Oroxylum indicum Seeds—Analysis of Flavonoids by Micellar Electrokinetic Chromatography


Abstract
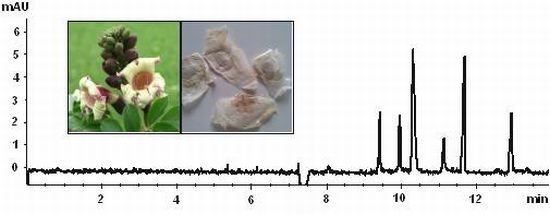
:
1. Introduction
2. Experimental Section
2.1. Chemicals, References and Plant Material
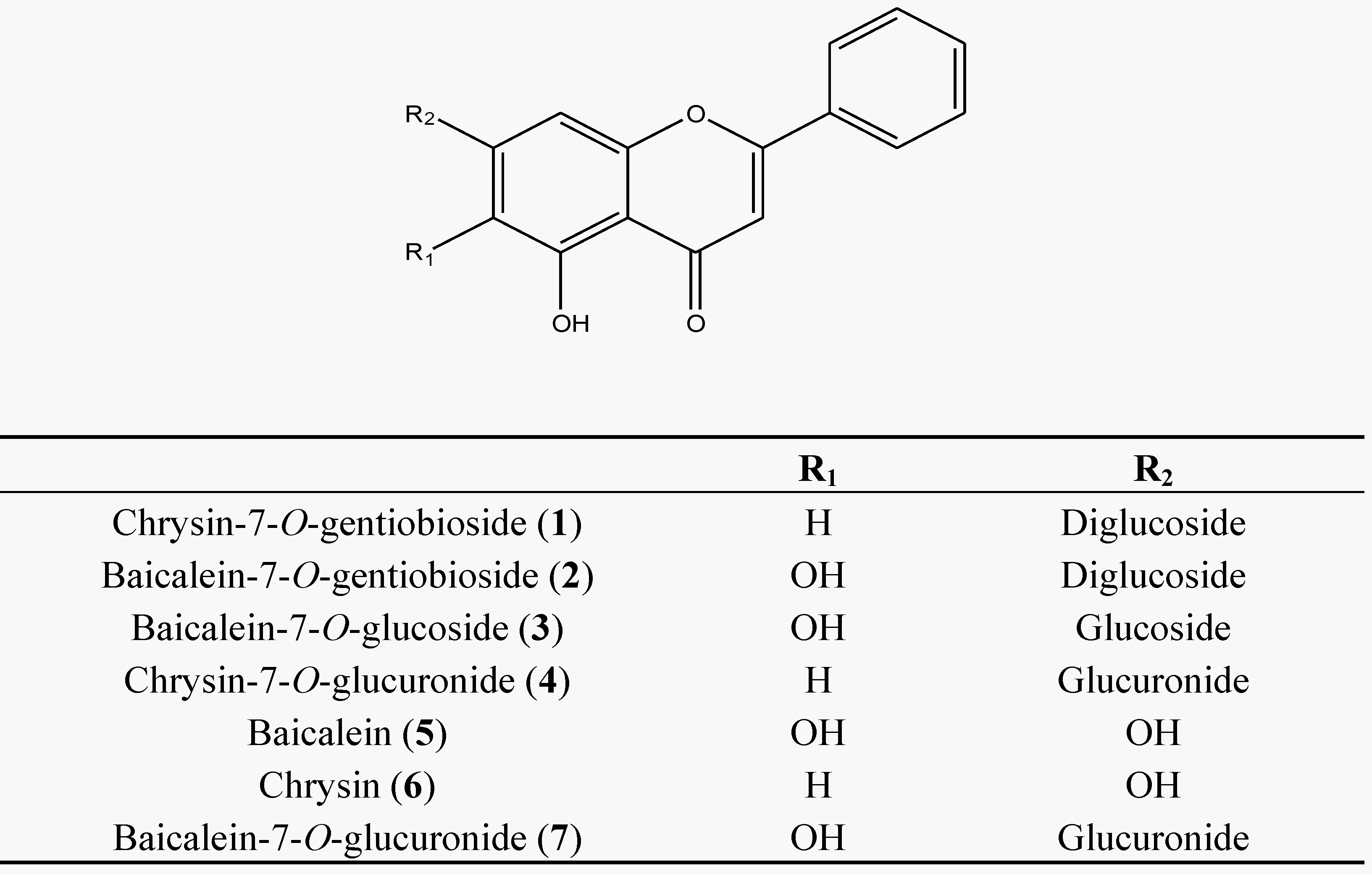
2.2. Method Validation
2.3. Sample Preparation
2.4. CE-Method
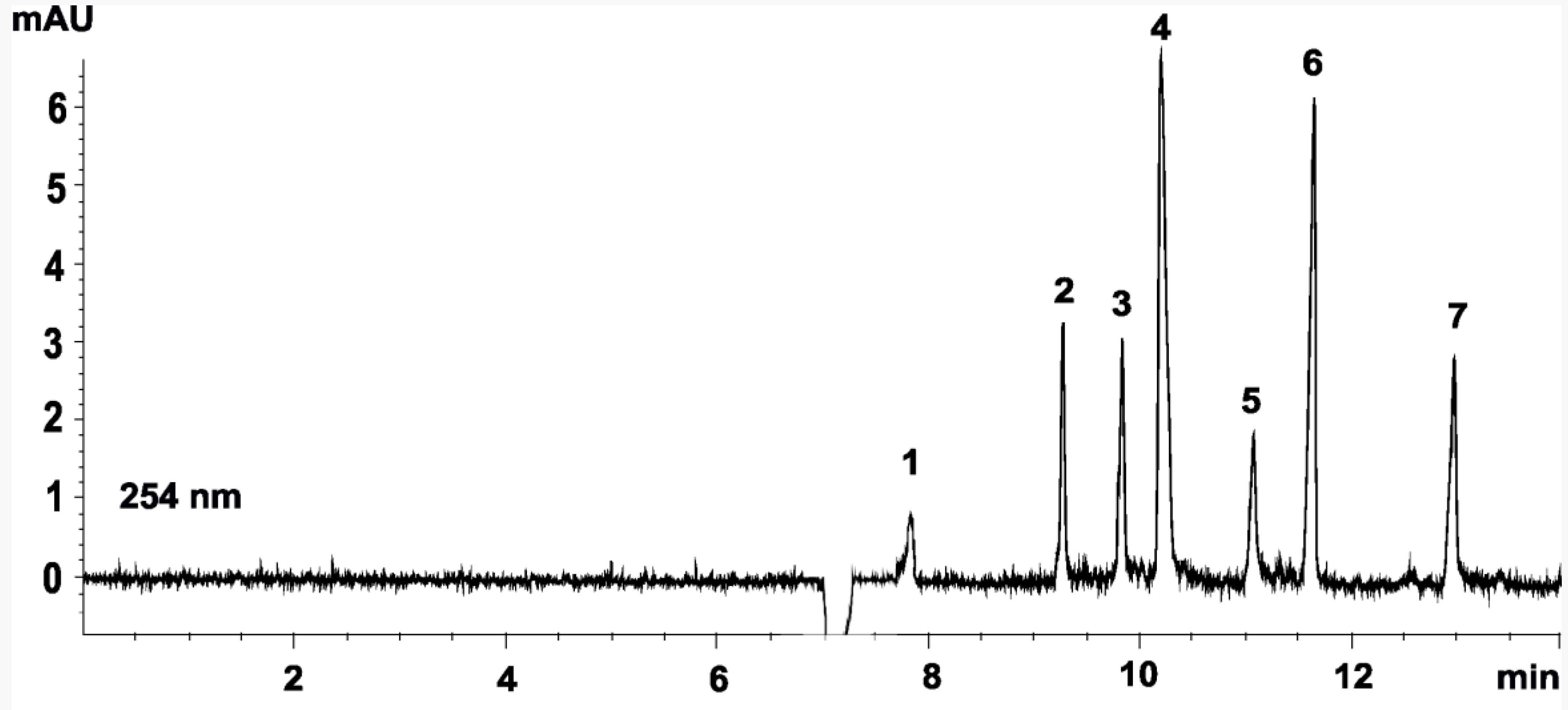
3. Results and Discussion
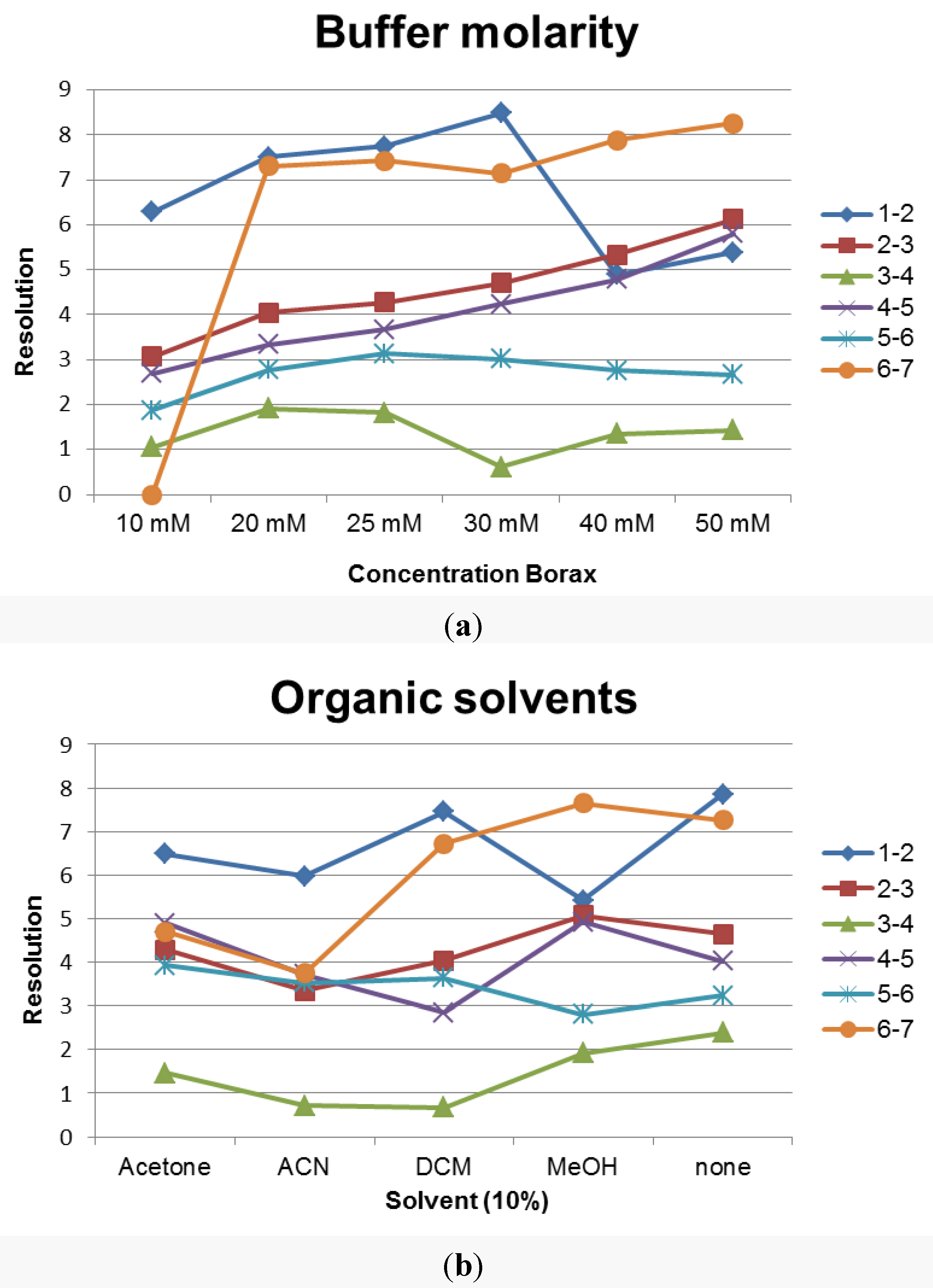
3.1. Method Development

3.2. Method Validation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regression equation | R2 | Range * | |
|---|---|---|---|
| 1 | y = 0.12384x | 0.9991 | 2.56–621.96 |
| 2 | y = 0.07970x | 0.9997 | 2.40–673.49 |
| 3 | y = 0.21337x | 0.9998 | 3.69–896.34 |
| 4 | y = 0.11283x | 0.9995 | 8.69–704.20 |
| 5 | y = 0.42260x | 0.9994 | 1.33–973.08 |
| 6 | y = 0.60629x | 0.9994 | 1.37–999.52 |
| 7 | y = 0.32222x | 0.9998 | 6.99–782.89 |
| LOD * | LOQ * | Precision (intra-day) | Precision (inter-day) | Accuracy (high) | Accuracy (medium) | Accuracy (low) | |
|---|---|---|---|---|---|---|---|
| 1 | 3.20 | 9.69 | 0.35% | 6.90% | - | - | - |
| 2 | 5.31 | 15.03 | 1.44% | 2.72% | - | - | - |
| 3 | 1.86 | 5.62 | 0.70% | 4.09% | - | - | - |
| 4 | 3.51 | 10.64 | 0.53% | 5.78% | - | - | - |
| 5 | 0.94 | 2.84 | 3.32% | 6.11% | 99.1% | 99.1% | 97.6% |
| 6 | 0.52 | 2.35 | 1.86% | 5.83% | 100.9% | 97.5% | 99.3% |
| 7 | 1.24 | 4.82 | 1.54% | 6.64% | - | - | - |
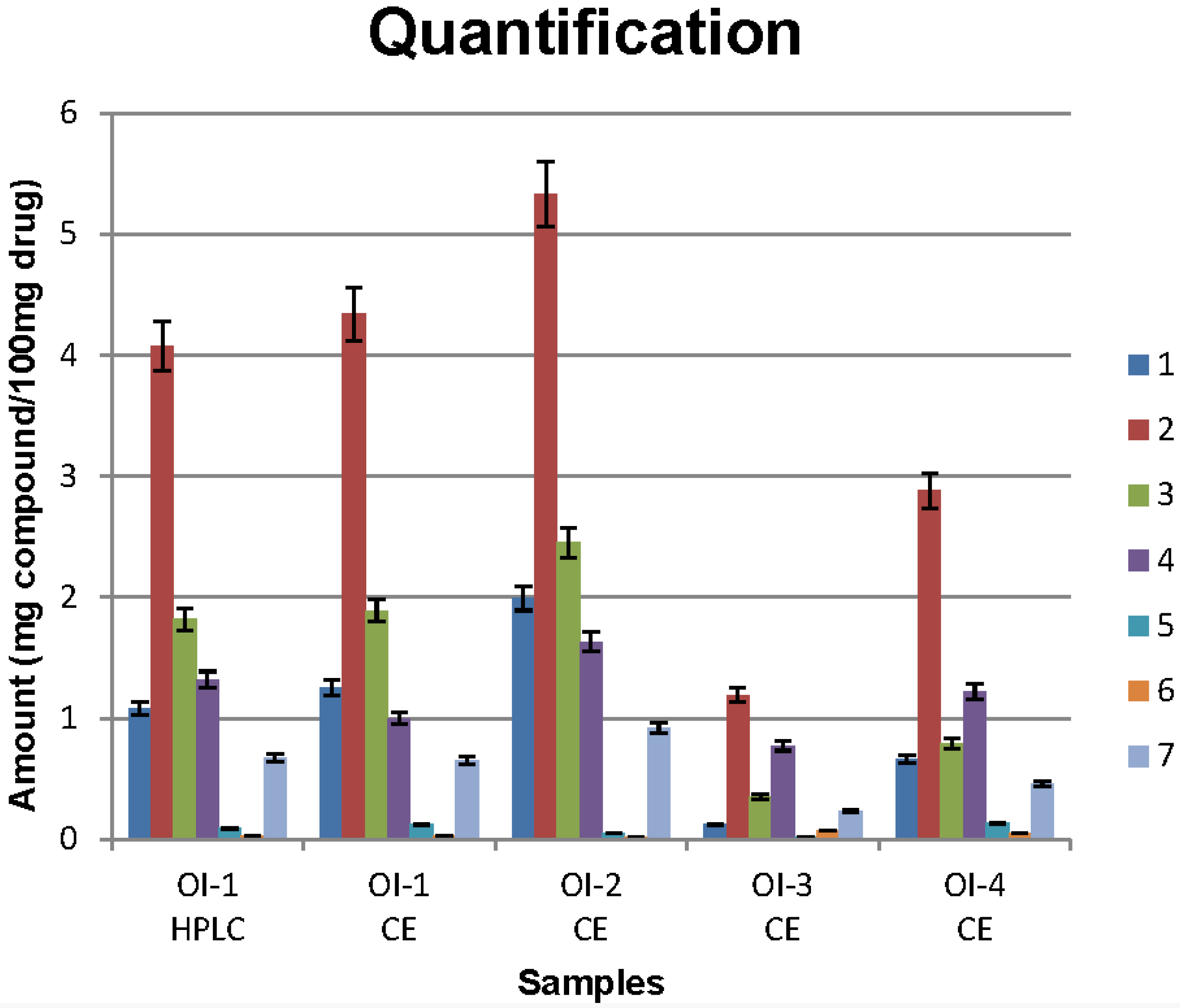
3.3. Sample Analysis

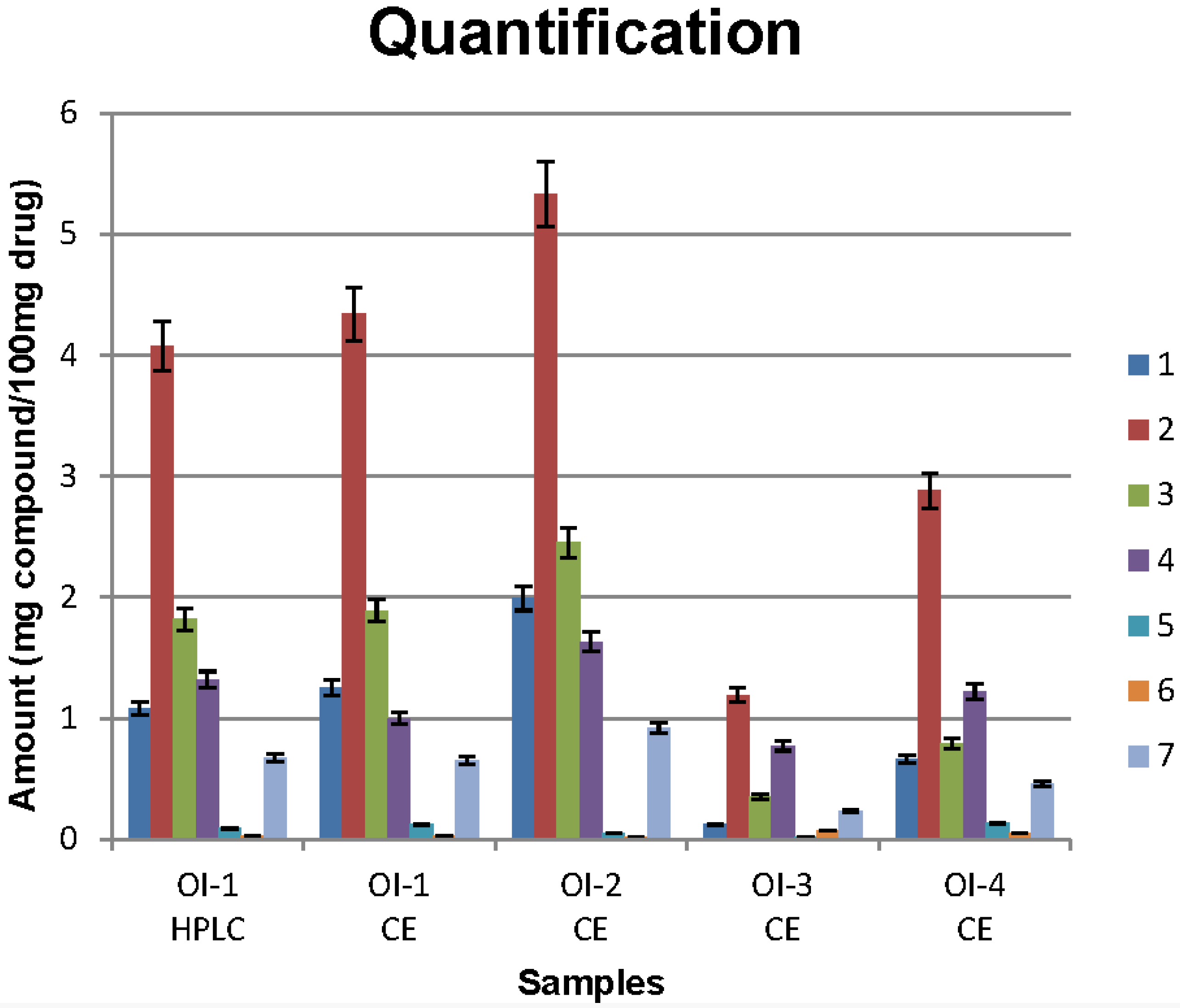
4. Conclusions
Conflict of Interest
References
- Harminder, S.V.; Chaudhary, A.K. A review on the taxonomy, ethnobotany, chemistry and pharmacology of Oroxylum indicum vent. Indian J. Pharm. Sci. 2011, 73, 483–490. [Google Scholar] [CrossRef]
- Bensky, D.; Clavey, S.; Stöger, E. Materia Medica, 3rd ed; Eastland Press: Seattle, WA, USA, 2004; pp. 456–457. [Google Scholar]
- The State Pharmacopoeia Commission of P.R. China, Pharmacopoeia of People’s Republic of China; China Medical Science Press: Beijing, China, 2010; p. 304.
- Yan, R.Y.; Cao, Y.Y.; Chen, Y.C.; Dai, H.Q.; Yu, S.X.; Wei, Y.L.; Li, H.; Yang, B. Antioxidant flavonoids from the seed of Oroxylum indicum. Fitoterapia 2011, 82, 841–848. [Google Scholar] [CrossRef]
- Radhika, L.G.; Meena, C.V.; Peter, S.; Rajesh, K.S.; Rosamma, M.P. Phytochemical and antimicrobial study of Oroxylum indicum. Ancient Sci. Life 2011, 30, 114–120. [Google Scholar]
- Laupattarakasem, P.; Houghton, P.J.; Hoult, J.R.; Itharat, A. An evaluation of the activity related to inflammation of four plants used in Thailand to treat arthritis. J. Ethnopharamcol. 2003, 85, 207–215. [Google Scholar] [CrossRef]
- Krüger, A.; Ganzera, M. Oroxylum indicum seeds—Analysis of flavonoids by HPLC-MS. J. Pharm. Biomed. Anal. 2012, 70, 553–556. [Google Scholar] [CrossRef]
- Ganzera, M.; Egger, C.; Zidorn, C.; Stuppner, H. Quantitative analysis of flavonoids and phenolic acids in Arnica montana L. by micellar electrokinetic capillary chromatography. Anal. Chim. Acta 2008, 614, 196–200. [Google Scholar] [CrossRef]
- Zehrmann, N.; Zidorn, C.; Ganzera, M. Analysis of rare flavonoid C-glycosides in Celtis australis L. by micellar electrokinetic chromatography. J. Pharm. Biomed. Anal. 2010, 51, 1165–1168. [Google Scholar] [CrossRef]
- De Rosa, S.; de Stefano, S. Chrysin 7-gentiobioside from the flowers of Spartium junceum. Phytochemistry 1983, 22, 2323–2324. [Google Scholar] [CrossRef]
- Terabe, S.; Otsuka, K.; Ichikawa, K.; Tsuchiya, A.; Ando, T. Electrokinetic separations with micellar solutions and open-tubular capillaries. Anal. Chem. 1984, 56, 111–113. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krüger, A.; Ganzera, M. Oroxylum indicum Seeds—Analysis of Flavonoids by Micellar Electrokinetic Chromatography. Chromatography 2014, 1, 1-8. https://doi.org/10.3390/chromatography1010001
Krüger A, Ganzera M. Oroxylum indicum Seeds—Analysis of Flavonoids by Micellar Electrokinetic Chromatography. Chromatography. 2014; 1(1):1-8. https://doi.org/10.3390/chromatography1010001
Chicago/Turabian StyleKrüger, Anja, and Markus Ganzera. 2014. "Oroxylum indicum Seeds—Analysis of Flavonoids by Micellar Electrokinetic Chromatography" Chromatography 1, no. 1: 1-8. https://doi.org/10.3390/chromatography1010001
APA StyleKrüger, A., & Ganzera, M. (2014). Oroxylum indicum Seeds—Analysis of Flavonoids by Micellar Electrokinetic Chromatography. Chromatography, 1(1), 1-8. https://doi.org/10.3390/chromatography1010001