
Restricted Pax3 Deletion within the Neural Tube Results in Congenital Hydrocephalus

Abstract
:1. Introduction
2. Experimental Section
2.1. Mice
2.2. Histology, Immunohistochemistry, X-gal Staining, and in Situ Hybridization
2.3. Skeletal Preparations
2.4. Western Blot Analysis
3. Results
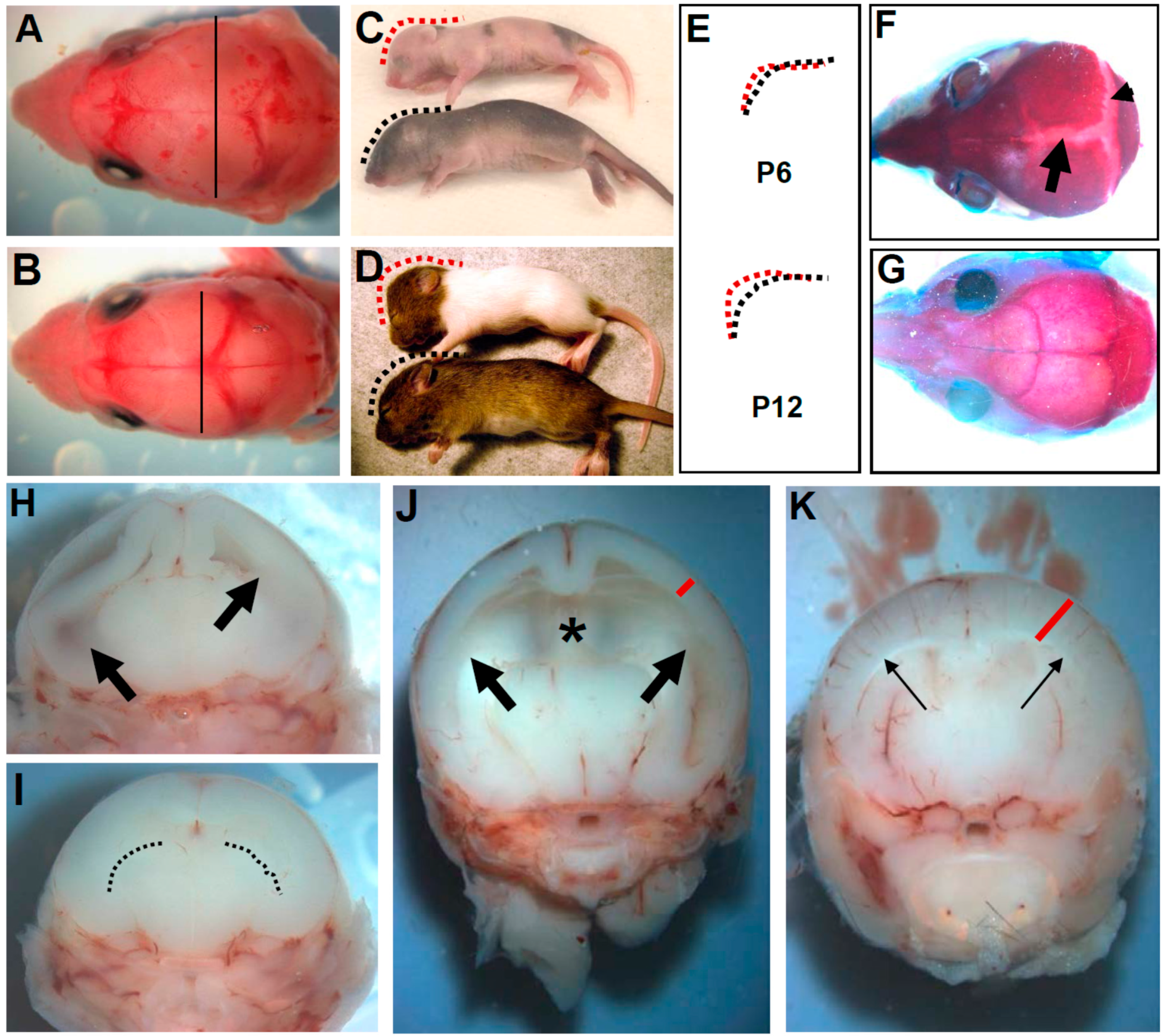
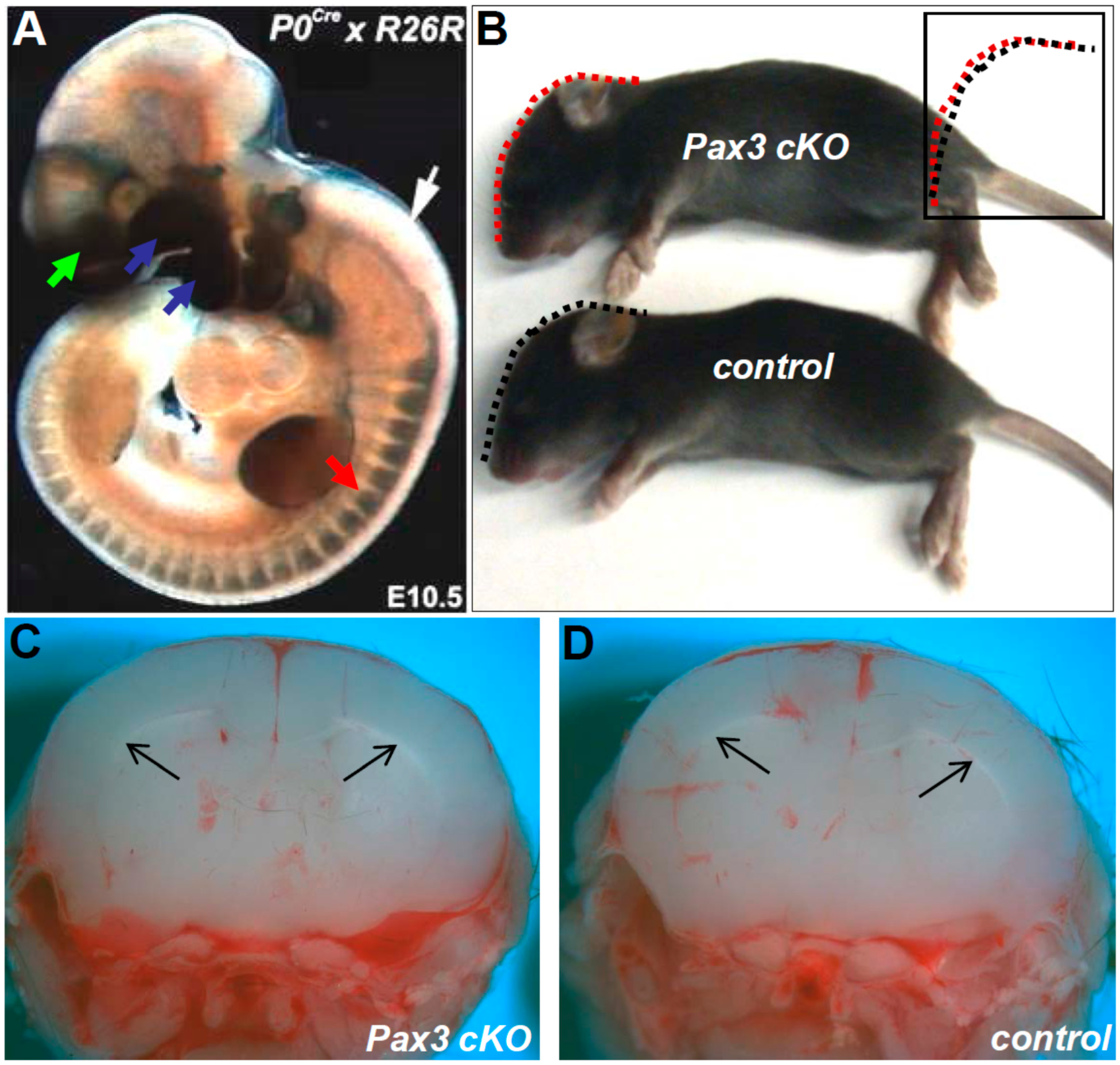
3.1. Wnt1-Cre Restricted Pax3 Deletion Causes Perinatal Hydrocephalus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant Line * | %Hydrocephalus ** |
|---|---|
| Wnt1-Cre:Pax3flox/flox | 100 (50/50) |
| P0-Cre:Pax3flox/flox | 0 (0/20) |
| Pax7−Cre:Pax3flox/flox | 100 (20/20) |
| Pax3+/Δ5:Pax7+/Δ2 | 40 (20/50) |
| Pax3+/Δ5:Pax7+/+ | 0 (0/100) |
| Pax3+/+:Pax7+/Δ2 | 0 (0/100) |
| Pax3+/+:Pax7Δ2/Δ2 | 0 (0/30) |
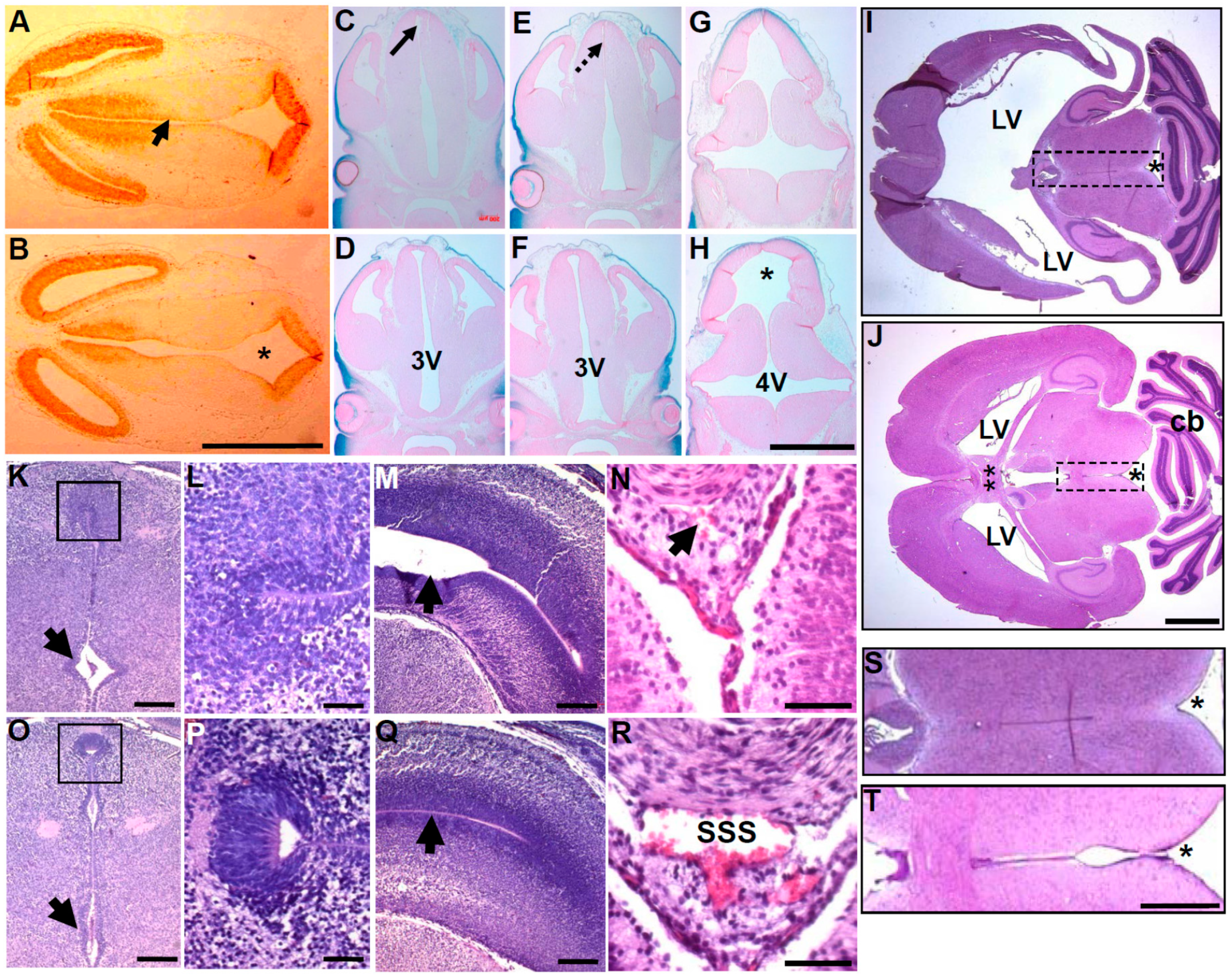
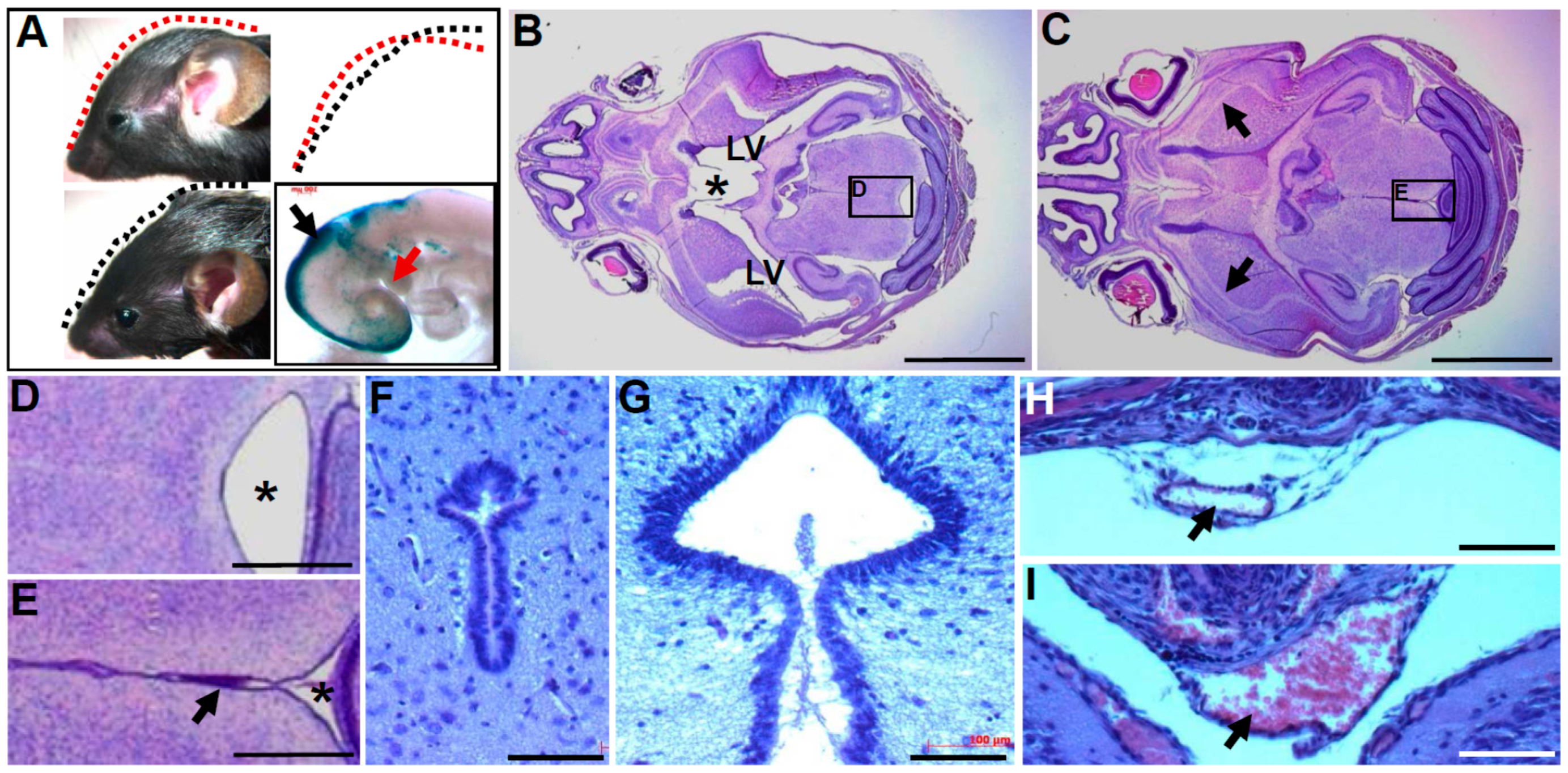
3.2. In Utero Tissue Loss and Progressive Ventricular Dilation in Hydrocephalic Wnt1-Cre-Mediated Pax3 Mutants
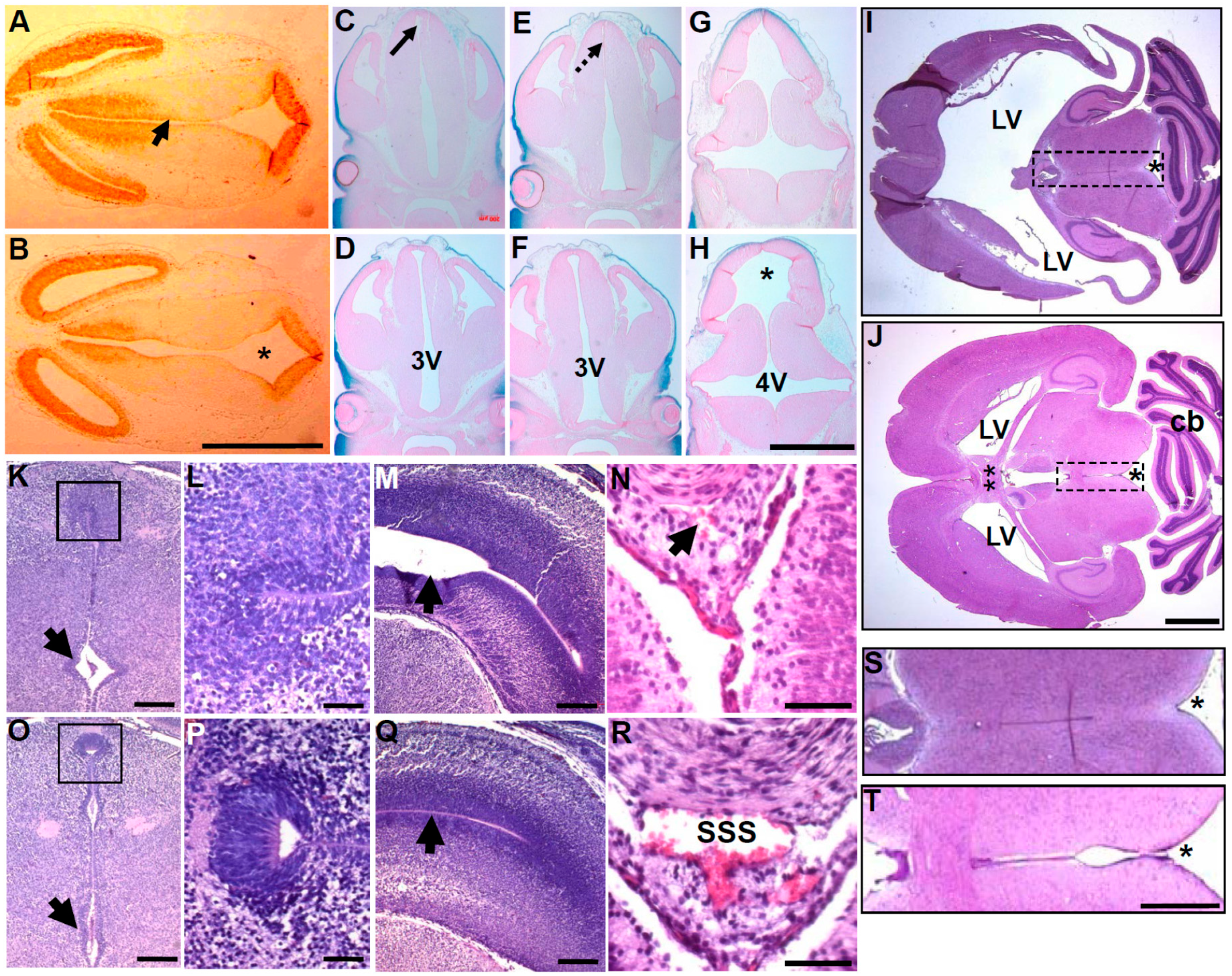
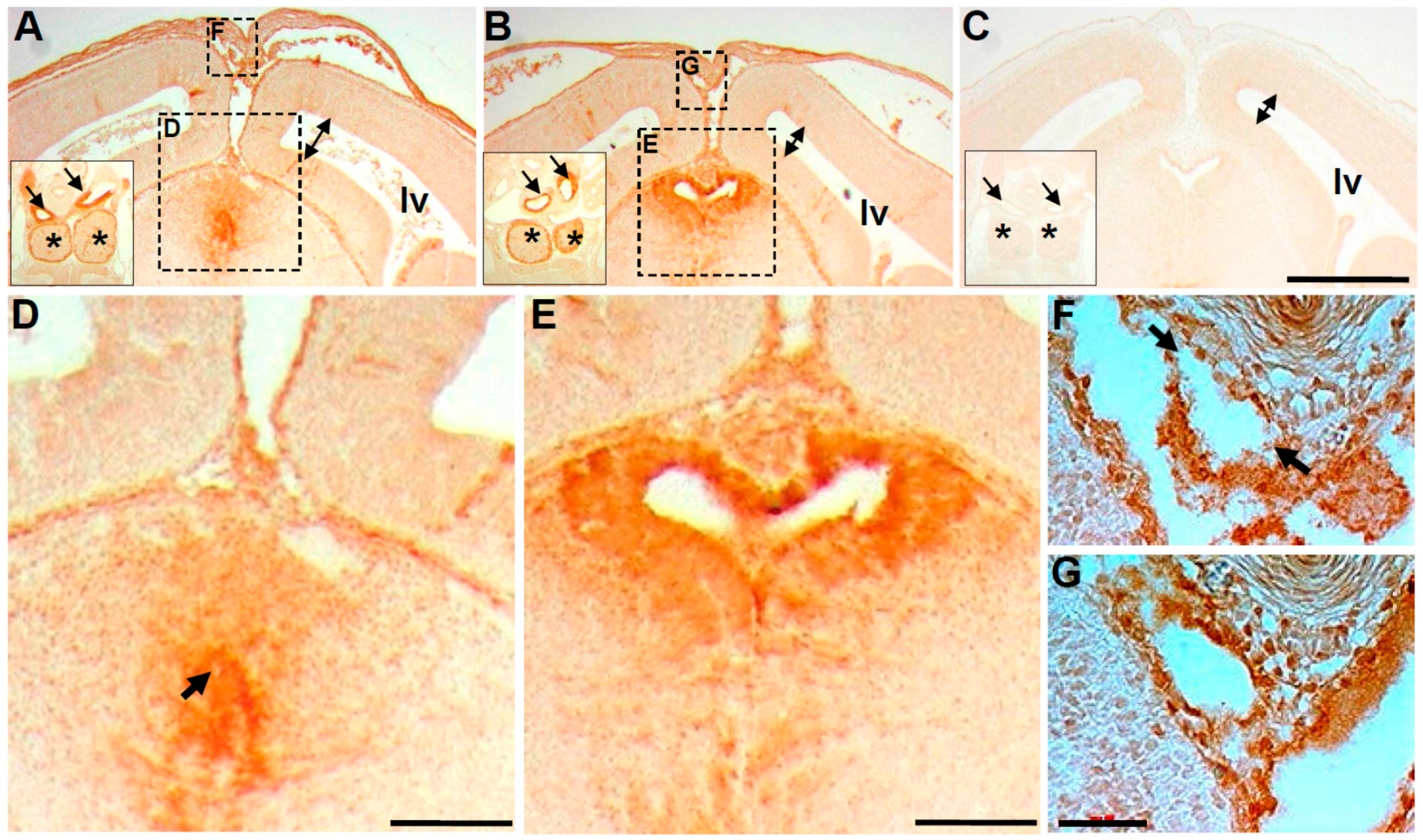
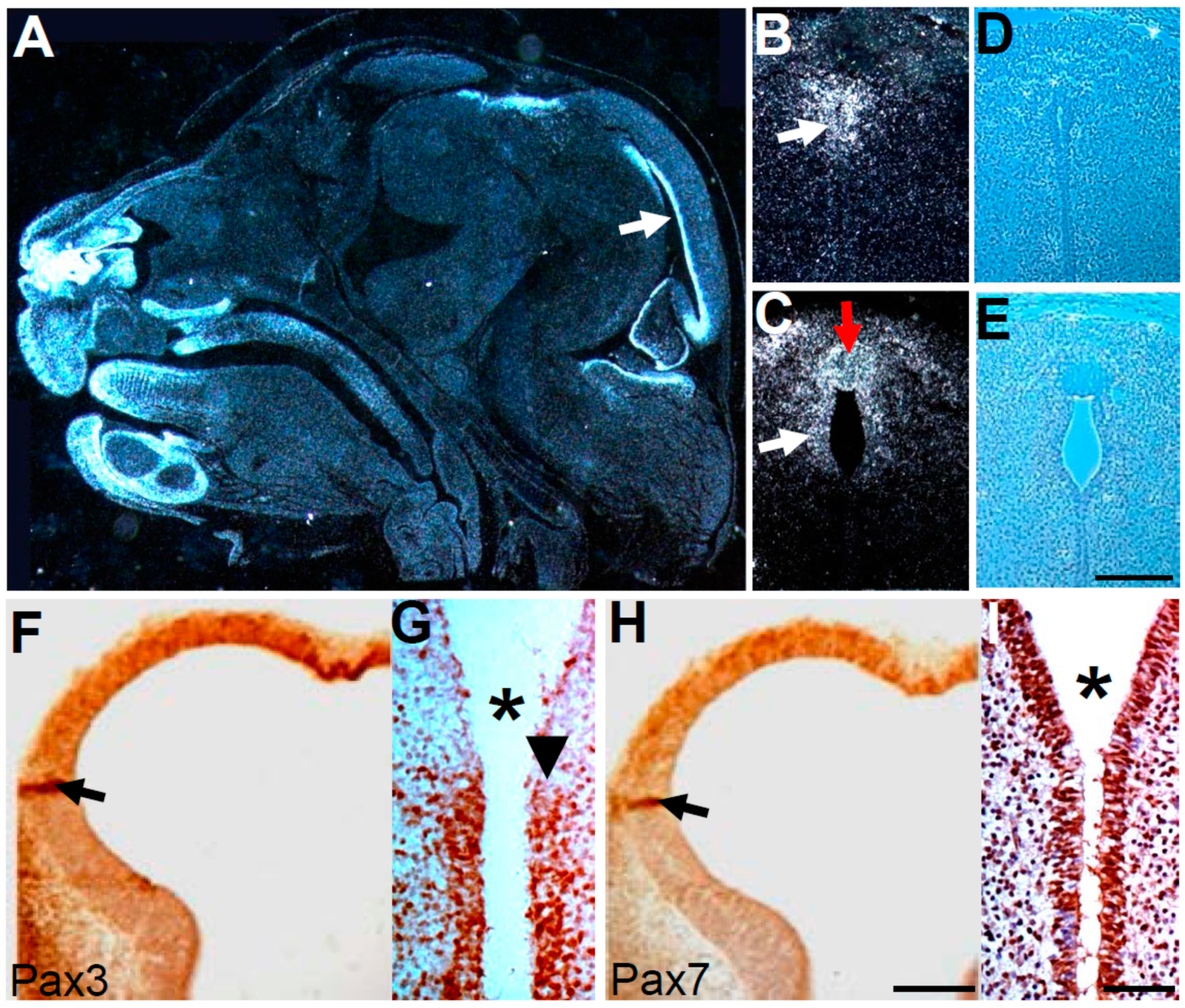
3.3. Wnt1-Cre Lineage Mapping Reveal that Ventricular Neuroepithelium and Subcommissural Organ Morphogenesis Requires Pax3
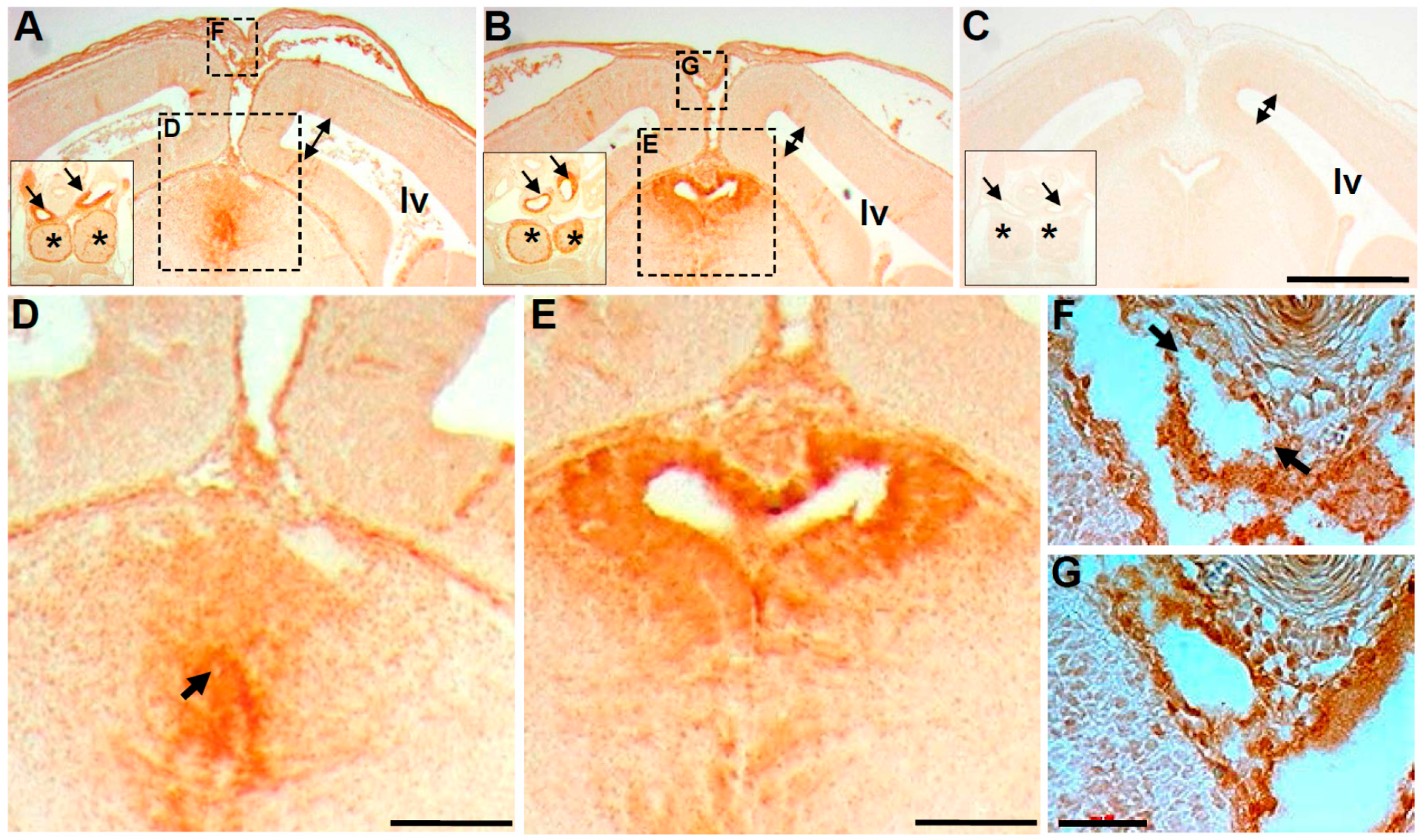
3.4. Molecular Marker Analysis Reveals Neuroepithelial Dysmorphia

3.5. Restricted Loss of Pax3 within Migratory Neural Crest Does not Cause Hydrocephalus
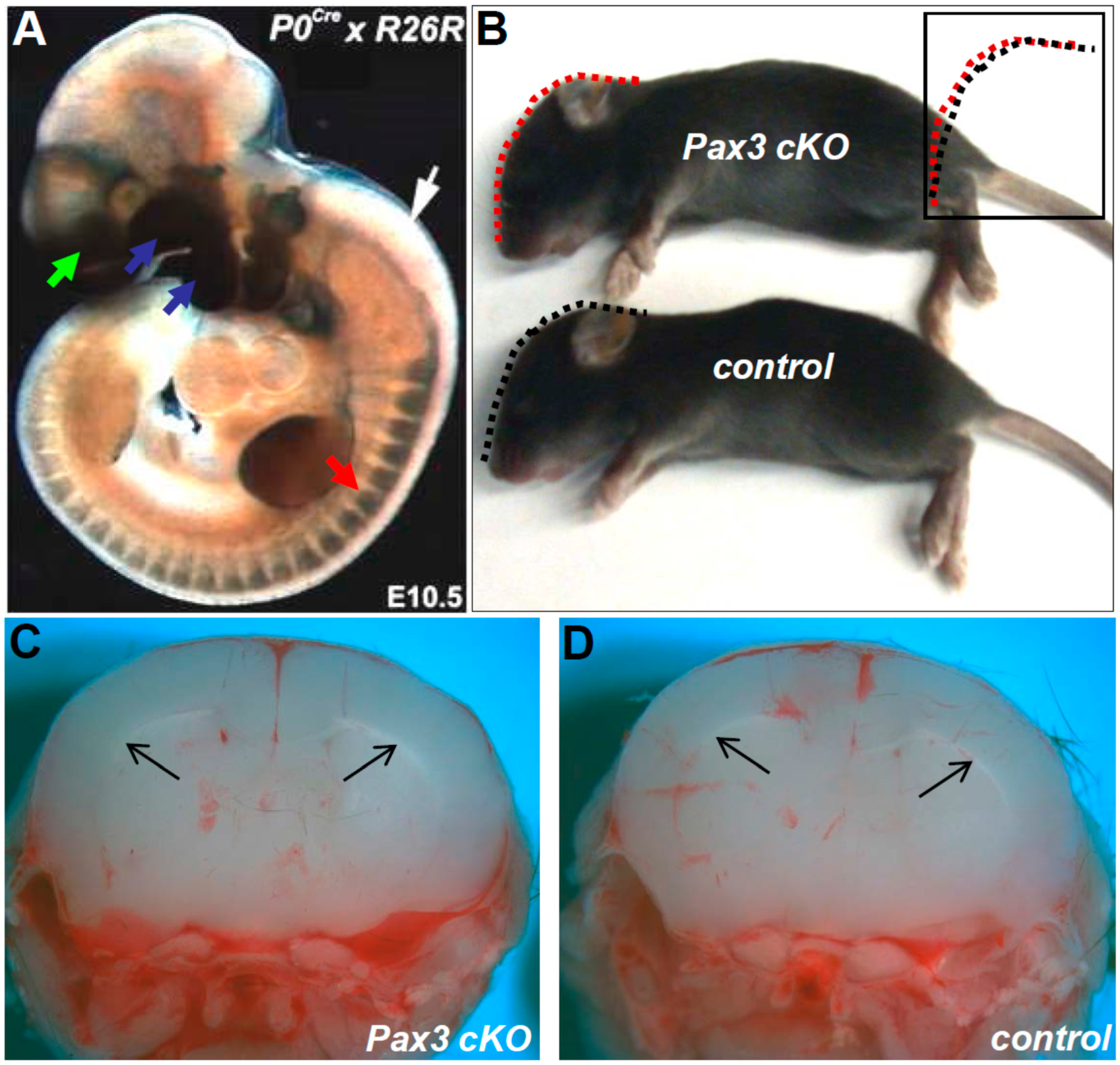
3.6. Neuroepithelial Deletion of Pax3 is Sufficient to Cause Hydrocephalus
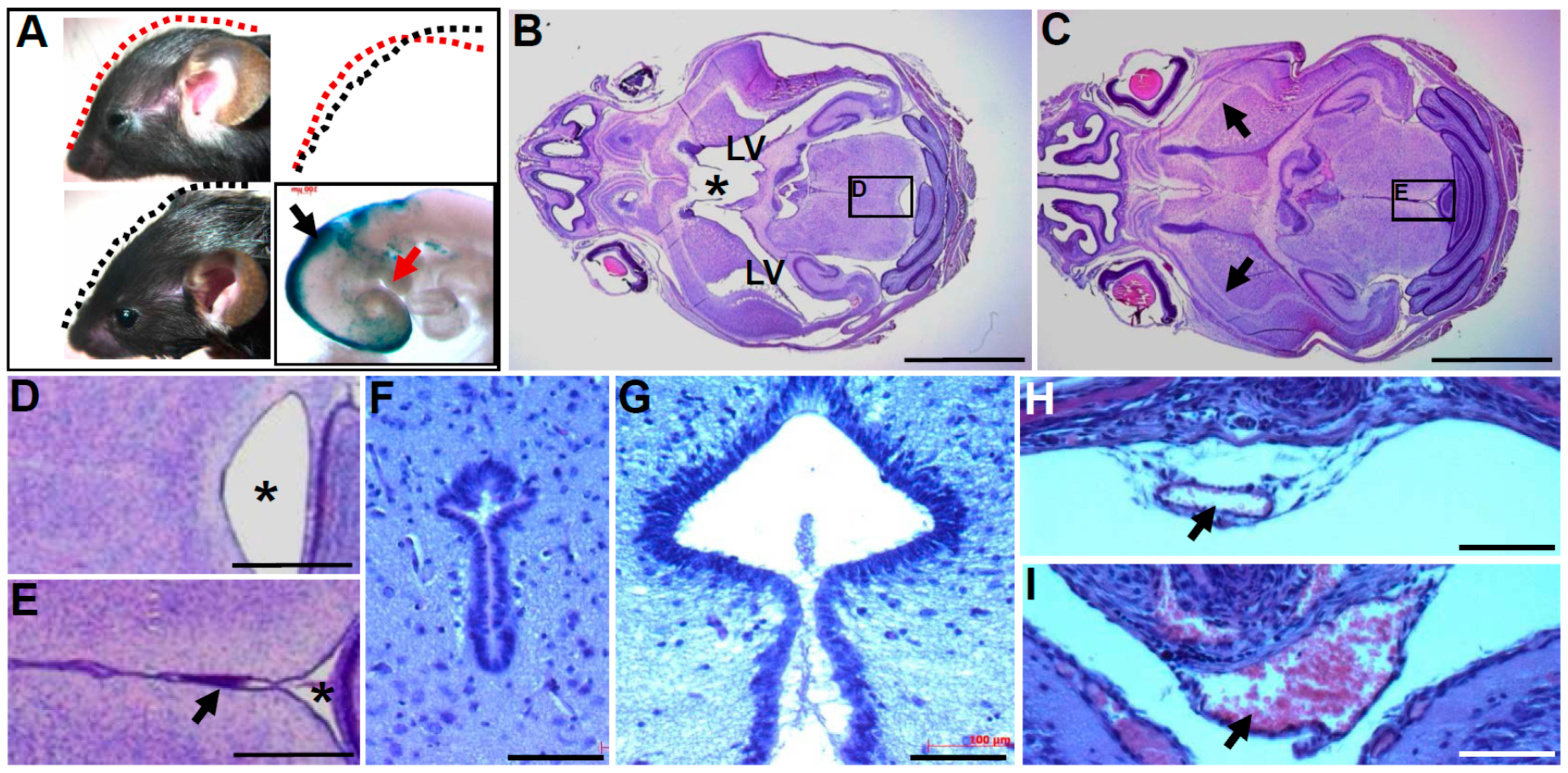
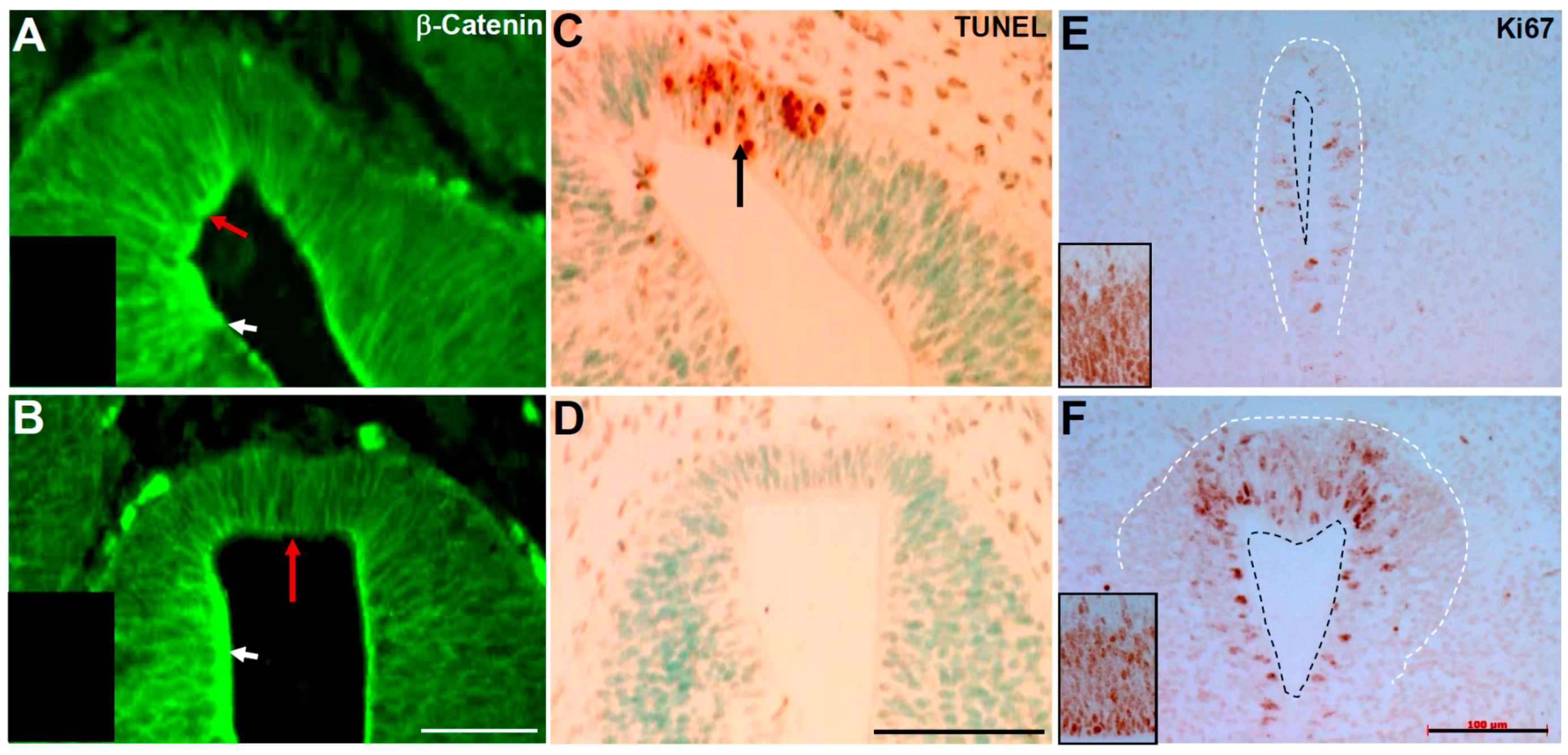
3.7. Pax3 May Play a Role in Regulating Cell Proliferation within the Epithelium
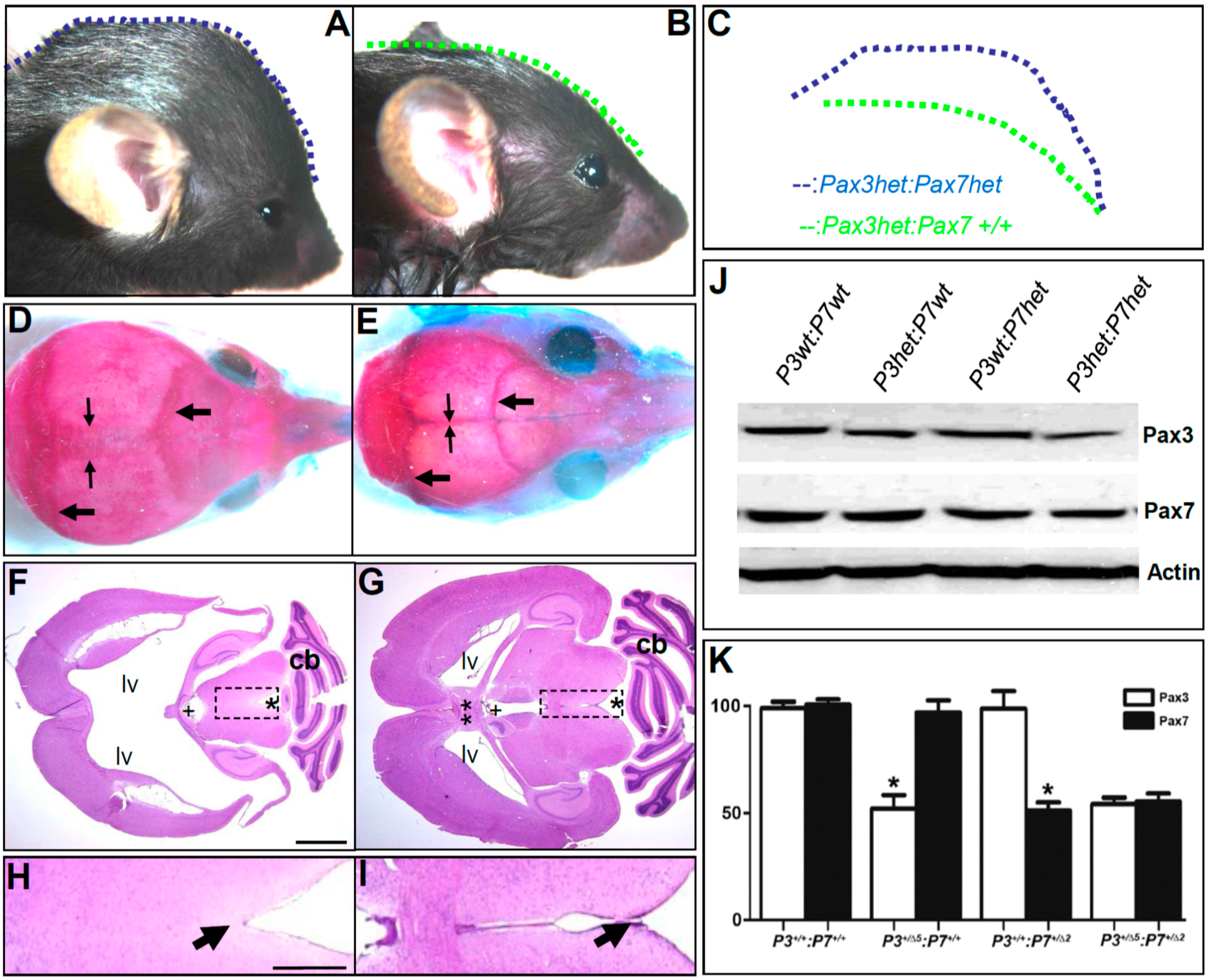
3.8. Pax3 and Pax7 Play a Combinatorial Role during Third Ventricle Morphogenesis
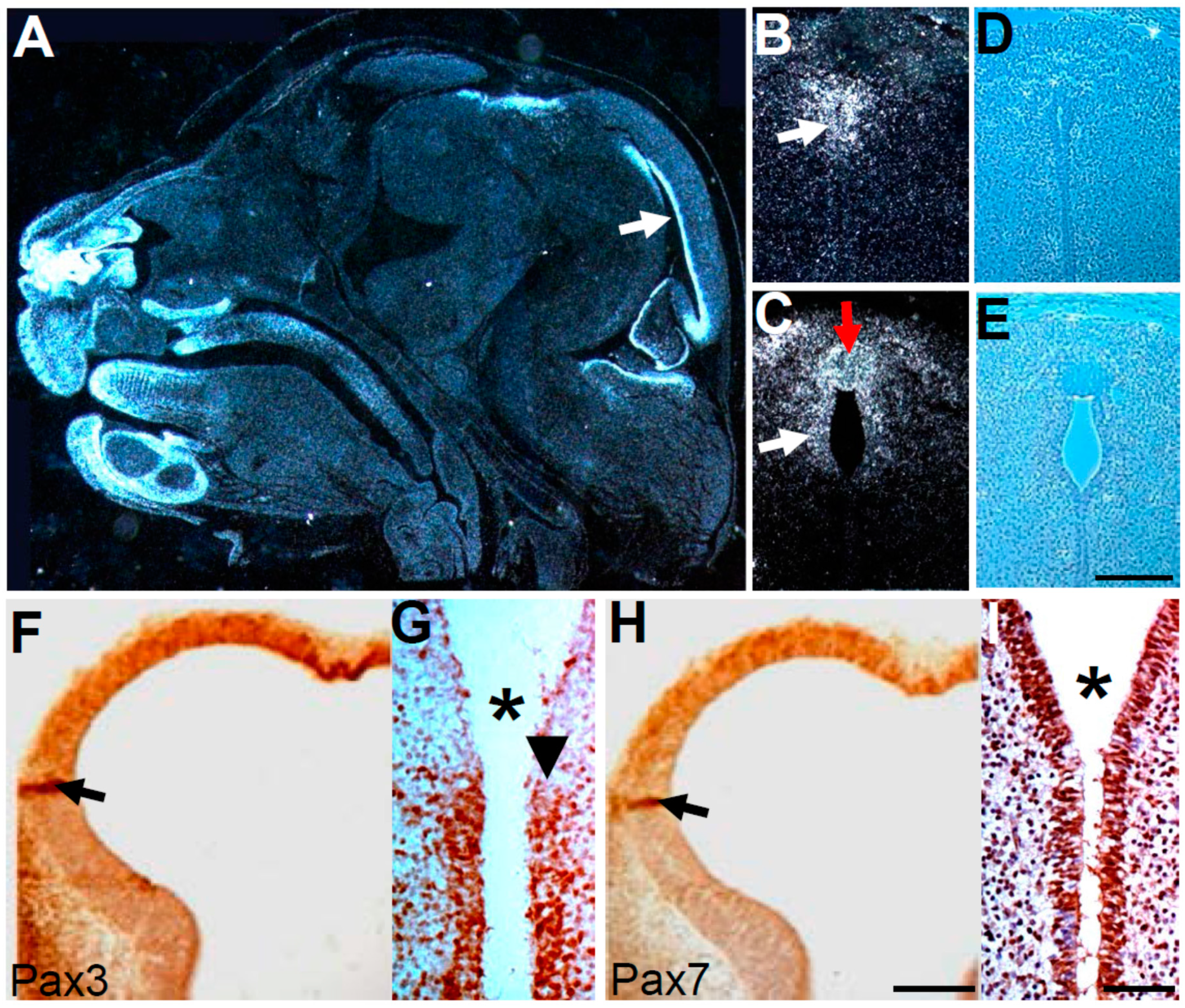

4. Discussion
4.1. Pax Family Genes are Required in Utero for Neuroepithelial Morphogenesis
4.2. Functional Effects of Pax3 Mutation in Congenital Hydrocephalus Pathogenesis
4.3. Congenital Hydrocephalus in Patients
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Conway, S.J.; Henderson, D.J.; Copp, A.J. Pax3 is required for cardiac neural crest migration in the mouse: Evidence from the Splotch (Sp2H) mutant. Development 1997, 124, 505–514. [Google Scholar] [PubMed]
- Epstein, J.A.; Li, J.; Lang, D.; Chen, F.; Brown, C.B.; Jin, F.; Lu, M.M.; Thomas, M.; Liu, E.; Wessels, A.; et al. Migration of cardiac neural crest cells in Splotch embryos. Development 2000, 127, 1869–1878. [Google Scholar] [PubMed]
- Zhou, H.M.; Wang, J.; Rogers, R.; Conway, S.J. Lineage-specific responses to reduced embryonic Pax3 expression levels. Dev. Biol. 2008, 315, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004, 18, 1088–1105. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.J.; Bundy, J.; Chen, J.; Dickman, E.; Rogers, R.; Will, B.M. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc. Res. 2000, 47, 314–328. [Google Scholar] [CrossRef]
- Etchevers, H.C.; Couly, G.; Vincent, C.; le Douarin, N.M. Anterior cephalic neural crest is required for forebrain viability. Development 1999, 126, 3533–3543. [Google Scholar] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Koushik, S.V.; Chen, H.; Wang, J.; Conway, S.J. Generation of a conditional loxP allele of the Pax3 transcription factor that enables selective deletion of the homeodomain. Genesis 2002, 32, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Olaopa, M.; Zhou, H.M.; Snider, P.; Wang, J.; Schwartz, R.; Moon, A.M.; Conway, S.J. Pax3 is essential for normal cardiac neural crest morphogenesis but is not required during migration nor outflow tract septation. Dev. Biol. 2011, 356, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Hansen, M.S.; Coffin, C.M.; Capecchi, M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004, 18, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Abe, K.; Mantani, A.; Hitoshi, Y.; Suzuki, M.; Osuzu, F.; Kuratani, S.; Yamamura, K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 1999, 212, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Hol, F.A.; Hamel, B.C.; Geurds, M.P.; Mullaart, R.A.; Barr, F.G.; Macina, R.A.; Mariman, E.C. A frameshift mutation in the gene for PAX3 in a girl with spina bifida and mild signs of Waardenburg syndrome. J. Med. Genet. 1995, 32, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Chatkupt, S.; Chatkupt, S.; Johnson, W.G. Waardenburg syndrome and myelomeningocele in a family. J. Med. Genet. 1993, 30, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Lepper, C.; Conway, S.J.; Fan, C.M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 2009, 460, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Hinton, R.; Moreno-Rodriguez, R.A.; Wang, J.; Rogers, R.; Lindsley, A.; Li, F.; Ingram, D.A.; Menick, D.; Field, L.; et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 2008, 102, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Fix, J.L.; Rogers, R.; Peabody-Dowling, G.; Ingram, D.; Lilly, B.; Conway, S.J. Generation and characterization of Csrp1 enhancer-driven tissue-restricted Cre-recombinase mice. Genesis. 2008, 46, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Dickman, E.; Rogers, R.; Conway, S.J. Abnormal skeletogenesis occurs coincident with increased apoptosis in the Splotch (Sp2H) mutant: Putative roles for Pax3 and PDGFRalpha in rib patterning. Anat. Rec. 1999, 255, 353–361. [Google Scholar] [CrossRef]
- Baas, D.; Meiniel, A.; Benadiba, C.; Bonnafe, E.; Meiniel, O.; Reith, W.; Durand, B. A deficiency in RFX3 causes hydrocephalus associated with abnormal differentiation of ependymal cells. Eur. J. Neurosci. 2006, 24, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Shanmugasundaram, R.; Shuyu, E.; Dragatsis, I. Congenital hydrocephalus associated with abnormal subcommissural organ in mice lacking huntingtin in Wnt1 cell lineages. Hum. Mol. Genet. 2009, 18, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Evangelista, M.; Luoh, S.M.; Frantz, G.D.; Chalasani, S.; Carano, R.A.; van Hoy, M.; Ramirez, J.; Ogasawara, A.K.; McFarland, L.M.; et al. Loss of the serine/threonine kinase fused results in postnatal growth defects and lethality due to progressive hydrocephalus. Mol. Cell. Biol. 2005, 25, 7054–7068. [Google Scholar] [CrossRef] [PubMed]
- Naruse, I.; Ueta, E. Hydrocephalus manifestation in the genetic polydactyly/arhinencephaly mouse (Pdn/Pdn). Congenit. Anom. 2002, 42, 27–31. [Google Scholar] [CrossRef]
- Meiniel, A. The secretory ependymal cells of the subcommissural organ: Which role in hydrocephalus? Int. J. Biochem. Cell. Biol. 2007, 39, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Fernandez, T.E.; Vasioukhin, V. Failure of epithelial tube maintenance causes hydrocephalus and renal cysts in Dlg5−/−mice. Dev. Cell. 2007, 13, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Brault, V.; Moore, R.; Kutsch, S.; Ishibashi, M.; Rowitch, D.H.; McMahon, A.; Sommer, L.; Boussadia, O.; Kemler, R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 2001, 128, 1253–1264. [Google Scholar] [PubMed]
- Joksimovic, M.; Patel, M.; Taketo, M.M.; Johnson, R.; Awatramani, R. Ectopic Wnt/beta-catenin signaling induces neurogenesis in the spinal cord and hindbrain floor plate. PLoS ONE 2012, 7, e30266. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Gan, Q.; Stokes, A.; Lassiter, R.; Wang, Y.; Chan, J.; Han, J.X.; Pleasure, D.E.; Epstein, J.A.; Zhou, C.J. β-catenin regulates Pax3 and Cdx2 for caudal neural tube closure and elongation. Development 2014, 141, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.A.; Ito, M.; Loeken, M.R. Neural tube defects in embryos of diabetic mice: Role of the Pax-3 gene and apoptosis. Diabetes 1997, 46, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Keller-Peck, C.R.; Mullen, R.J. Altered cell proliferation in the spinal cord of mouse neural tube mutants curly tail and Pax3 splotch-delayed. Brain Res. Dev. Brain Res. 1997, 102, 177–188. [Google Scholar] [CrossRef]
- Jostes, B.; Walther, C.; Gruss, P. The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech. Dev. 1990, 33, 27–37. [Google Scholar] [CrossRef]
- Goulding, M.; Chalepakis, G.; Deutsch, U.; Erselius, J.R.; Gruss, P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991, 10, 1135–1147. [Google Scholar] [PubMed]
- Wu, M.; Li, J.; Engleka, K.A.; Zhou, B.; Lu, M.M.; Plotkin, J.B.; Epstein, J.A. Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice. J. Clin. Invest. 2008, 118, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Williams, M.A.; Rigamonti, D. Genetics of human hydrocephalus. J. Neurol. 2006, 253, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Lötscher, P.; Engler, S.; Baggiolini, A.; Varum Tavares, S.; Brügger, V.; John, N.; Büchmann-Møller, S.; Snider, P.L.; Conway, S.J.; et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J. Neurosci. 2014, 34, 6112–6122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.D.; Morgan, S.C.; Loeken, M.R. Pax3 stimulates p53 ubiquitination and degradation independent of transcription. PLoS ONE. 2011, 6, e29379. [Google Scholar] [CrossRef] [PubMed]
- Van Landingham, M.; Nguyen, T.V.; Roberts, A.; Parent, A.D.; Zhang, J. Risk factors of congenital hydrocephalus: A 10 Years Retrospective Study. J. Neurol. Neurosurg. Psychiatry. 2009, 80, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Loeken, M.R. Advances in understanding the molecular causes of diabetes-induced birth defects. J. Soc. Gynecol. Investig. 2006, 13, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H.; Wyandt, H.E.; McDonald-McGinn, D.M.; Zackai, E.Z.; Milunsky, A. Molecular cytogenetic characterization of multiple intrachromosomal rearrangements of chromosome 2q in a patient with Waardenburg's syndrome and other congenital defects. Clin. Genet. 2004, 66, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Borg, I.; Squire, M.; Menzel, C.; Stout, K.; Morgan, D.; Willatt, L.; O’Brien, P.C.; Ferguson-Smith, M.A.; Ropers, H.H.; Tommerup, N.; et al. A cryptic deletion of 2q35 including part of the PAX3 gene detected by breakpoint mapping in a child with autism and a de novo 2;8 translocation. J. Med. Genet. 2002, 39, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Read, A.P.; Newton, V.E. Waardenburg syndrome. J. Med. Genet. 1997, 34, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Aymé, S.; Philip, N. Possible homozygous Waardenburg syndrome in a fetus with exencephaly. Am. J. Med. Genet. 1995, 59, 263–265. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.-M.; Conway, S.J. Restricted Pax3 Deletion within the Neural Tube Results in Congenital Hydrocephalus. J. Dev. Biol. 2016, 4, 7. https://doi.org/10.3390/jdb4010007
Zhou H-M, Conway SJ. Restricted Pax3 Deletion within the Neural Tube Results in Congenital Hydrocephalus. Journal of Developmental Biology. 2016; 4(1):7. https://doi.org/10.3390/jdb4010007
Chicago/Turabian StyleZhou, Hong-Ming, and Simon J. Conway. 2016. "Restricted Pax3 Deletion within the Neural Tube Results in Congenital Hydrocephalus" Journal of Developmental Biology 4, no. 1: 7. https://doi.org/10.3390/jdb4010007