
Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration

Abstract
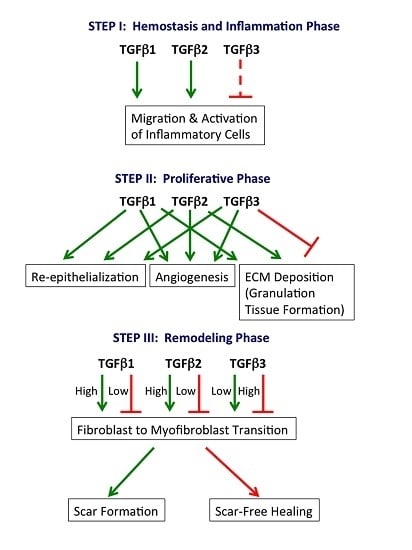
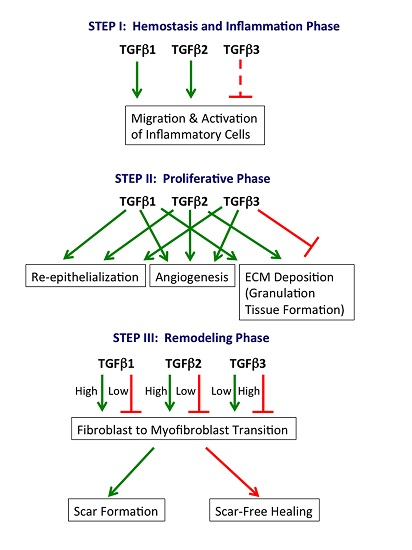
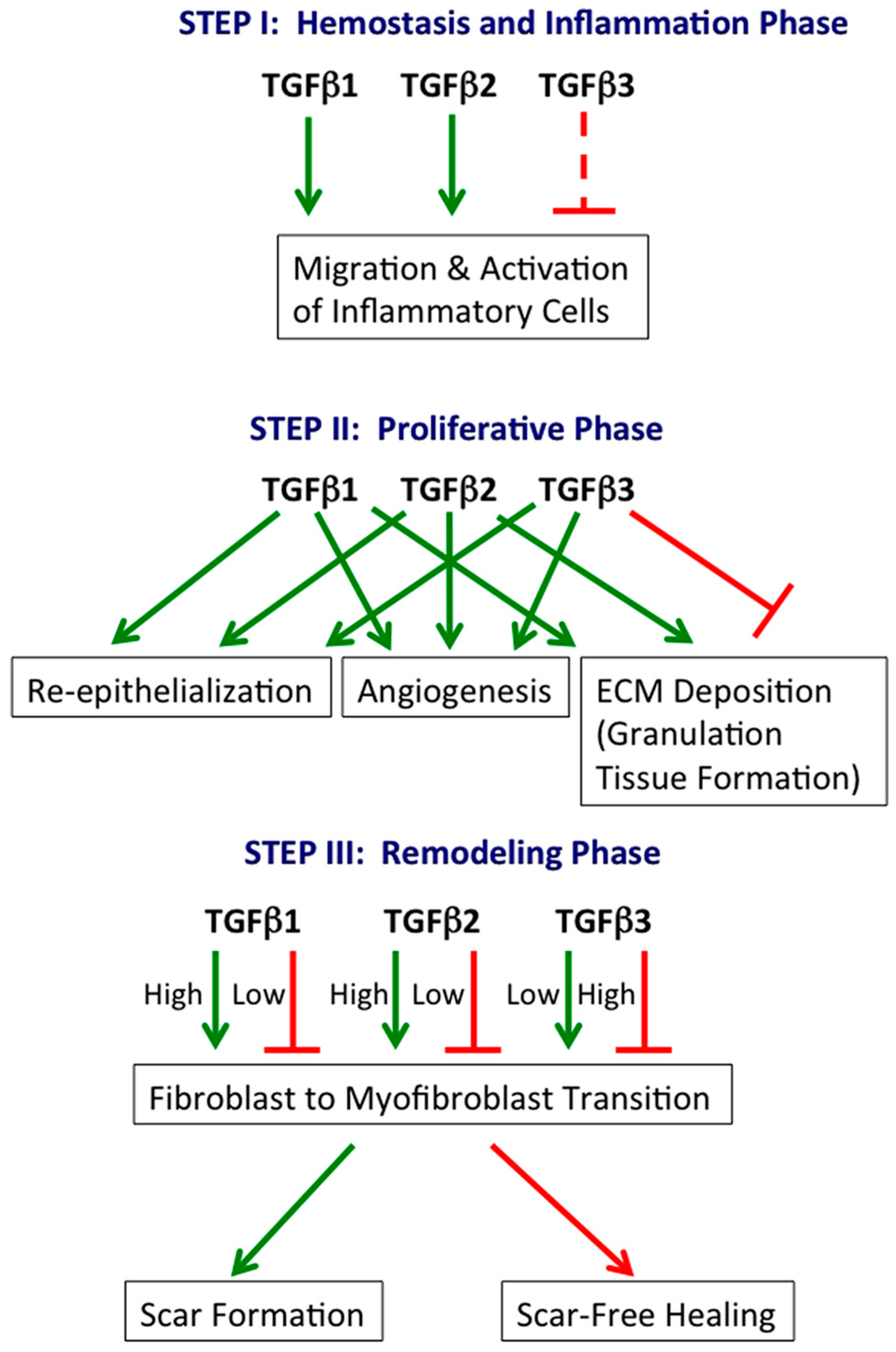
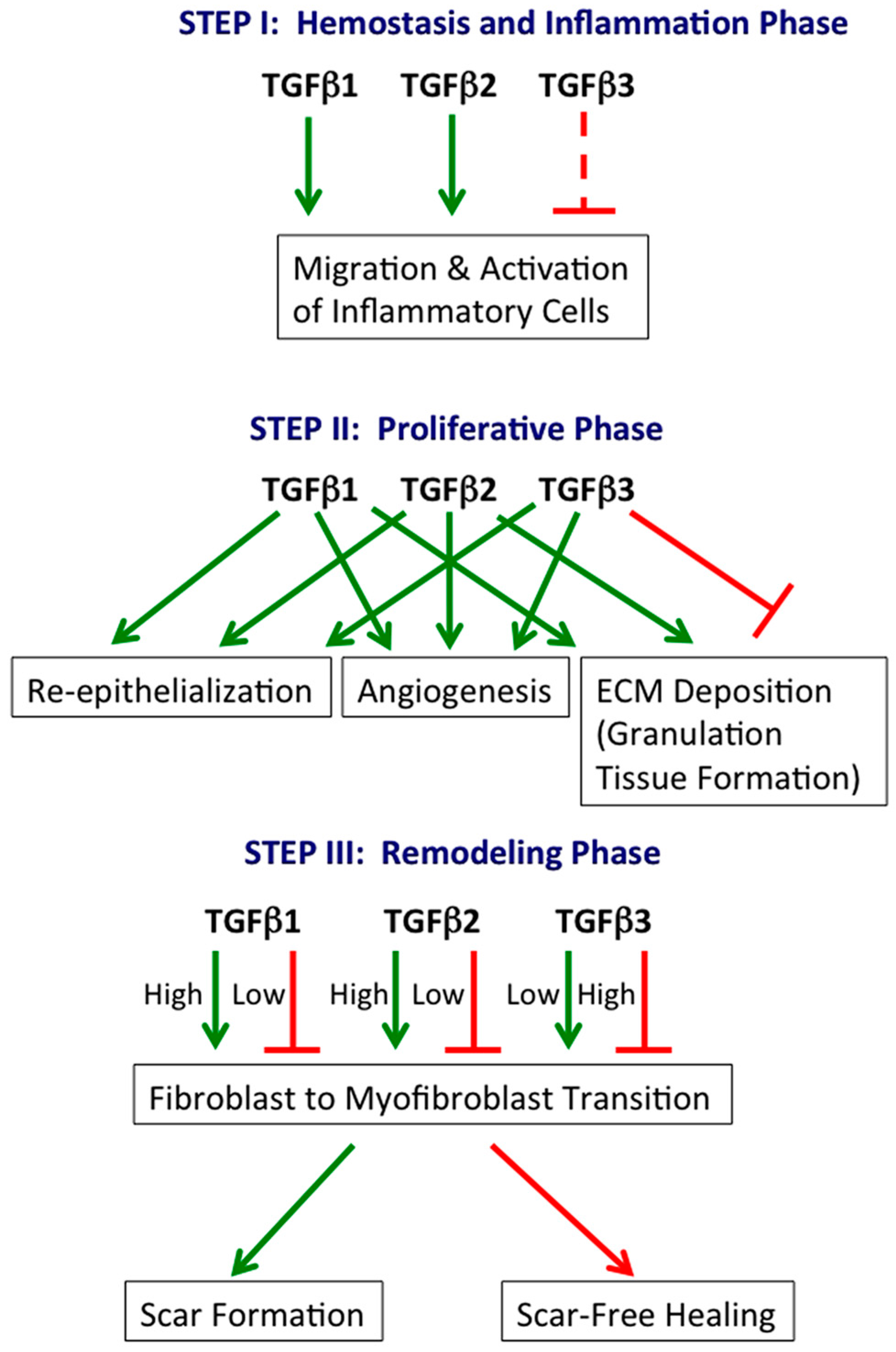
:
{kind=link}
{kind=link}
1. Transforming Growth Factor Beta (TGFβ) Signalling
2. Cutaneous Wound Repair
2.1. Hemostasis and Inflammation Phase
2.2. Proliferative Phase
2.3. Remodeling Phase
2.4. Exceptions to Scar Formation in Mammals
3. Multi-Tissue Regeneration
3.1. Blastema Formation
3.2. TGFβ in Multi-Tissue Regeneration
3.2.1. Murphy Roths Large (MRL) Mice
3.2.2. TGFβ1 Receptor Mutant Mice
4. TGFβ Signalling Targeting in Wound Healing and Tissue Regeneration
4.1. Small Molecule Inhibitors
4.2. Monoclonal Antibodies
4.3. Ligand Traps/Decoy Receptors
4.4. Antisense Oligonucleotides
4.5. Indirect TGFβ-targeting Agents
4.6. Recombinant TGFβ
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| αSMA | Alpha Smooth Muscle Actin |
| AER | Apical Ectodermal Ridge |
| ALK | Activin-Like Kinase |
| ASON | Anti-Sense Oligonucleotides |
| BMP | Bone Morphogenic Protein |
| CACG | Chronic Angle Closure Glaucoma |
| ECM | Extracellular Matrix |
| EEA-1 | Early Endosome Antigen 1 |
| EMT | Epithelial to Mesenchymal Transition |
| EndoMT | Endothelial to Mesenchymal Transition |
| Erk | Extracellular Signal Regulated Kinases |
| GDF | Growth and Differentiation Factor |
| JNK | C-Jun N-Terminal Kinases |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinases |
| MMP | Matrix Metalloproteinase |
| MRL | Murphy Roths Large |
| MRTF | Myocardin-Related Transcription Factor |
| MIS | MullerianInhibiting Substances |
| PAI | Plasminogen Activator Inactivator |
| PFD | Pirfenidone |
| PI3K | Phosphatidylinositide 3 Kinases |
| PMN | Polymorphonuclear Cells |
| POAG | Primary Open Angle Glaucoma |
| SAPK | Stress-Activated Protein Kinases |
| SMAD | Small Mothers against Decapentaplegic |
| SMI | Small Molecule Inhibitors |
| TAZ | Transcriptional Coactivator with PDZ-Binding Motif |
| TβRI | Transforming Growth Factor Beta Receptor I |
| TβRII | Transforming Growth Factor Beta Receptor II |
| TβRIII | Transforming Growth Factor Beta Receptor III |
| TGFβ | Transforming Growth Factor Beta |
| VEGF | Vascular Endothelial Growth Factor |
| YAP | Yes-Associated Protein |
References
- Viloria-Petit, A.; Richard, A.; Zours, S.; Jarad, M.; Coomber, B.L. Role of transforming growth factor beta in angiogenesis. In Biochemical Basis and Therapeutic Implications of Angiogenesis, Advances in Biochemistry in Health and Disease; Mehta, J., Dhalla, N.S., Eds.; Springer: New York, NY, USA, 2013; Volume 6, pp. 23–45. [Google Scholar]
- Antsiferova, M.; Werner, S. The bright and the dark sides of activin in wound healing and cancer. J. Cell Sci. 2012, 125, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Tgfb signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Simakov, O.; Kawashima, T.; Marletaz, F.; Jenkins, J.; Koyanagi, R.; Mitros, T.; Hisata, K.; Bredeson, J.; Shoguchi, E.; Gyoja, F.; et al. Hemichordate genomes and deuterostome origins. Nature 2015, 527, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. The regulation of tgfb signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Fuchs, E. The harmonies played by tgf-beta in stem cell biology. Cell Stem Cell 2012, 11, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent tgfb activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross talk among tgf-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harbor Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Unchaining the beast; insights from structural and evolutionary studies on tgfb secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013, 24, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Landstrom, M.; Moustakas, A. Mechanism of tgf-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. Tgf-beta activates erk map kinase signalling through direct phosphorylation of shca. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Huminiecki, L.; Goldovsky, L.; Freilich, S.; Moustakas, A.; Ouzounis, C.; Heldin, C.H. Emergence, development and diversification of the tgf-beta signalling pathway within the animal kingdom. BMC Evol. Biol. 2009, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. Tgf-beta signaling in vascular biology and dysfunction. Cell. Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the tgf-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Goumans, M.J.; ten Dijke, P. Tgf-beta receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr. Pharm. Biotechnol. 2011, 12, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Maring, J.A.; Trojanowska, M.; ten Dijke, P. Role of endoglin in fibrosis and scleroderma. Int. Rev. Cell Mol. Biol. 2012, 297, 295–308. [Google Scholar] [PubMed]
- Bilandzic, M.; Stenvers, K.L. Reprint of: Betaglycan: A multifunctional accessory. Mol. Cell. Endocrinol. 2012, 359, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of smad function in tgf-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef]
- Tang, W.B.; Ling, G.H.; Sun, L.; Liu, F.Y. Smad anchor for receptor activation (SARA) in TGF-beta signaling. Front. Biosci. 2010, 2, 857–860. [Google Scholar]
- Bakkebo, M.; Huse, K.; Hilden, V.I.; Forfang, L.; Myklebust, J.H.; Smeland, E.B.; Oksvold, M.P. SARA is dispensable for functional TGF-beta signaling. FEBS Lett. 2012, 586, 3367–3372. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. Taz controls smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 2004, 23, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Liang, Y.Y.; Wrighton, K.; Ungermannova, D.; Wang, X.P.; Brunicardi, F.C.; Liu, X.; Feng, X.H.; Lin, X. Ubiquitination and proteolysis of cancer-derived smad4 mutants by scfskp2. Mol. Cell. Biol. 2004, 24, 7524–7537. [Google Scholar] [CrossRef] [PubMed]
- Kardassis, D.; Murphy, C.; Fotsis, T.; Moustakas, A.; Stournaras, C. Control of transforming growth factor beta signal transduction by small gtpases. FEBS J. 2009, 276, 2947–2965. [Google Scholar] [CrossRef] [PubMed]
- Di Guglielmo, G.M.; le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate tgf-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; McLean, S.; le Roy, C.; O’Connor-McCourt, M.; Wrana, J.L.; di Guglielmo, G.M. The extracellular domain of the tgfbeta type II receptor regulates membrane raft partitioning. Biochem. J. 2009, 421, 119–131. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; di Guglielmo, G.M. Tgf beta (transforming growth factor beta) receptor type III directs clathrin-mediated endocytosis of tgf beta receptor types I and II. Biochem. J. 2010, 429, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I tgf-beta receptor engages traf6 to activate tak1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein par6 by tgfb receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-smad pathways in tgf-beta signaling. Cell. Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Okada, Y.; Miyamoto, T.; Yamanaka, O.; Ohnishi, Y.; Ooshima, A.; Liu, C.Y.; Weng, D.; Kao, W.W. Role of p38 map kinase in regulation of cell migration and proliferation in healing corneal epithelium. Investig. Ophthalmol. Vis. Sci. 2004, 45, 100–109. [Google Scholar] [CrossRef]
- Secker, G.A.; Shortt, A.J.; Sampson, E.; Schwarz, Q.P.; Schultz, G.S.; Daniels, J.T. Tgfb stimulated re-epithelialisation is regulated by ctgf and ras/mek/erk signalling. Exp. Cell Res. 2008, 314, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Laverty, H.G.; Wakefield, L.M.; Occleston, N.L.; O’Kane, S.; Ferguson, M.W. Tgf-beta3 and cancer: A review. Cytokine Growth Factor Rev. 2009, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Kim, S.J.; Noma, T.; Glick, A.B.; Lafyatis, R.; Lechleider, R.; Jakowlew, S.B.; Geiser, A.; O'Reilly, M.A.; Danielpour, D.; et al. Multiple forms of tgf-beta: Distinct promoters and differential expression. Ciba Found. Symp. 1991, 157, 7–15; discussion 15–28. [Google Scholar] [PubMed]
- Mercado-Pimentel, M.E.; Runyan, R.B. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007, 185, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Nawshad, A.; LaGamba, D.; Hay, E.D. Transforming growth factor beta (tgfb) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (emt). Arch. Oral Biol. 2004, 49, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sebe, A.; Peterfi, Z.; Masszi, A.; Thirone, A.C.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szaszi, K.; Mucsi, I.; et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol. Biol. Cell. 2007, 18, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.W.; Chen, Q.K.; Gjorevski, N.; Nelson, C.M. Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. J. Cell. Biochem. 2010, 110, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.D.; Narani, N.; McCulloch, C.A. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am. J. Pathol. 1999, 154, 871–882. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent tgf-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.; Nakano, H.; Kelley, T.J.; Hinz, B.; Kapus, A. Differential topical susceptibility to tgfb in intact and injured regions of the epithelium: Key role in myofibroblast transition. Mol. Biol. Cell 2013, 24, 3326–3336. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.; Kofler, M.; Szaszi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated mrtf and taz and tgfbeta-regulated smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of yap/taz in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Barbul, A.; Efron, D.T.; Kavalukas, S.L. Schwartz’s Manual of Surgery, 8th ed.; McGraw-Hill Medical Pub. Division: New York, NY, USA, 2010. [Google Scholar]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Valluru, M.; Staton, C.A.; Reed, M.W.; Brown, N.J. Transforming growth factor-beta and endoglin signaling orchestrate wound healing. Front. Physiol. 2011, 2, 89. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet tgf-beta1 contributions to plasma tgf-beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012, 119, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [PubMed]
- Grainger, D.J.; Mosedale, D.E.; Metcalfe, J.C. Tgf-beta in blood: A complex problem. Cytokine Growth Factor Rev. 2000, 11, 133–145. [Google Scholar] [CrossRef]
- Brandes, M.E.; Mai, U.E.; Ohura, K.; Wahl, S.M. Type I transforming growth factor-beta receptors on neutrophils mediate chemotaxis to transforming growth factor-beta. J. Immunol. 1991, 147, 1600–1606. [Google Scholar] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Reibman, J.; Meixler, S.; Lee, T.C.; Gold, L.I.; Cronstein, B.N.; Haines, K.A.; Kolasinski, S.L.; Weissmann, G. Transforming growth factor beta 1, a potent chemoattractant for human neutrophils, bypasses classic signal-transduction pathways. Proc. Natl. Acad. Sci. USA 1991, 88, 6805–6809. [Google Scholar] [CrossRef] [PubMed]
- Mahdavian Delavary, B.; van der Veer, W.M.; van Egmond, M.; Niessen, F.B.; Beelen, R.H. Macrophages in skin injury and repair. Immunobiology 2011, 216, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M.; Hunt, D.A.; Wakefield, L.M.; McCartney-Francis, N.; Wahl, L.M.; Roberts, A.B.; Sporn, M.B. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. USA 1987, 84, 5788–5792. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, D.M.; Polverini, P.J.; Kamp, D.W.; Leibovich, S.J. Transforming growth factor-beta (tgf beta) is chemotactic for human monocytes and induces their expression of angiogenic activity. Biochem. Biophys. Res. Commun. 1988, 157, 793–800. [Google Scholar] [CrossRef]
- Chen, C.C.; Manning, A.M. Tgf-beta 1, IL-10 and IL-4 differentially modulate the cytokine-induced expression of IL-6 and IL-8 in human endothelial cells. Cytokine 1996, 8, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.C. Wound healing in adult skin: Aiming for perfect regeneration. Int. J. Dermatol. 2011, 50, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in wound healing: A comprehensive review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Guasch, G.; Schober, M.; Pasolli, H.A.; Conn, E.B.; Polak, L.; Fuchs, E. Loss of tgfb signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 2007, 12, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Pietenpol, J.A.; Holt, J.T.; Stein, R.W.; Moses, H.L. Transforming growth factor beta 1 suppression of c-myc gene transcription: Role in inhibition of keratinocyte proliferation. Proc. Natl. Acad. Sci. USA 1990, 87, 3758–3762. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, H.; Patel, S.B.; Pastar, I. The role of tgfb signaling in wound epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Siddiqui, A.; Morris, D.E.; Cox, D.A.; Roth, S.I.; Mustoe, T.A. Transforming growth factor beta 3 (tgf beta 3) accelerates wound healing without alteration of scar prominence. Histologic and competitive reverse-transcription-polymerase chain reaction studies. Arch. Surg. 1997, 132, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Le, M.; Naridze, R.; Morrison, J.; Biggs, L.C.; Rhea, L.; Schutte, B.C.; Kaartinen, V.; Dunnwald, M. Transforming growth factor beta 3 is required for excisional wound repair in vivo. PLoS ONE 2012, 7, e48040. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saulis, A.S.; Liu, W.R.; Roy, N.K.; Chao, J.D.; Ledbetter, S.; Mustoe, T.A. The temporal effects of anti-tgf-beta1, 2, and 3 monoclonal antibody on wound healing and hypertrophic scar formation. J. Am. Coll. Surg. 2005, 201, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, T.A.; Pierce, G.F.; Thomason, A.; Gramates, P.; Sporn, M.B.; Deuel, T.F. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science 1987, 237, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Puolakkainen, P.A.; Reed, M.J.; Gombotz, W.R.; Twardzik, D.R.; Abrass, I.B.; Sage, H.E. Acceleration of wound healing in aged rats by topical application of transforming growth factor-beta(1). Wound Repair Regen. 1995, 3, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin. Cancer Biol. 2012, 22, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, F.J.; Lehmann, K.; Warne, P.H.; Hill, C.S.; Downward, J. Epithelial to mesenchymal transition in madin-darby canine kidney cells is accompanied by down-regulation of smad3 expression, leading to resistance to transforming growth factor-beta-induced growth arrest. J. Biol. Chem. 2003, 278, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Yang, X.; Glick, A.B.; Weinstein, M.; Letterio, J.L.; Mizel, D.E.; Anzano, M.; Greenwell-Wild, T.; Wahl, S.M.; Deng, C.; et al. Mice lacking smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999, 1, 260–266. [Google Scholar] [PubMed]
- Jinno, K.; Takahashi, T.; Tsuchida, K.; Tanaka, E.; Moriyama, K. Acceleration of palatal wound healing in smad3-deficient mice. J. Dent. Res. 2009, 88, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The crumbs complex couples cell density sensing to hippo-dependent control of the tgf-beta-smad pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates yap/taz to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; D’Amore, P.A. Blood vessel formation: What is its molecular basis? Cell 1996, 87, 1153–1155. [Google Scholar] [CrossRef]
- Nissen, N.N.; Polverini, P.J.; Koch, A.E.; Volin, M.V.; Gamelli, R.L.; DiPietro, L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am. J. Pathol. 1998, 152, 1445–1452. [Google Scholar] [PubMed]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [PubMed]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Pentinmikko, N.; Ilmonen, M.; Salven, P. Dual action of tgf-beta induces vascular growth in vivo through recruitment of angiogenic vegf-producing hematopoietic effector cells. Angiogenesis 2012, 15, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.A.; Tombetti, E.; Maugeri, N.; Rovere-Querini, P.; Brunelli, S.; Manfredi, A.A. Vascular remodelling and mesenchymal transition in systemic sclerosis. Stem Cells Int. 2016, 2016, 4636859. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to mesenchymal transition (endomt) in the pathogenesis of human fibrotic diseases. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Ehsan, S.M.; Wang, K.; Wu, N.; Newman, A.C.; Romero-Lopez, M.; Fong, A.H.; George, S.C.; Edwards, R.A.; Hughes, C.C. Angiogenic sprouting is regulated by endothelial cell expression of slug. J. Cell Sci. 2014, 127, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Opalenik, S.R.; Davidson, J.M. Fibroblast differentiation of bone marrow-derived cells during wound repair. FASEB J. 2005, 19, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Donnelly, S.C.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. Tgf-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Chin, G.S.; Liu, W.; Peled, Z.; Lee, T.Y.; Steinbrech, D.S.; Hsu, M.; Longaker, M.T. Differential expression of transforming growth factor-beta receptors I and II and activation of smad 3 in keloid fibroblasts. Plastic Reconstr. Surg. 2001, 108, 423–429. [Google Scholar] [CrossRef]
- Querfeld, C.; Eckes, B.; Huerkamp, C.; Krieg, T.; Sollberg, S. Expression of tgf-beta 1, -beta 2 and -beta 3 in localized and systemic scleroderma. J. Dermatol. Sci. 1999, 21, 13–22. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Duncan, J.; Bond, J.; Bush, J.; Durani, P.; So, K.; Taylor, L.; Chantrey, J.; Mason, T.; James, G.; et al. Prophylactic administration of avotermin for improvement of skin scarring: Three double-blind, placebo-controlled, phase I/II studies. Lancet 2009, 373, 1264–1274. [Google Scholar] [CrossRef]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralisation of tgf-beta 1 and tgf-beta 2 or exogenous addition of tgf-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108(Pt. 3), 985–1002. [Google Scholar] [PubMed]
- Murata, H.; Zhou, L.; Ochoa, S.; Hasan, A.; Badiavas, E.; Falanga, V. Tgf-beta3 stimulates and regulates collagen synthesis through tgf-beta1-dependent and independent mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Gabbiana, G. Modulation of alpha-smooth muscle actin expression in fibroblasts by transforming growth factor-beta isoforms: An in vivo and in vitro study. Wound Repair Regen. 1996, 4, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Corr, D.T.; Gallant-Behm, C.L.; Shrive, N.G.; Hart, D.A. Biomechanical behavior of scar tissue and uninjured skin in a porcine model. Wound Repair Regen. 2009, 17, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.W.; Kiama, S.G.; Seifert, M.G.; Goheen, J.R.; Palmer, T.M.; Maden, M. Skin shedding and tissue regeneration in african spiny mice (acomys). Nature 2012, 489, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Brant, J.O.; Lopez, M.C.; Baker, H.V.; Barbazuk, W.B.; Maden, M. A comparative analysis of gene expression profiles during skin regeneration in mus and acomys. PLoS ONE 2015, 10, e0142931. [Google Scholar] [CrossRef] [PubMed]
- Cass, D.L.; Bullard, K.M.; Sylvester, K.G.; Yang, E.Y.; Longaker, M.T.; Adzick, N.S. Wound size and gestational age modulate scar formation in fetal wound repair. J. Pediatr. Surg. 1997, 32, 411–415. [Google Scholar] [CrossRef]
- Rowlatt, U. Intrauterine wound healing in a 20 week human fetus. Virchows Archiv. A Pathol. Anat. Histol. 1979, 381, 353–361. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Soo, C.; Hu, F.Y.; Zhang, X.; Wang, Y.; Beanes, S.R.; Lorenz, H.P.; Hedrick, M.H.; Mackool, R.J.; Plaas, A.; Kim, S.J.; et al. Differential expression of fibromodulin, a transforming growth factor-beta modulator, in fetal skin development and scarless repair. Am. J. Pathol. 2000, 157, 423–433. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Lorenz, H.P.; Meuli, M.; Lin, R.Y.; Adzick, N.S. A model of scarless human fetal wound repair is deficient in transforming growth factor beta. J. Pediatr. Surg. 1995, 30, 198–202. [Google Scholar] [CrossRef]
- Tanaka, E.M.; Reddien, P.W. The cellular basis for animal regeneration. Dev. Cell 2011, 21, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Kragl, M.; Knapp, D.; Nacu, E.; Khattak, S.; Maden, M.; Epperlein, H.H.; Tanaka, E.M. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature 2009, 460, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Lehoczky, J.A.; Robert, B.; Tabin, C.J. Mouse digit tip regeneration is mediated by fate-restricted progenitor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20609–20614. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Lindau, P.; Ueno, H.; Longaker, M.T.; Weissman, I.L. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature 2011, 476, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Stoick-Cooper, C.L.; Moon, R.T.; Weidinger, G. Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev. 2007, 21, 1292–1315. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.N.; Tassava, R.A. Apical epithelial cap morphology and fibronectin gene expression in regenerating axolotl limbs. Dev. Dyn. 2000, 217, 216–224. [Google Scholar] [CrossRef]
- Nacu, E.; Tanaka, E.M. Limb regeneration: A new development? Annu. Rev. Cell Dev. Biol. 2011, 27, 409–440. [Google Scholar] [CrossRef] [PubMed]
- Globus, M.; Vethamany-Globus, S.; Lee, Y.C. Effect of apical epidermal cap on mitotic cycle and cartilage differentiation in regeneration blastemata in the newt, notophthalmus viridescens. Dev. Biol. 1980, 75, 358–372. [Google Scholar] [CrossRef]
- Neufeld, D.A.; Day, F.A.; Settles, H.E. Stabilizing role of the basement membrane and dermal fibers during newt limb regeneration. Anat. Rec. 1996, 245, 122–127. [Google Scholar] [CrossRef]
- Campbell, L.J.; Crews, C.M. Wound epidermis formation and function in urodele amphibian limb regeneration. Cell. Mol. Life Sci. 2008, 65, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.J.; Suarez-Castillo, E.C.; Ortiz-Zuazaga, H.; Knapp, D.; Tanaka, E.M.; Crews, C.M. Gene expression profile of the regeneration epithelium during axolotl limb regeneration. Dev. Dyn. 2011, 240, 1826–1840. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, J.R.; Athippozhy, A.; Seifert, A.W.; Putta, S.; Stromberg, A.J.; Maden, M.; Gardiner, D.M.; Voss, S.R. Gene expression patterns specific to the regenerating limb of the mexican axolotl. Biol. Open 2012, 1, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.S. The effect of apical cap removal on limb regeneration in amblystoma larvae. J. Exp. Zool. 1957, 134, 357–381. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. Sb-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (alk) receptors alk4, alk5, and alk7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Levesque, M.; Gatien, S.; Finnson, K.; Desmeules, S.; Villiard, E.; Pilote, M.; Philip, A.; Roy, S. Transforming growth factor: Beta signaling is essential for limb regeneration in axolotls. PLoS ONE 2007, 2, e1227. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.M.; Whitman, M. Tgf-beta signaling is required for multiple processes during xenopus tail regeneration. Dev. Biol. 2008, 315, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Jazwinska, A.; Badakov, R.; Keating, M.T. Activin-betaa signaling is required for zebrafish fin regeneration. Curr. Biol. 2007, 17, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Veit, J.; Rainer, G.; Jazwinska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on tgfb signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.W.; Vickaryous, M.K.; Viloria-Petit, A.M. Characterization of tgfb signaling during tail regeneration in the leopard gecko (eublepharis macularius). Dev. Dyn. 2013, 242, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.D.; Clark, R.K.; Heber-Katz, E. A new murine model for mammalian wound repair and regeneration. Clin. Immunol. Immunopathol. 1998, 88, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Heber-Katz, E. The regenerating mouse ear. Semin. Cell Dev. Biol. 1999, 10, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kreft, B.; Yokoyama, H.; Naito, T.; Kelley, V.R. Dysregulated transforming growth factor-beta in neonatal and adult autoimmune mrl-lpr mice. J. Autoimmun. 1996, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Kench, J.A.; Russell, D.M.; Fadok, V.A.; Young, S.K.; Worthen, G.S.; Jones-Carson, J.; Henson, J.E.; Henson, P.M.; Nemazee, D. Aberrant wound healing and tgf-beta production in the autoimmune-prone mrl/+ mouse. Clin. Immunol. 1999, 92, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Rao, J.K.; Gilkeson, G.S.; Ruiz, P.; Eicher, E.M.; Pisetsky, D.S.; Matsuzawa, A.; Rochelle, J.M.; Seldin, M.F. Genetic analysis of mrl-lpr mice: Relationship of the fas apoptosis gene to disease manifestations and renal disease-modifying loci. J. Exp. Med. 1992, 176, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Tolba, R.H.; Schildberg, F.A.; Decker, D.; Abdullah, Z.; Buttner, R.; Minor, T.; von Ruecker, A. Mechanisms of improved wound healing in murphy roths large (mrl) mice after skin transplantation. Wound Repair Regen. 2010, 18, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Johnson, K.; Li, J.; Piamonte, V.; Steffy, B.M.; Hsieh, M.H.; Ng, N.; Zhang, J.; Walker, J.R.; Ding, S.; et al. Regenerative phenotype in mice with a point mutation in transforming growth factor beta type I receptor (tgfbr1). Proc. Natl. Acad. Sci. USA 2011, 108, 14560–14565. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the tgfb signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Ten Dijke, P. Exploring anti-tgf-beta therapies in cancer and fibrosis. Growth Factors 2011, 29, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Carlson, M.E.; Conboy, M.J.; Hsu, M.; Barchas, L.; Jeong, J.; Agrawal, A.; Mikels, A.J.; Agrawal, S.; Schaffer, D.V.; Conboy, I.M. Relative roles of tgf-beta1 and wnt in the systemic regulation and aging of satellite cell responses. Aging Cell 2009, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.P.; Liu, Y.H.; Ho, Y.J.; Wu, S.M. Pharmacological inhibition of tgfb receptor improves nkx2.5 cardiomyoblast-mediated regeneration. Cardiovasc. Res. 2015, 105, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Q.; Liu, K.; Shen, J.F.; Xu, G.T.; Ye, W. Sb-431542 inhibition of scar formation after filtration surgery and its potential mechanism. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, S.B.; Barry, S.T.; Delves, C.J.; Horgan, C.M. The integrin alphavbeta3 is a receptor for the latency-associated peptides of transforming growth factors beta1 and beta3. Biochem. J. 2003, 369, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra279. [Google Scholar] [CrossRef] [PubMed]
- Lonning, S.; Mannick, J.; McPherson, J.M. Antibody targeting of tgf-beta in cancer patients. Curr. Pharm. Biotechnol. 2011, 12, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.L.; Wong, T.T.; Cordeiro, M.F.; Anderson, I.K.; Khaw, P.T. Evaluation of anti-tgf-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3394–3401. [Google Scholar] [CrossRef]
- Siriwardena, D.; Khaw, P.T.; King, A.J.; Donaldson, M.L.; Overton, B.M.; Migdal, C.; Cordeiro, M.F. Human antitransforming growth factor beta(2) monoclonal antibody--a new modulator of wound healing in trabeculectomy: A randomized placebo controlled clinical study. Ophthalmology 2002, 109, 427–431. [Google Scholar] [CrossRef]
- Khaw, P.; Grehn, F.; Hollo, G.; Overton, B.; Wilson, R.; Vogel, R.; Smith, Z. A phase III study of subconjunctival human anti-transforming growth factor beta(2) monoclonal antibody (cat-152) to prevent scarring after first-time trabeculectomy. Ophthalmology 2007, 114, 1822–1830. [Google Scholar] [PubMed]
- Bonafoux, D.; Lee, W.C. Strategies for tgf-beta modulation: A review of recent patents. Expert Opin. Ther. Pat. 2009, 19, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Ezquerro, I.J.; Lasarte, J.J.; Dotor, J.; Castilla-Cortazar, I.; Bustos, M.; Penuelas, I.; Blanco, G.; Rodriguez, C.; Lechuga Mdel, C.; Greenwel, P.; et al. A synthetic peptide from transforming growth factor beta type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine 2003, 22, 12–20. [Google Scholar] [CrossRef]
- Santiago, B.; Gutierrez-Canas, I.; Dotor, J.; Palao, G.; Lasarte, J.J.; Ruiz, J.; Prieto, J.; Borras-Cuesta, F.; Pablos, J.L. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J. Investig. Dermatol. 2005, 125, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.S.; Dotor, J.; Hontanilla, B. Effect of p144(r) (anti-tgf-beta) in an “in vivo” human hypertrophic scar model in nude mice. PLoS ONE 2015, 10, e0144489. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.F.; Mead, A.; Ali, R.R.; Alexander, R.A.; Murray, S.; Chen, C.; York-Defalco, C.; Dean, N.M.; Schultz, G.S.; Khaw, P.T. Novel antisense oligonucleotides targeting tgf-beta inhibit in vivo scarring and improve surgical outcome. Gene Ther. 2003, 10, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Loiselle, A.E.; Yukata, K.; Geary, M.B.; Kondabolu, S.; Shi, S.; Jonason, J.H.; Awad, H.A.; O’Keefe, R.J. Development of antisense oligonucleotide (aso) technology against tgf-beta signaling to prevent scarring during flexor tendon repair. J. Orthop. Res. 2015, 33, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Thangapazham, R.L.; Sharad, S.; Maheshwari, R.K. Skin regenerative potentials of curcumin. BioFactors 2013, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, T.A.; Ruoslahti, E. Targeted antiscarring therapy for tissue injuries. Adv. Wound Care 2013, 2, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Spector, I. Halofuginone—The multifaceted molecule. Molecules 2015, 20, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (tgf-beta) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L. Angiotensin receptor blockers: A panacea for marfan syndrome and related disorders? Drug Discov. Today 2015, 20, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates tgf-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Uehara, K.; Ota, S.; Tobita, K.; Ambrosio, F.; Cummins, J.H.; Terada, S.; Fu, F.H.; Huard, J. The timing of administration of a clinically relevant dose of losartan influences the healing process after contusion induced muscle injury. J. Appl. Physiol. 2013, 114, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Kamber, M.; Papalazarou, V.; Rouni, G.; Papageorgopoulou, E.; Papalois, A.; Kostourou, V. Angiotensin II inhibitor facilitates epidermal wound regeneration in diabetic mice. Front. Physiol. 2015, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, N.; Shea, B.S.; Tager, A.M. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am. J. Respir. Crit. Care Med. 2014, 190, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Guha, R.; Trivedi, R.; Kompella, U.B.; Konar, A.; Hazra, S. Pirfenidone nanoparticles improve corneal wound healing and prevent scarring following alkali burn. PLoS ONE 2013, 8, e70528. [Google Scholar] [CrossRef] [PubMed]
- Mandapalli, P.K.; Labala, S.; Bojja, J.; Venuganti, V.V. Effect of pirfenidone delivered using layer-by-layer thin film on excisional wound healing. Eur. J. Pharm. Sci. 2016, 83, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Occleston, N.L.; O’Kane, S.; Laverty, H.G.; Cooper, M.; Fairlamb, D.; Mason, T.; Bush, J.A.; Ferguson, M.W. Discovery and development of avotermin (recombinant human transforming growth factor beta 3): A new class of prophylactic therapeutic for the improvement of scarring. Wound Repair Regen. 2011, 19, s38–s48. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.; Duncan, J.A.; Bond, J.S.; Durani, P.; So, K.; Mason, T.; O’Kane, S.; Ferguson, M.W. Scar-improving efficacy of avotermin administered into the wound margins of skin incisions as evaluated by a randomized, double-blind, placebo-controlled, phase ii clinical trial. Plastic Reconstr. Surg. 2010, 126, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- McCollum, P.T.; Bush, J.A.; James, G.; Mason, T.; O’Kane, S.; McCollum, C.; Krievins, D.; Shiralkar, S.; Ferguson, M.W. Randomized phase ii clinical trial of avotermin versus placebo for scar improvement. Br. J. Surg. 2011, 98, 925–934. [Google Scholar] [CrossRef] [PubMed]
- So, K.; McGrouther, D.A.; Bush, J.A.; Durani, P.; Taylor, L.; Skotny, G.; Mason, T.; Metcalfe, A.; O’Kane, S.; Ferguson, M.W. Avotermin for scar improvement following scar revision surgery: A randomized, double-blind, within-patient, placebo-controlled, phase ii clinical trial. Plastic Reconstr. Surg. 2011, 128, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.O.; Shakib, K.; Heliotis, M.; Tsiridis, E.; Mantalaris, A.; Ripamonti, U.; Tsiridis, E. Tgf-beta3: A potential biological therapy for enhancing chondrogenesis. Expert Opin. Biol. Ther. 2009, 9, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Almeida, H.V.; Liu, Y.; Cunniffe, G.M.; Mulhall, K.J.; Matsiko, A.; Buckley, C.T.; O’Brien, F.J.; Kelly, D.J. Controlled release of transforming growth factor-beta3 from cartilage-extra-cellular-matrix-derived scaffolds to promote chondrogenesis of human-joint-tissue-derived stem cells. Acta Biomater. 2014, 10, 4400–4409. [Google Scholar] [CrossRef] [PubMed]
- Shakir, S.; MacIsaac, Z.M.; Naran, S.; Smith, D.M.; Bykowski, M.R.; Cray, J.J.; Craft, T.K.; Wang, D.; Weiss, L.; Campbell, P.G.; et al. Transforming growth factor beta 1 augments calvarial defect healing and promotes suture regeneration. Tissue Eng. Part A 2015, 21, 939–947. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilbert, R.W.D.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. https://doi.org/10.3390/jdb4020021
Gilbert RWD, Vickaryous MK, Viloria-Petit AM. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. Journal of Developmental Biology. 2016; 4(2):21. https://doi.org/10.3390/jdb4020021
Chicago/Turabian StyleGilbert, Richard W.D., Matthew K. Vickaryous, and Alicia M. Viloria-Petit. 2016. "Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration" Journal of Developmental Biology 4, no. 2: 21. https://doi.org/10.3390/jdb4020021
APA StyleGilbert, R. W. D., Vickaryous, M. K., & Viloria-Petit, A. M. (2016). Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. Journal of Developmental Biology, 4(2), 21. https://doi.org/10.3390/jdb4020021