Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics


Abstract
:1. Introduction
2. Experimental
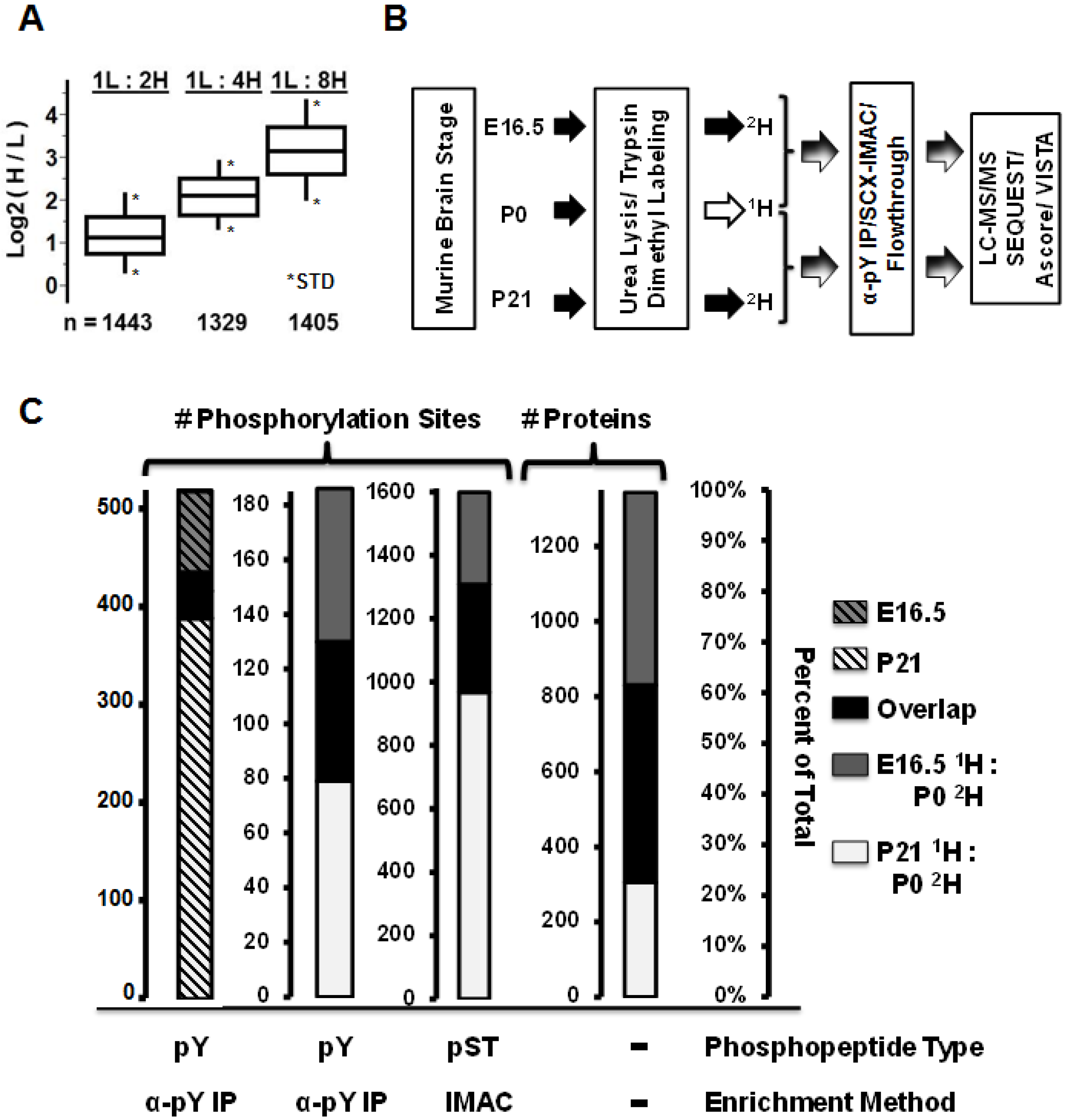
2.1. Tissue Harvesting and Preparation of Tryptic Peptides
2.2. Dimethyl Labeling of Tryptic Peptides
2.3. Anti-Phosphotyrosine Peptide Immunoprecipitations
2.4. SCX-IMAC
2.5. Mass Spectrometry and Data Analysis
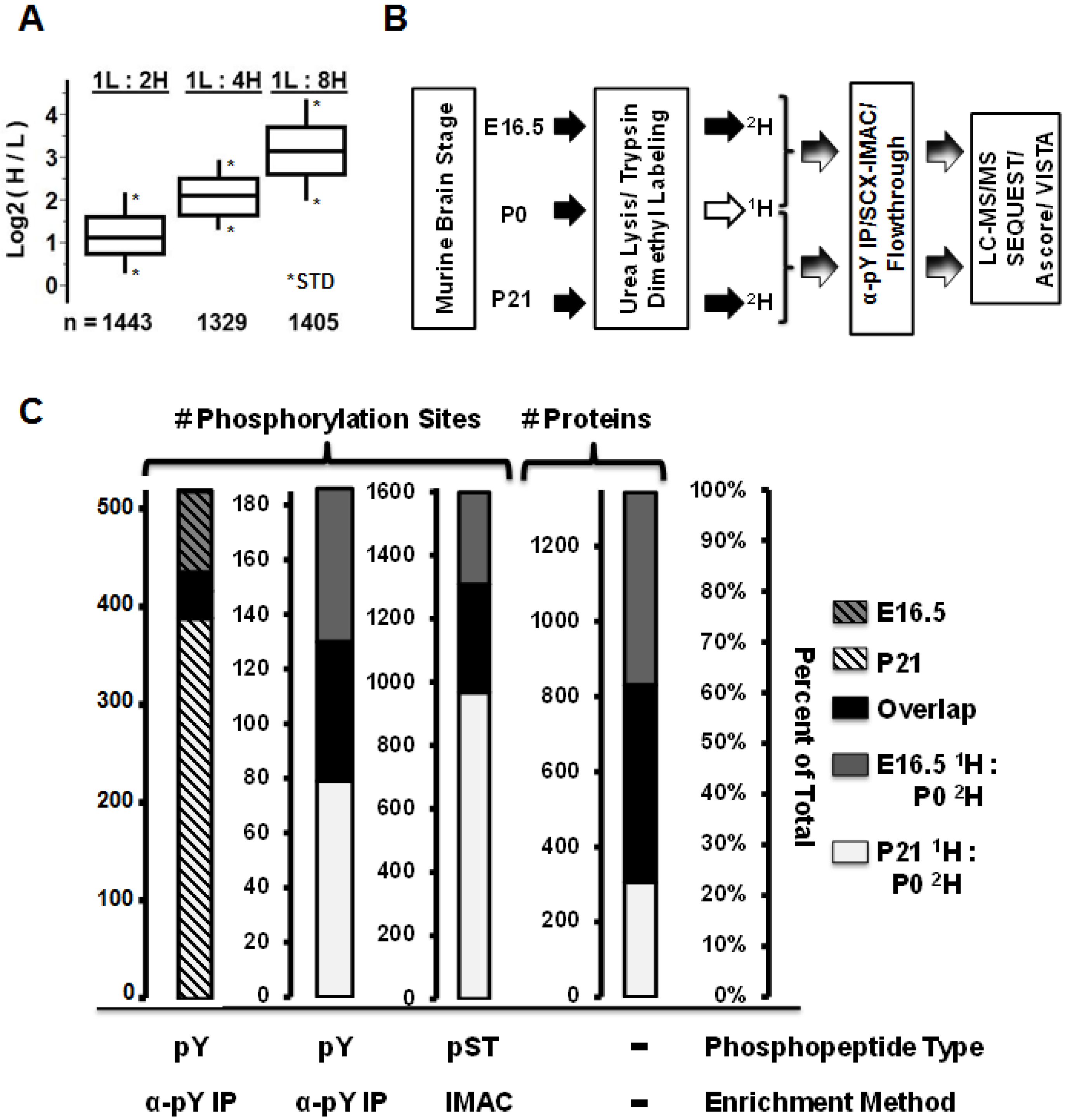
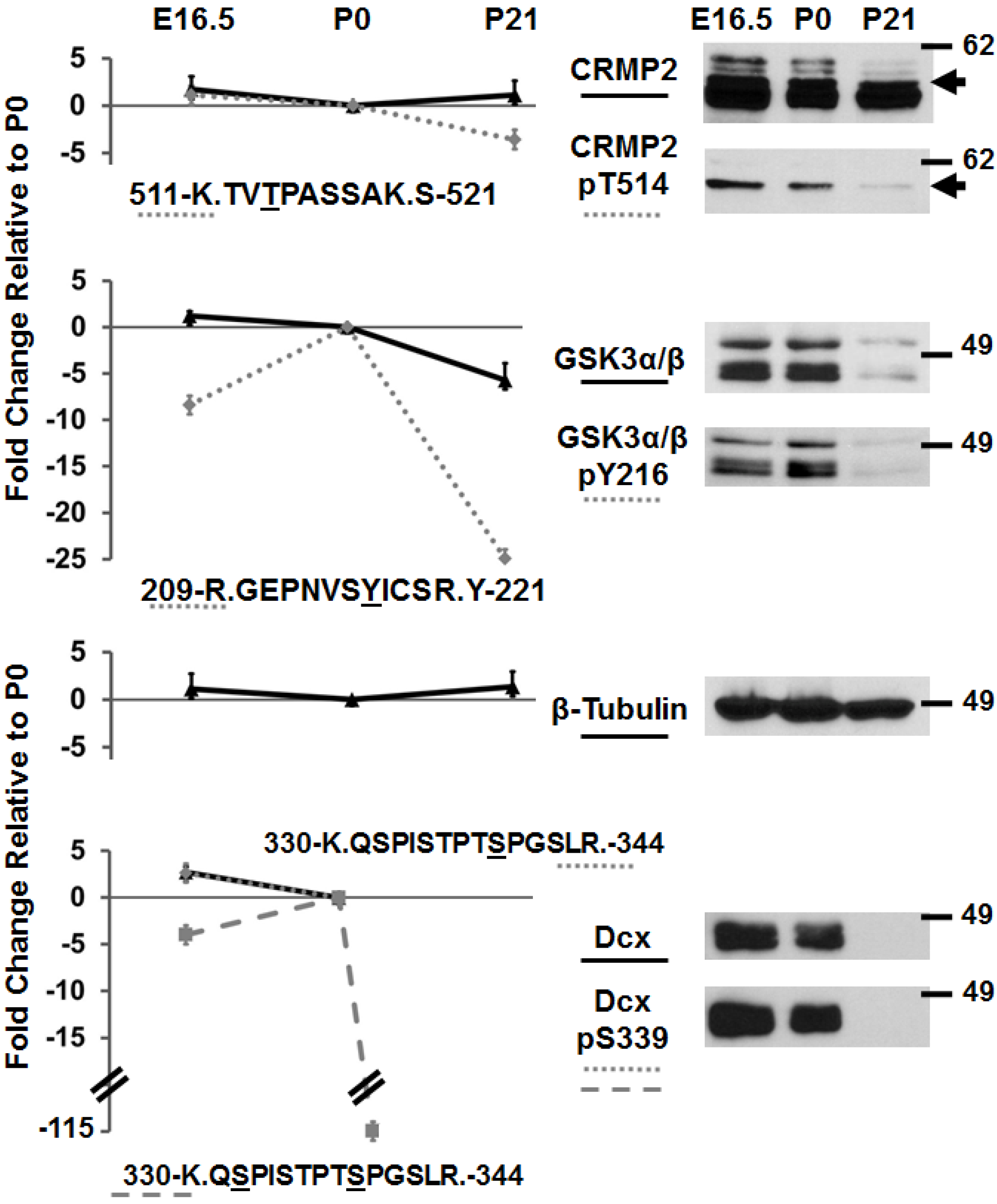
2.6. Immunoblots and Antibodies
2.7. Bioinformatics
3. Results and Discussion
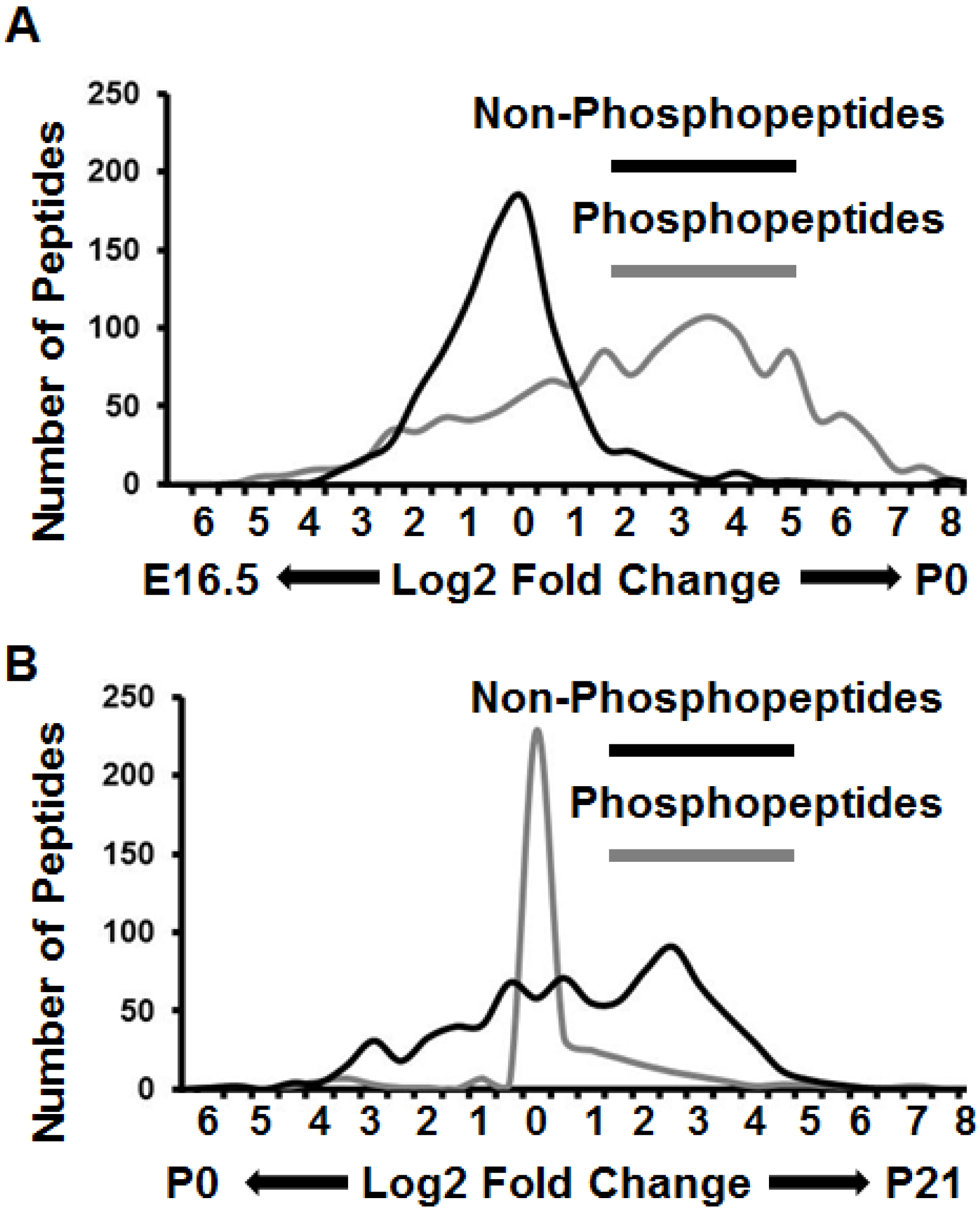
3.1. Comparative Murine Brain Proteomics Reveals Developmentally-Dynamic Proteomes and Phosphoproteomes
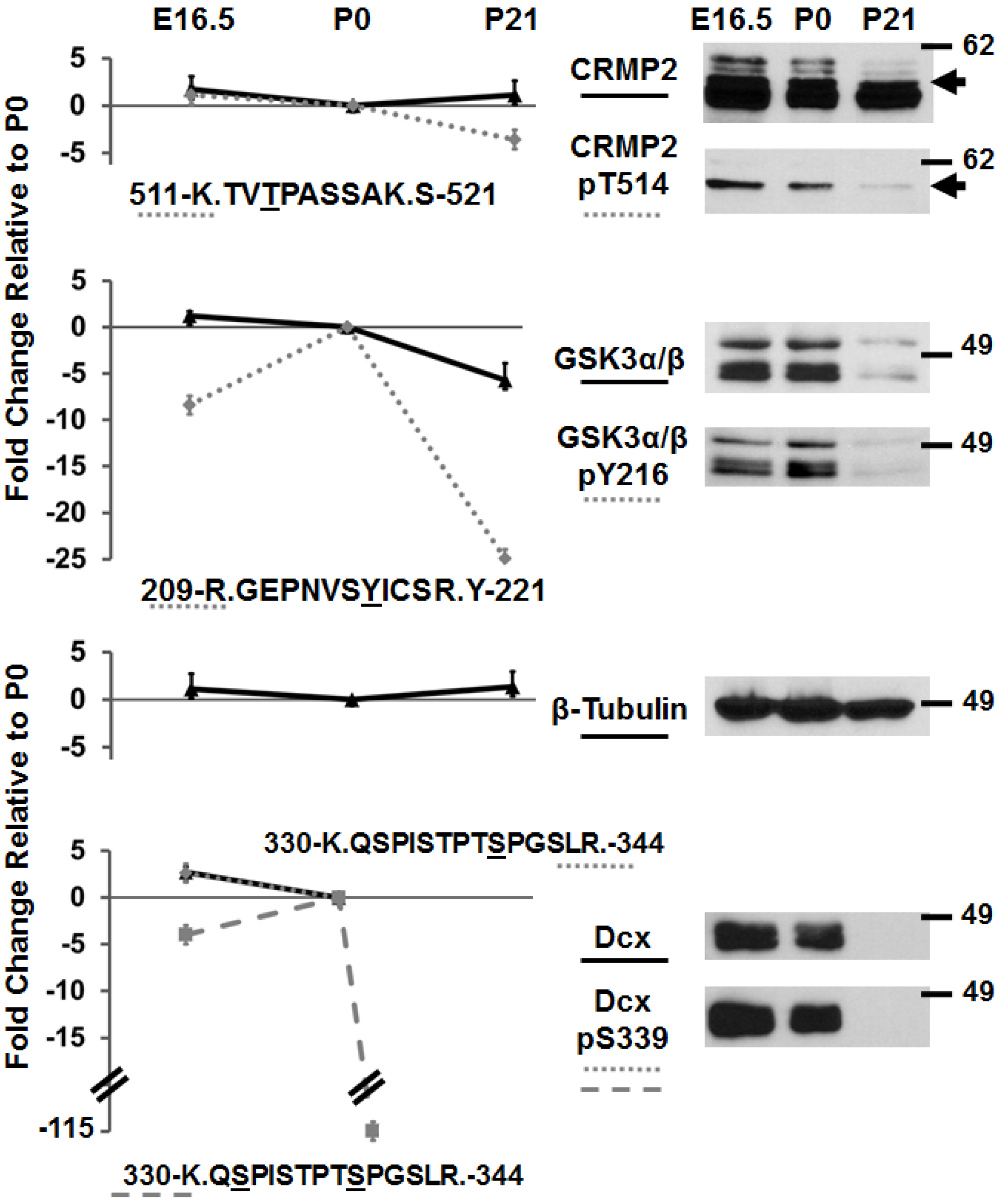
3.2. Immunoblotting of Developmentally-Important Phosphoproteins Shows Agreement with Quantitative MS Data
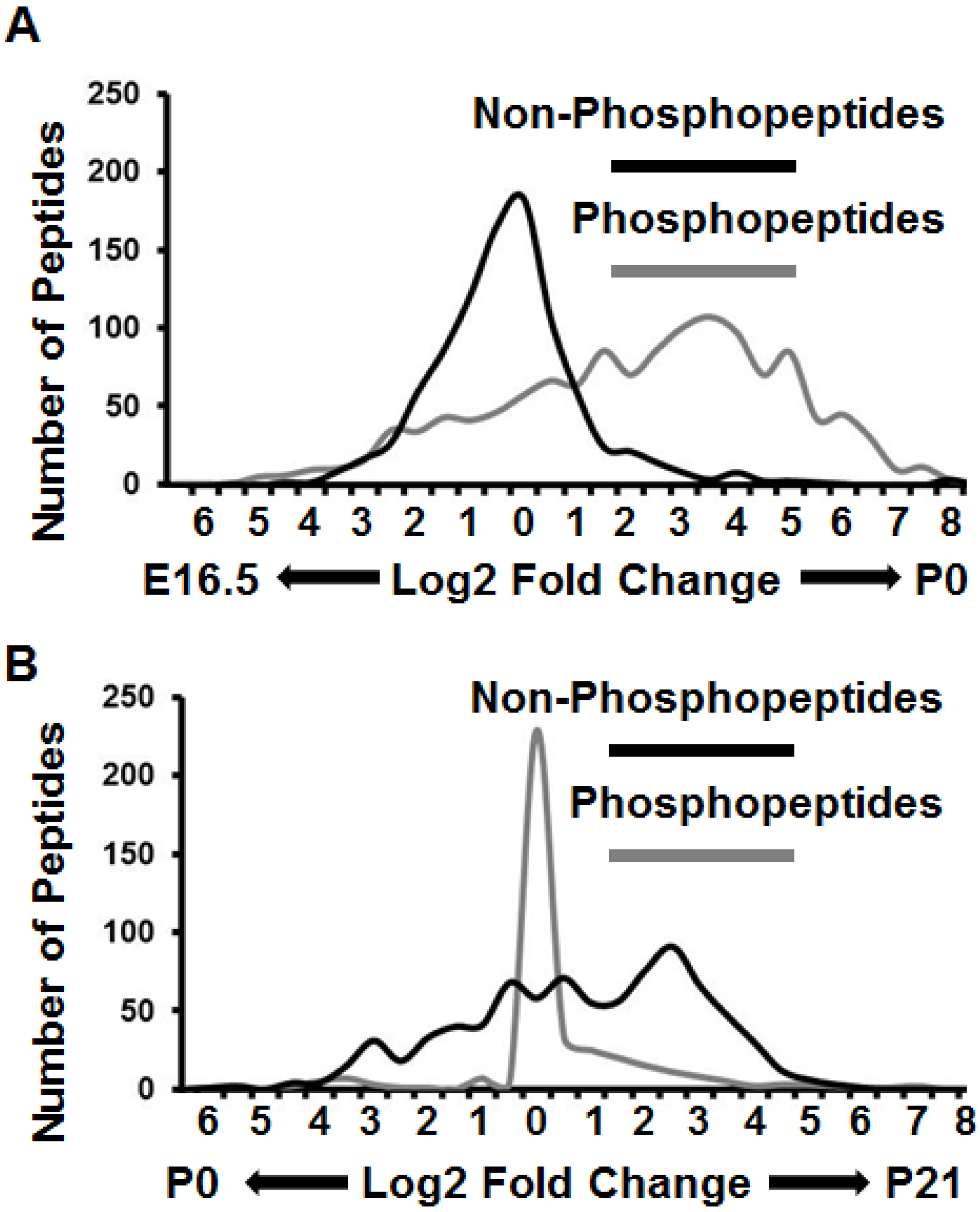
3.3. Proteomes and Phosphoproteomes Display Distinct Profiles at Different Stages of Brain Development
3.4. Phosphotyrosine Sites Showing the Most Extreme Differences between E16.5 and P21 Brain Offer Avenues for Distinct Hypothesis Testing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | pY Site | Peptide (Y# is pY) | Log2 Fold Higher E16.5 over P0 | Fold Higher E16.5 over P0 | Protein Name |
| Mbp | 199 | TTHY#GSLPQK | 6.61 | 98 | Myelin basic protein |
| Dlg4 | 283 | NTYDVVY#LK | 6.35 | 81 | Discs large homolog 4, Postsynaptic density protein 95 |
| Syt1 | 229 | TLVMAVY#DFDR | 5.51 | 45 | Synaptotagmin I |
| Grin2b | 1039 | HSQLSDLY#GK | 5.39 | 42 | GluRepsilon2/ N-methyl D-aspartate receptor 2B |
| Dlg2 | 340 | HMLGEDDY#TRPPEPVYSTVNK | 4.92 | 30 | Discs large homolog 2, Postsynaptic density protein 93 |
| Syt1 | 380 | VFVGY#NSTGAELR | 4.54 | 23 | Synaptotagmin I |
| Ckmt1 | 154 | SGY#FDER | 4.53 | 23 | Creatine kinase, mitochondrial 1 |
| Mbp | 169 | GAY#DAQGTLSK | 3.64 | 12 | Myelin basic protein |
| Dlg4 | 647 | FIEAGQY#NSHLYGTSVQSVR | 3.50 | 11 | Discs large homolog 4, Postsynaptic density protein 95 |
| Gene Symbol | pY Site | Peptide (Y# is pY) | Log2 Fold Higher P0 over P21 | Fold Higher P0 over P21 | Protein Name |
| Dab1 | 232 | EGVY#DVPK | 3.91 | 15 | Disabled 1 |
| Prpf4b | 338 | LCDFGSASHVADNDITPY#LVSR | 3.99 | 16 | Pre-mRNA-processing factor 4b |
| Afap1l2 | 413 | VAQQPLSLVGCDVLPDPSPDHLY#SFR | 4.21 | 18 | Actin filament-associated protein 1-like 2 |
| Hipk3 | 359 | TVCSTY#LQSR | 4.58 | 24 | Homeodomain interacting protein kinase 3 |
| Gsk3b | 216 | GEPNVSY#ICSR | 4.64 | 25 | Glycogen synthase kinase-3 beta |
| Hipk1 | 352 | AVCSTY#LQSR | 5.23 | 38 | Homeodomain interacting protein kinase 1 |
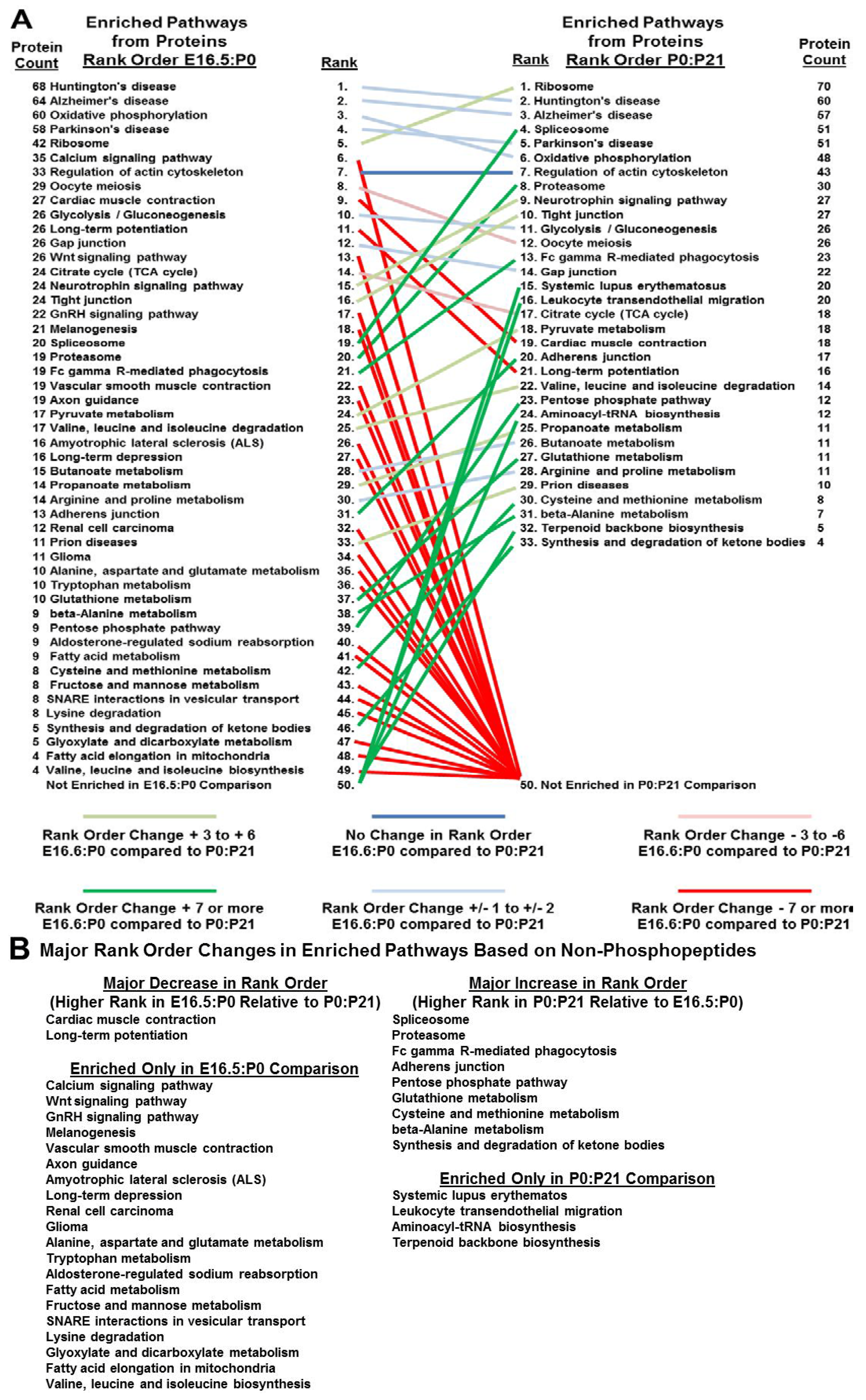
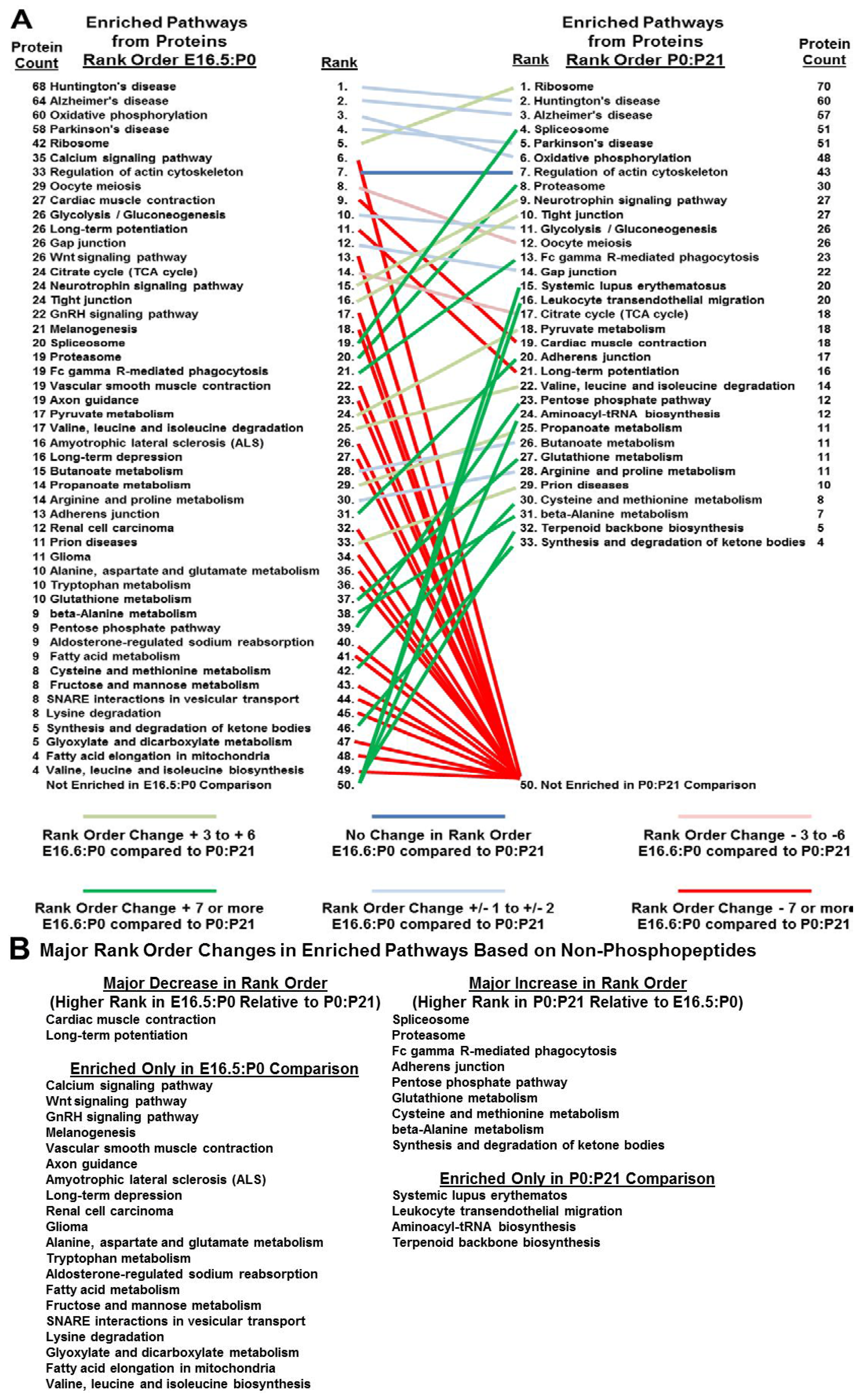
3.5. Bioinformatics Reveals Functional and Developmental Dynamics in Brain Proteomes and Phosphoproteomes

| Enriched Gene Ontology Categories of Phosphoproteins Showing Major Fold Changes in the E16.5:P0 Phosphopeptide Comparison | |
| Enriched in E16.5 | Enriched in P0 |
| Cytoskeleton | Protein Complexes Cytoskeleton Non-Membrane Bound Organelles |
| Neuronal Projection | |
| Synaptic Transmission | |
| Cell Signaling | |
| Splicing | |
| Enriched Gene Ontology Categories of Phosphoproteins Showing Major Fold Changes in the P0:P21 Phosphopeptide Comparison | |
| Enriched in P0 | Enrichedin P21 |
| Neurogenesis and Differentiation | Synaptic Transmission |
| Developmental Growth | Ion Transport |
| Morphogenesis | Cognition |
| Cell Signaling | Cell Signaling |
| Various Metabolism | Non-Membrane Bound Organelles |
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Patterson, S.D.; Aebersold, R.H. Proteomics: The first decade and beyond. Nat. Genet. 2003, 33, 311–323. [Google Scholar] [CrossRef]
- Goswami, T.; Ballif, B.A. Methods for the isolation of phosphoproteins and phosphopeptides for mass spectrometry analysis: Toward increased functional phosphoproteomics. In Sample Preparation in Biological Mass Spectrometry; Ivanov, A.R., Lazarev, A.V., Eds.; Springer: New York, NY, USA, 2011; pp. 627–655. [Google Scholar]
- Ballif, B.A.; Villen, J.; Beausoleil, S.A.; Schwartz, D.; Gygi, S.P. Phosphoproteomic analysis of the developing mouse brain. Mol. Cell. Proteomics 2004, 3, 1093–1101. [Google Scholar] [CrossRef]
- Goswami, T.; Li, X.; Smith, A.M.; Luderowski, E.M.; Vincent, J.J.; Rush, J.; Ballif, B.A. Comparative phosphoproteomic analysis of neonatal and adult murine brain. Proteomics 2012, 12, 2185–2189. [Google Scholar] [CrossRef]
- Ballif, B.A.; Carey, G.R.; Sunyaev, S.R.; Gygi, S.P. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J. Proteome Res. 2008, 7, 311–318. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Dephoure, N.; Gygi, S.P. A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods 2011, 54, 379–386. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Bakalarski, C.E.; Elias, J.E.; Villen, J.; Haas, W.; Gerber, S.A.; Everley, P.A.; Gygi, S.P. The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J. Proteome Res. 2008, 7, 4756–4765. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Ballif, B.A.; Arnaud, L.; Cooper, J.A. Tyrosine phosphorylation of disabled-1 is essential for reelin-stimulated activation of Akt and Src family kinases. Brain Res. Mol. Brain Res. 2003, 117, 152–159. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in kegg. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Khidekel, N.; Ficarro, S.B.; Clark, P.M.; Bryan, M.C.; Swaney, D.L.; Rexach, J.E.; Sun, Y.E.; Coon, J.J.; Peters, E.C.; Hsieh-Wilson, L.C. Probing the dynamics of o-glcnac glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 2007, 3, 339–348. [Google Scholar] [CrossRef]
- Ballif, B.A.; Arnaud, L.; Arthur, W.T.; Guris, D.; Imamoto, A.; Cooper, J.A. Activation of a dab1/crkl/c3g/rap1 pathway in reelin-stimulated neurons. Curr. Biol. 2004, 14, 606–610. [Google Scholar] [CrossRef]
- Buel, G.R.; Rush, J.; Ballif, B.A. Fyn promotes phosphorylation of collapsin response mediator protein 1 at tyrosine 504, a novel, isoform-specific regulatory site. J. Cell. Biochem. 2010, 111, 20–28. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3a signalling is mediated via sequential cdk5 and gsk3beta phosphorylation of crmp2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar]
- Graham, M.E.; Ruma-Haynes, P.; Capes-Davis, A.G.; Dunn, J.M.; Tan, T.C.; Valova, V.A.; Robinson, P.J.; Jeffrey, P.L. Multisite phosphorylation of doublecortin by cyclin-dependent kinase 5. Biochem. J. 2004, 381, 471–481. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Rush, J.; Gerber, S.A. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 2011, 6, 175–186. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. Phosphositeplus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at prosite. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef]
- Lopez, A.J. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu. Rev. Genet. 1998, 32, 279–305. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Doubleday, P.F.; Ballif, B.A. Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics. Proteomes 2014, 2, 191-207. https://doi.org/10.3390/proteomes2020191
Doubleday PF, Ballif BA. Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics. Proteomes. 2014; 2(2):191-207. https://doi.org/10.3390/proteomes2020191
Chicago/Turabian StyleDoubleday, Peter F., and Bryan A. Ballif. 2014. "Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics" Proteomes 2, no. 2: 191-207. https://doi.org/10.3390/proteomes2020191