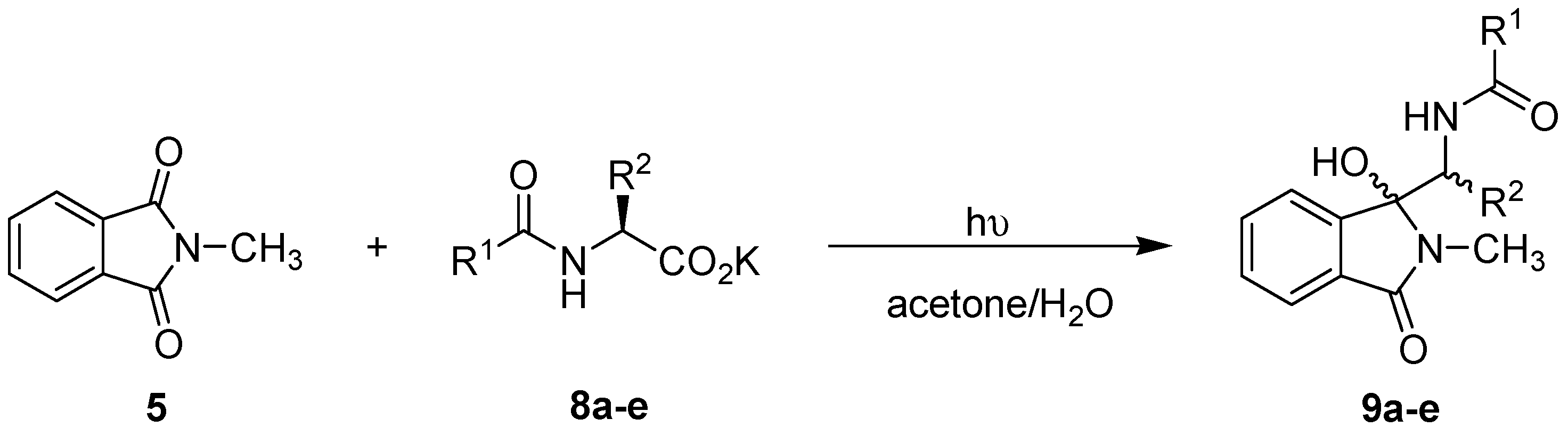
2.2. General Procedure for Photodecarboxylative Additions to N-methylphthalimide
3. Results and Discussion
3.1. Intramolecular Photodecarboxylative Cyclization Reactions

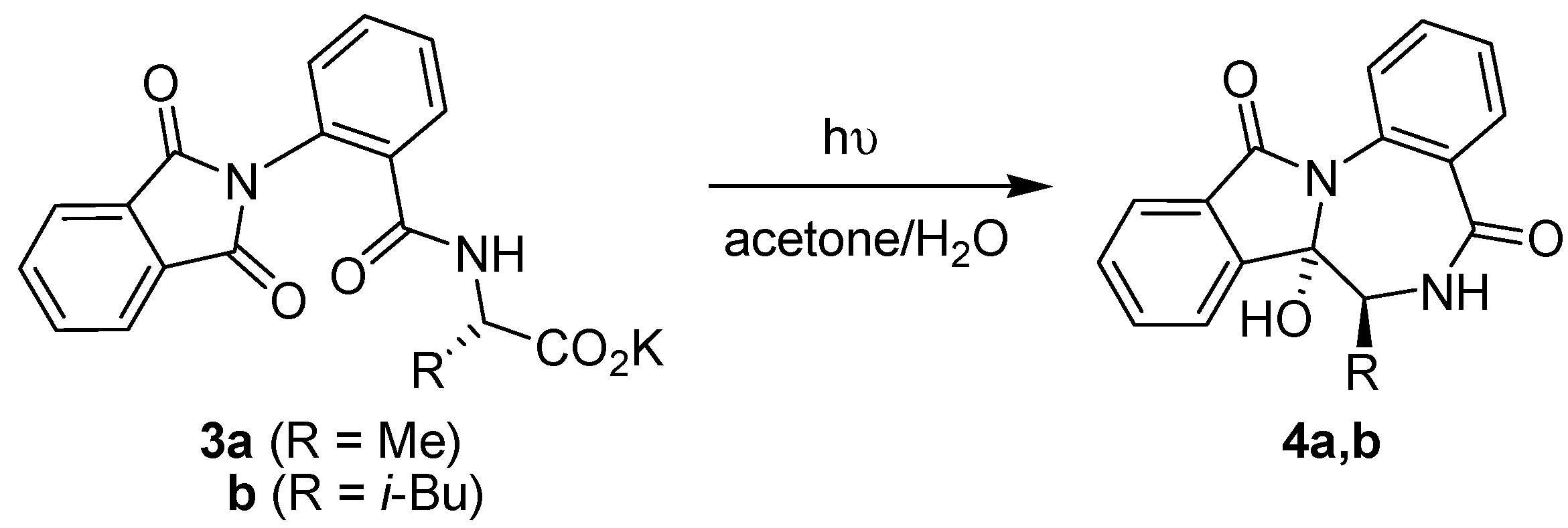

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Batch reactor | μ-Reactor | ||
|---|---|---|---|---|
| Time (h) | Yield of 4 (%) | Time (min) | Yield of 4 (%) | |
| 3a | 4 | 40 | 168 a | 35 |
| 3b | 2.5 | 58 | 56 a | 87 |
3.2. Intermolecular Photodecarboxylative Addition Reactions

| Entry | R1 | R2 | R3 | Batch reactor | μ-Reactor | ||
|---|---|---|---|---|---|---|---|
| Time (h) | Yield of 7 (%) | Time (min) | Yield of 7 (%) | ||||
| 6a | H | H | H | 1 | 80 | 42 | 98 a |
| 6b | CH3 | H | H | 4 | 53 | 240 | 93 a |
| 6c | F | H | H | 4 | 39 | 240 | 53 |
| 6d | H | H | Br | 5 | 35 | 280 | 60 |
| 6e | H | H | I | 4 | 51 | 168 b | 23 |
| 6f | Cl | Cl | H | 3 | 53 | 180 | 93 a |

| Entry | R1 | R2 | Batch reactor | μ-Reactor | ||
|---|---|---|---|---|---|---|
| Time (h) | Yield of 9 (%) | Time (min) | Yield of 9 (%) | |||
| 8a | CH3 | i-Pr | 3.5 | 52 | 210 | 58 |
| 8b | CH3 | i-Bu | 3 | 26 | 42 a | 18 |
| 8c | CH3 | sec-Bu | 3 | 78 | 21 a | 27 |
| 8d | t-BuO | CH3 | 3 | 65 | 168 a | 93 |
| 8e | t-BuO | Bn | 3 | 34 | 168 a | 53 |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Oelgemöller, M. Highlights of photochemical reactions in microflow reactors. Chem. Eng. Technol. 2012, 35, 1144–1152. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Shvydkiv, O. Recent advances in microflow photochemistry. Molecules 2011, 16, 7522–7550. [Google Scholar] [CrossRef]
- Coyle, E.E.; Oelgemöller, M. Micro-photochemistry: Photochemistry in microstructured reactors. The new photochemistry of the future? Photochem. Photobiol. Sci. 2008, 7, 1313–1322. [Google Scholar] [CrossRef]
- Sugimoto, A.; Fukuyama, T.; Sumino, Y.; Takagi, M.; Ryu, I. Microflow photo-radical reaction using a compact light source: Application to the Barton reaction leading to a key intermediate for myriceric acid A. Tetrahedron 2009, 65, 1593–1598. [Google Scholar] [CrossRef]
- Aillet, T.; Loubiere, K.; Dechy-Cabaret, O.; Prat, L. Photochemical synthesis of a “cage” compound in a microreactor: Rigorous comparison with a batch photoreactor. Chem. Eng. Proc. 2013, 64, 38–47. [Google Scholar] [CrossRef]
- Maeda, H.; Nashihara, S.; Mukae, H.; Yoshimi, Y.; Mizuno, K. Improved efficiency and product selectivity in the photo-Claisen-type rearrangement of an aryl naphthylmethyl ether using a microreactor/flow system. Res. Chem. Intermed. 2013, 39, 301–310. [Google Scholar] [CrossRef]
- Terao, K.; Nishiyama, Y.; Tanimoto, H.; Morimoto, T.; Oelgemöller, M.; Kakiuchi, K. Diastereoselective [2 + 2] photocycloaddition of a chiral cyclohexenone with ethylene in a continuous flow microcapillary reactor. J. Flow Chem. 2012, 2, 73–76. [Google Scholar] [CrossRef]
- Bach, T.; Hehn, J.P. Photochemical reactions as key steps in natural product synthesis. Angew. Chem. Int. Ed. 2011, 50, 1000–1045. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical reactions as key steps in organic synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef]
- Iriondo-Alberdi, J.; Greaney, M.F. Photocycloaddition in natural product synthesis. Eur. J. Org. Chem. 2007, 4801–4815. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Synthetic applications of photoinduced electron transfer decarboxylation reactions. Synlett 1999, 1999, 1169–1178. [Google Scholar] [CrossRef]
- Kramer, W.; Griesbeck, A.G.; Nerowski, F.; Oelgemöller, M. Synthetic potential of the PET-decarboxylation of ω-phthalimido carboxylic acids. J. Inf. Rec. 1998, 24, 81–85. [Google Scholar]
- Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C.A.; Oelgemöller, M. Photodecarboxylative benzylations of phthalimide in pH 7 buffer: A simple access to 3-arylmethyleneisoindolin-1-ones. Tetrahedron Lett. 2010, 51, 4738–4741. [Google Scholar] [CrossRef]
- Hatoum, F.; Engler, J.; Zelmer, C.; Wißen, J.; Motti, C.A.; Lex, J.; Oelgemöller, M. Photodecarboxylative addition of carboxylates to phthalimides: A concise access to biologically active 3-(aryl and alkyl)methylene-1H-isoindolin-1-ones. Tetrahedron Lett. 2012, 53, 5573–5577. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Photoinduced decarboxylation reactions. Radical chemistry in water. Green Chem. 1999, 1, 205–207. [Google Scholar] [CrossRef]
- Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. From conventional to microphotochemistry: Photodecarboxylation reactions involving phthalimides. Org. Lett. 2010, 12, 5170–5173. [Google Scholar] [CrossRef]
- Shvydkiv, O.; Nolan, K.; Oelgemöller, M. Microphotochemistry—4,4'-dimethoxybenzophenone mediated photodecarboxylation reactions involving phthalimides. Beilstein J. Org. Chem. 2011, 7, 1055–1063. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Henz, A.; Kramer, W.; Lex, J.; Nerowski, F.; Oelgemöller, M.; Peters, K.; Peters, E.-M. Synthesis of medium- and large-ring compounds initiated by photochemical decarboxylation of ω-phthalimidoalkanoates. Helv. Chim. Acta 1997, 80, 912–933. [Google Scholar] [CrossRef]
- Hatoum, F.; Gallagher, S.; Baragwanath, L.; Lex, J.; Oelgemöller, M. Photodecarboxylative benzylations of phthalimides. Tetrahedron Lett. 2009, 50, 6335–6338. [Google Scholar] [CrossRef]
- Sato, Y.; Nakai, H.; Mizoguchi, T.; Kawanishi, M.; Kanaoka, Y. Photochemistry of the phthalimide system. I. Photodecarboxylation of N-phthaloyl-α-amino acids. Chem. Pharm. Bull. 1973, 21, 1164–1166. [Google Scholar]
- Griesbeck, A.G.; Henz, A.; Peters, K.; Peters, E.-M.; von Schnering, H.G. Photo electron transfer induced macrocyclization of N-phthaloyl-ω-amino-carboxylic acids. Angew. Chem. Int. Ed. 1995, 34, 474–476. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Lex, J. Stereoselective synthesis of 1,4-benzodiazepins via photoinduced decarboxylation of N-phthaloylanthranilic acid amides. Synthesis 2001, 1159–1166. [Google Scholar] [CrossRef]
- Wagner, P.J. Conformational flexibility and photochemistry. Acc. Chem. Res. 1983, 16, 461–467. [Google Scholar] [CrossRef]
- Huang, F.; Nau, W.M. A conformational flexibility scale for amino acids in peptides. Angew. Chem. Int. Ed. 2003, 42, 2269–2272. [Google Scholar] [CrossRef]
- Yokoi, H.; Nakano, T.; Fujita, W.; Ishiguro, K.; Sawaki, Y. In-cage formation of carbanions in photoinduced electron-transfer reaction of carboxylate ions. J. Am. Chem. Soc. 1998, 120, 12453–12458. [Google Scholar] [CrossRef]
- Jackson, R.A.; Sharifi, M. Stabilization of benzylic radicals by substituents: An EPR study of para-substituted benzyl radicals. J. Chem. Soc., Perkin Trans. 1996, 2, 775–778. [Google Scholar] [CrossRef]
- Singh, N.K.; Popelier, P.L.A.; O’Malley, P.J. Substituent effects on the stability of para substituted benzyl radicals. Chem. Phys. Lett. 2006, 426, 219–221. [Google Scholar] [CrossRef]
- Suryan, M.M.; Stein, S.E. Stabilities of substituted benzyl radicals: Dissociation rates of amino-, hydroxy-, and cyanoethylbenzenes. J. Phys. Chem. 1989, 93, 7362–7365. [Google Scholar] [CrossRef]
- Gallagher, S.; Hatoum, F.; Zientek, N.; Oelgemöller, M. Photodecarboxylative additions of N-protected α-amino acids to N-methylphthalimide. Tetrahedron Lett. 2010, 51, 3639–3641. [Google Scholar] [CrossRef]
- Schaich, K.M. Free radical initiation in proteins and amino acids by ionizing and ultraviolet radiations and lipid oxidation—Part II: Ultraviolet radiation and photolysis. CRC Critical Rev. Food Sci. Nutr. 1980, 13, 131–159. [Google Scholar] [CrossRef]
- Ho, J.; Coote, M.L.; Easton, C.J. The distal effect of N-electron-withdrawing groups on the stability of peptide carbon radicals. Aust. J. Chem. 2011, 64, 403–408. [Google Scholar] [CrossRef]
- White, D.P.; Anthony, J.C.; Oyefeso, A.O. Computational measurement of steric effects: The size of organic substituents computed by Ligand Repulsive Energies. J. Org. Chem. 1999, 64, 7707–7716. [Google Scholar] [CrossRef]
- Gallagher, S. From conventional to microphotochemistry: A study of phthalimide and phthalonitrile derivatives. Ph.D. Thesis, Dublin City University, Dublin, Ireland, April 2011. [Google Scholar]
- Newman, S.G.; Jensen, K.F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. [Google Scholar] [CrossRef]
- Chin, P.; Barney, W.S.; Pindzola, B.A. Microstructured reactors as tools for the intensification of pharmaceutical reactions and processes. Curr. Opin. Drug Dis. Dev. 2009, 12, 848–861. [Google Scholar]
- Werner, S.; Seliger, R.; Rauter, H.; Wissmann, F. Quarzglas-mikrophotoreaktor und synthese von 10-hydroxycamptothecin und 7-alkyl-10-hydroxycamptothecin. EP-2065387A2, 2009. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oelgemöller, M.; Gallagher, S.; McCarthy, K. Microflow Photochemistry—Photodecarboxylations in Microformats. Processes 2014, 2, 158-166. https://doi.org/10.3390/pr2010158
Oelgemöller M, Gallagher S, McCarthy K. Microflow Photochemistry—Photodecarboxylations in Microformats. Processes. 2014; 2(1):158-166. https://doi.org/10.3390/pr2010158
Chicago/Turabian StyleOelgemöller, Michael, Sonia Gallagher, and Kevin McCarthy. 2014. "Microflow Photochemistry—Photodecarboxylations in Microformats" Processes 2, no. 1: 158-166. https://doi.org/10.3390/pr2010158