
The Physiological and Pathological Implications of the Formation of Hydrogels, with a Specific Focus on Amyloid Polypeptides


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hydrogels and Physical Polypeptide Hydrogels
2.1. Hydrogels
2.2. Physical Polypeptide Hydrogels
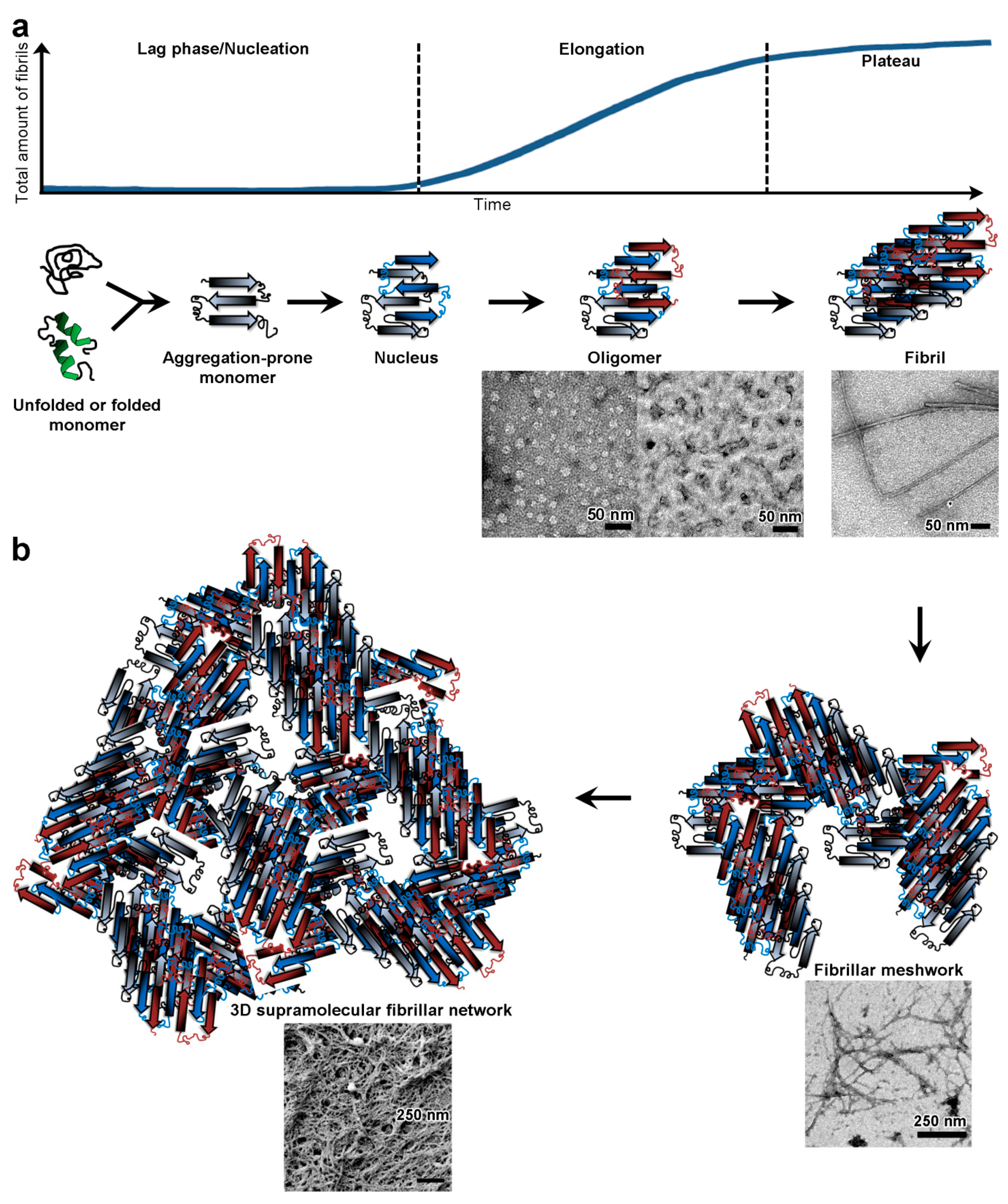
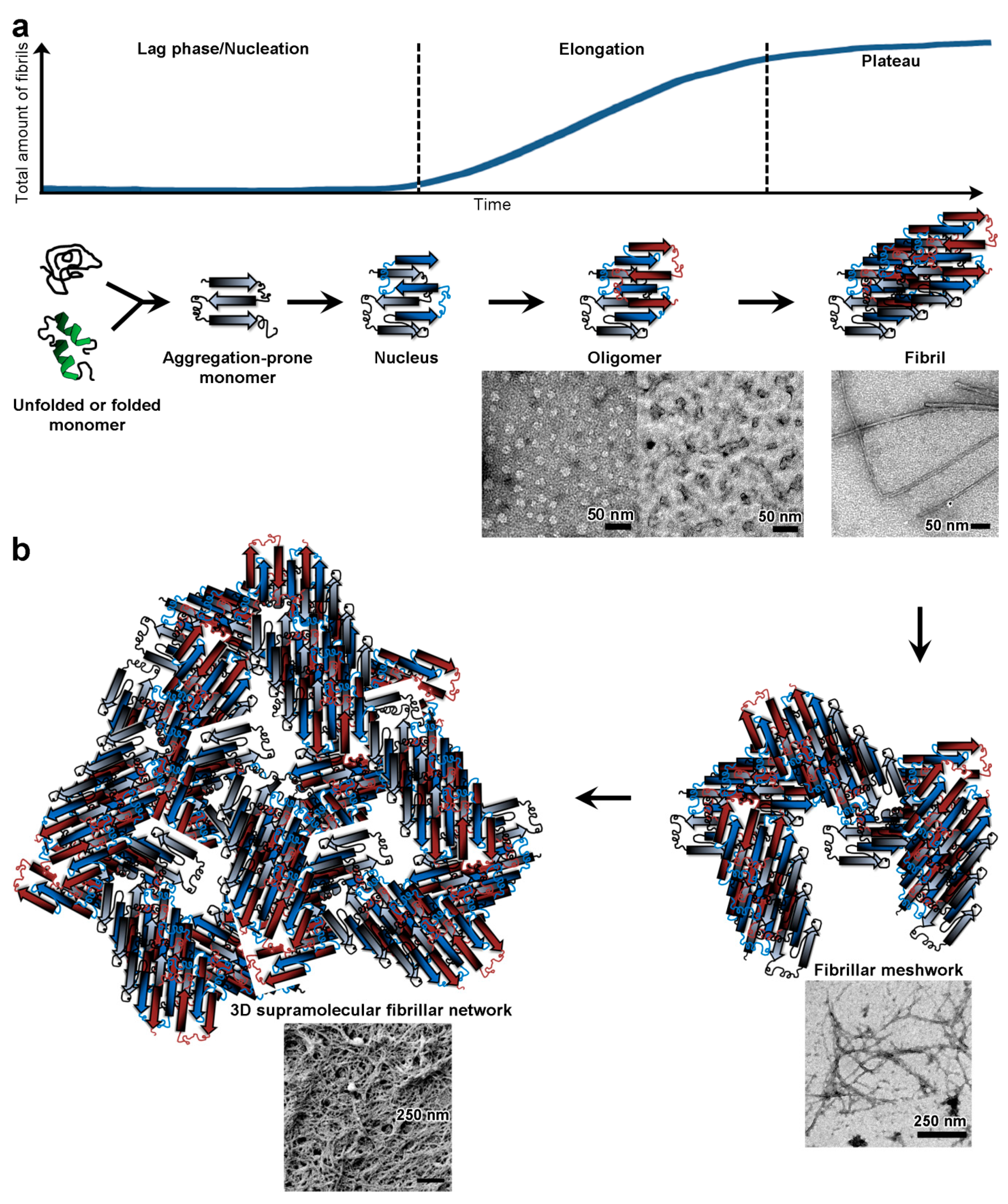
2.3. Study of Physical Polypeptide Hydrogels
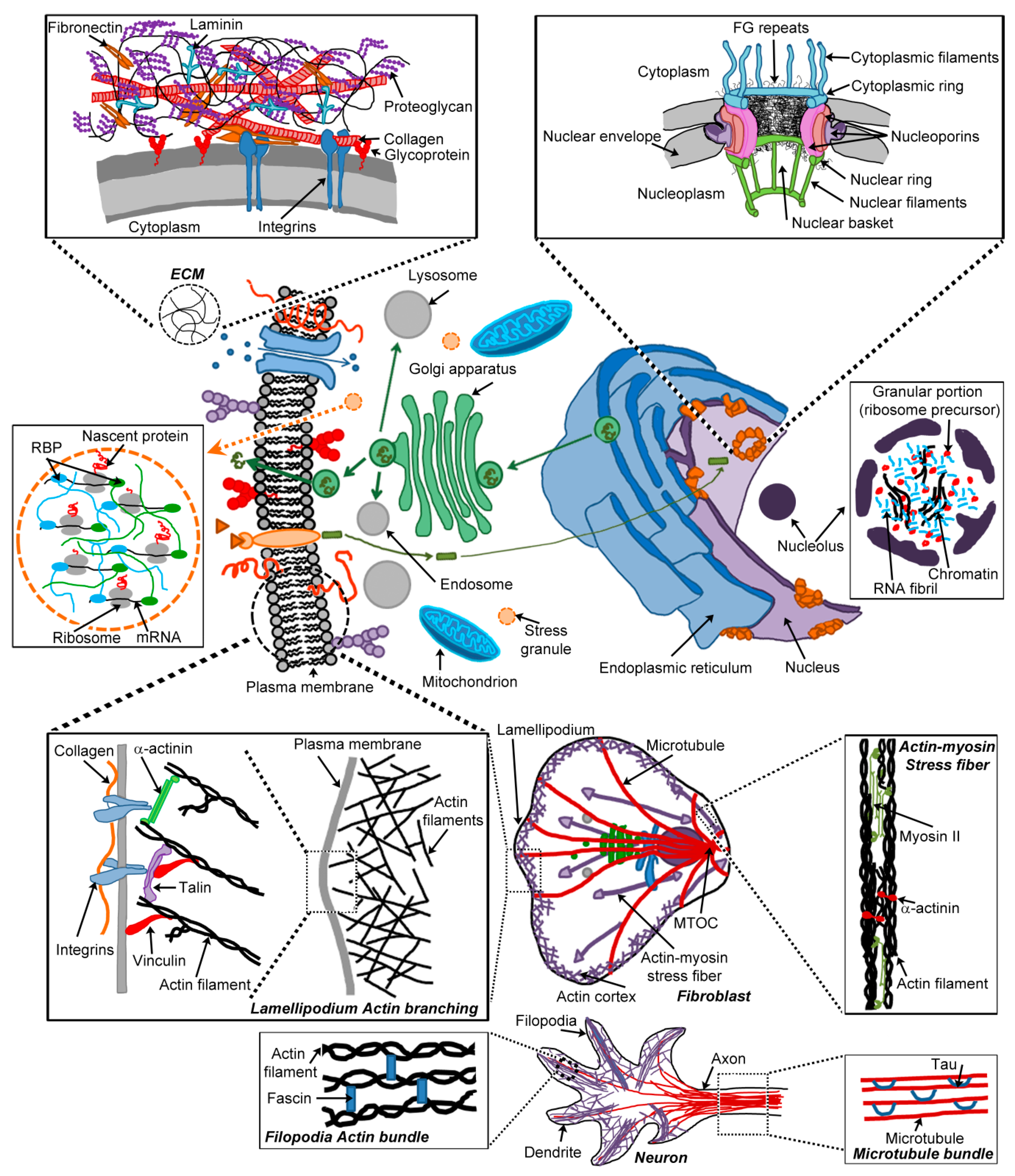
3. Cellular Hydrogels
3.1. Hydrogels as Diffusion Barriers
3.1.1. The Extracellular Matrix
3.1.2. Nuclear Pore Complexes
3.2. The Cytoskeleton
3.3. Membrane-Less Organelles
3.3.1. Nucleoli
3.3.2. Stress Granules
3.4. Secretory Granules and Peptide Hormones
4. Natural, Functional and Non-Pathological Hydrogels
4.1. Microbial Adhesins
4.1.1. Bacterial Biofilms
4.1.2. Hydrophobins
4.2. Silk
5. Pathological Amyloid Hydrogels
5.1. Islet Amyloid Polypeptide
5.2. Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
5.3. α-Synuclein
5.4. Amyloid-β Peptide and Tau
5.4.1. Amyloid-β Peptide
5.4.2. Tau
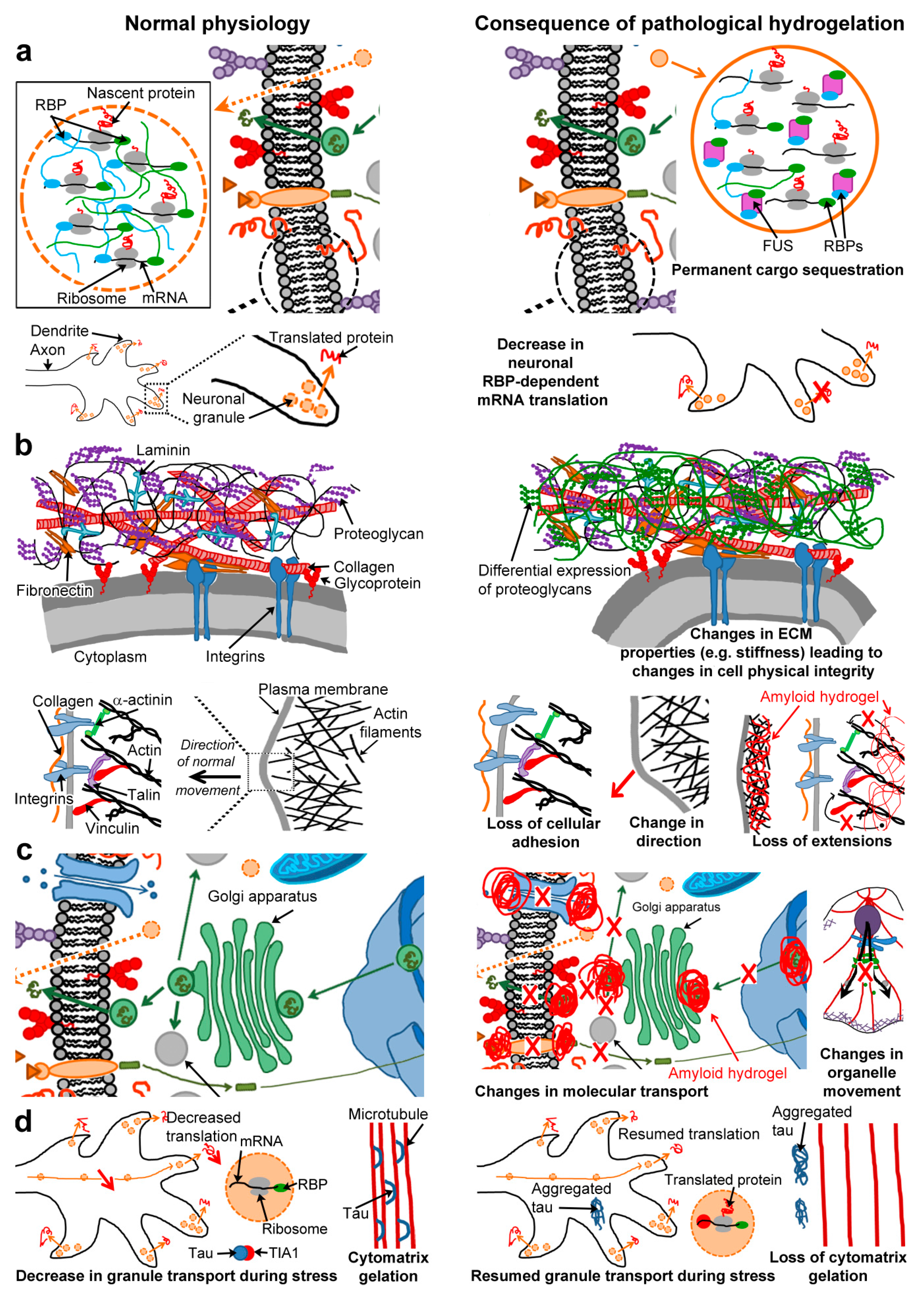
6. Consequences of Gelation by Pathological Amyloids
6.1. Permanent Cargo Sequestration (Figure 3a)
6.2. Cell Physical Integrity and Motility (Figure 3b)
6.3. Molecular Transport (Figure 3c)
6.4. Regulation of Non-Pathological Gelators (Figure 3d)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| air-water interface | AWI |
| Alzheimer’s disease | AD |
| amyloid-β peptide | Aβ |
| amyloid precursor protein | APP |
| amyotrophic lateral sclerosis | ALS |
| extracellular matrix | ECM |
| frontotemporal dementia | FTD |
| fused in sarcoma | FUS |
| heterogeneous nuclear ribonucleoprotein A1 | hnRNPA1 |
| human immunodeficiency virus | HIV |
| islet amyloid polypeptide | IAPP |
| liquid-liquid phase separation | LLPS |
| microtubule organising centre | MTOC |
| non-amyloid-β component of AD | NAC |
| nuclear pore complex | NPC |
| nuclear transport receptor | NTR |
| RNA-binding protein | RBP |
| T cell intracellular antigen 1 | TIA1 |
| 43-kD TARDNA–binding protein | TDP-43 |
References
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Maury, C.P.J. Self-propagating β-sheet polypeptide structures as prebiotic informational molecular entities: The amyloid world. Orig. Life Evol. Biosph. 2009, 39, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. On the possible amyloid origin of protein folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Low, A.; Chandrashekaran, I.R.; Adda, C.G.; Yao, S.; Sabo, J.K.; Zhang, X.; Soetopo, A.; Anders, R.F.; Norton, R.S. Merozoite surface protein 2 of Plasmodium falciparum: Expression, structure, dynamics, and fibril formation of the conserved N-terminal domain. Biopolymers 2007, 87, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Adda, C.G.; Murphy, V.J.; Sunde, M.; Waddington, L.J.; Schloegel, J.; Talbo, G.H.; Vingas, K.; Kienzle, V.; Masciantonio, R.; Howlett, G.J.; et al. Plasmodium falciparum merozoite surface protein 2 is unstructured and forms amyloid-like fibrils. Mol. Biochem. Parasitol. 2009, 166, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Mercado, G.; Martin-Benito, J.; Giraldo, R.; Monasterio, O.; Lagos, R.; Valpuesta, J.M. Structural characterization of microcin e492 amyloid formation: Identification of the precursors. J. Struct. Biol. 2012, 178, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Ingevechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Bieler, S.; Estrada, L.; Lagos, R.; Baeza, M.; Castilla, J.; Soto, C. Amyloid formation modulates the biological activity of a bacterial protein. J. Biol. Chem. 2005, 280, 26880–26885. [Google Scholar] [CrossRef] [PubMed]
- Graether, S.P.; Slupsky, C.M.; Sykes, B.D. Freezing of a fish antifreeze protein results in amyloid fibril formation. Biophys. J. 2003, 84, 552–557. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Vriend, G.; Hamodrakas, S.J. Amyloids protect the silkmoth oocyte and embryo. FEBS Lett. 2000, 479, 141–145. [Google Scholar] [CrossRef]
- Kenney, J.M.; Knight, D.; Wise, M.J.; Vollrath, F. Amyloidogenic nature of spider silk. Eur. J. Biochem. 2002, 269, 4159–4163. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in alzheimer's disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.W.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin). Protein Sci. 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, D.T.S.; Hoppener, J.W.M.; Posthuma, G.; Lips, C.J.M.; Liskamp, R.M.J. Inhibition of amyloid fibril formation of human amylin by N-alkylated amino acid and α-hydroxy acid residue containing peptides. Chem. Eur. J. 2002, 8, 4285–4291. [Google Scholar] [CrossRef]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Krysmann, M.J.; Castelletto, V.; Kelarakis, A.; Hamley, I.W.; Hule, R.A.; Pochan, D.J. Self-assembly and hydrogelation of an amyloid peptide fragment. Biochemistry 2008, 47, 4597–4605. [Google Scholar] [CrossRef] [PubMed]
- Manno, M.; Giacomazza, D.; Newman, J.; Martorana, V.; San Biagio, P.L. Amyloid gels: Precocious appearance of elastic properties during the formation of an insulin fibrillar network. Langmuir 2010, 26, 1424–1426. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Lee, C.F.; Hodder, P.; Hawkins, N.; Vaux, D.J. Dynamics of the formation of a hydrogel by a pathogenic amyloid peptide: Islet amyloid polypeptide. Sci. Rep. 2016, 6, 32124. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.R.; Cagnol, F.; Russell, A.B.; Izzard, M.J. Surface properties of class II hydrophobins from Trichoderma reesei and influence on bubble stability. Langmuir 2007, 23, 7995–8002. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Harnau, L.; Jung, J.M.; Mezzenga, R. Gelation, phase behavior, and dynamics of β-lactoglobulin amyloid fibrils at varying concentrations and ionic strengths. Biomacromolecules 2012, 13, 3241–3252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Dicko, C.; Bain, C.D.; Gong, Z.G.; Jacobs, R.M.J.; Shao, Z.Z.; Terry, A.E.; Vollrath, F. Behavior of silk protein at the air–water interface. Soft Matter 2012, 8, 9705–9712. [Google Scholar] [CrossRef]
- Lepere, M.; Chevallard, C.; Hernandez, J.F.; Mitraki, A.; Guenoun, P. Multiscale surface self-assembly of an amyloid-like peptide. Langmuir 2007, 23, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Cheong, D.W.; Accardo, A.; Di Fabrizio, E.; Riekel, C.; Hauser, C.A.E. Aliphatic peptides show similar self-assembly to amyloid core sequences, challenging the importance of aromatic interactions in amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: New York, NY, USA, 1980; p. 672. [Google Scholar]
- Chirani, N.; Yahia, H.; Gritsch, L.; Motta, F.L.; Chirani, S.; Fare, S. History and applications of hydrogels. J. Biomed. Sci. 2015, 4, 13. [Google Scholar]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.M.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Device 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Winter, H.H.; Mours, M. Rheology of polymers near liquid–solid transitions. Adv. Polym. Sci. 1997, 134, 165–234. [Google Scholar]
- Pappu, R.V.; Wang, X.; Vitalis, A.; Crick, S.L. A polymer physics perspective on driving forces and mechanisms for protein aggregation. Arch. Biochem. Biophys. 2008, 469, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Gorlich, D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell 2007, 130, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Ader, C.; Frey, S.; Maas, W.; Schmidt, H.B.; Gorlich, D.; Baldus, M. Amyloid-like interactions within nucleoporin FG hydrogels. Proc. Natl. Acad. Sci. USA 2010, 107, 6281–6285. [Google Scholar] [CrossRef] [PubMed]
- Crick, S.L.; Ruff, K.M.; Garai, K.; Frieden, C.; Pappu, R.V. Unmasking the roles ofnN- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 20075–20080. [Google Scholar] [CrossRef] [PubMed]
- Trappmann, B.; Gautrot, J.E.; Connelly, J.T.; Strange, D.G.T.; Li, Y.; Oyen, M.L.; Stuart, M.A.C.; Boehm, H.; Li, B.J.; Vogel, V.; et al. Extracellular-matrix tethering regulates stem-cell fate. Nat. Mater. 2012, 11, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, F.F.; Hackett, J.M.; Chaudhry, S.H.; Toll, F.N.; Toye, B.; Hodge, W.; Griffith, M. Collagen and glycopolymer based hydrogel for potential corneal application. Acta Biomater. 2010, 6, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Black, K.A.; Lin, B.F.; Wonder, E.A.; Desai, S.S.; Chung, E.J.; Ulery, B.D.; Katari, R.S.; Tirrell, M.V. Biocompatibility and characterization of a peptide amphiphile hydrogel for applications in peripheral nerve regeneration. Tissue Eng. Part A 2015, 21, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.G.; Rahim, M.; Ng, Q.; Segura, T. Hyaluronic acid and fibrin hydrogels with concentrated DNA/PEI polyplexes for local gene delivery. J. Control. Release 2011, 153, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Tous, E.; Minakawa, M.; Morita, M.; Robb, J.D.; Koomalsingh, K.J.; Gorman, J.H.; Gorman, R.C.; Burdick, J.A. Injectable hydrogel properties influence infarct expansion and extent of postinfarction left ventricular remodeling in an ovine model. Proc. Natl. Acad. Sci. USA 2010, 107, 11507–11512. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.M.R.; Stephanopoulos, N.; Sur, S.; Lee, S.S.; Newcomb, C.; Stupp, S.I. The powerful functions of peptide-based bioactive matrices for regenerative medicine. Ann. Biomed. Eng. 2015, 43, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.Q.; Pochan, D.J. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Kopecek, J.; Yang, J.Y. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009, 5, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.; Nunalee, M.L.; Lim, D.W.; Simnick, A.J.; Chilkoti, A. Peptide-based biopolymers in biomedicine and biotechnology. Mater. Sci. Eng. R Rep. 2008, 62, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Jonker, A.M.; Lowik, D.W.P.M.; van Hest, J.C.M. Peptide- and protein-based hydrogels. Chem. Mater. 2012, 24, 759–773. [Google Scholar] [CrossRef]
- Stendahl, J.C.; Rao, M.S.; Guler, M.O.; Stupp, S.I. Intermolecular forces in the self-assembly of peptide amphiphile nanofibers. Adv. Funct. Mater. 2006, 16, 499–508. [Google Scholar] [CrossRef]
- Cui, H.G.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Ozbas, B.; Kretsinger, J.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar] [CrossRef]
- Rodriguez, L.M.D.; Hemar, Y.; Cornish, J.; Brimble, M.A. Structure-mechanical property correlations of hydrogel forming β-sheet peptides. Chem. Soc. Rev. 2016, 45, 4797–4824. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, A.; Bell, M.; Boden, N.; Keen, J.N.; Knowles, P.F.; McLeish, T.C.B.; Pitkeathly, M.; Radford, S.E. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature 1997, 386, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Banwell, E.F.; Abelardo, E.S.; Adams, D.J.; Birchall, M.A.; Corrigan, A.; Donald, A.M.; Kirkland, M.; Serpell, L.C.; Butler, M.F.; Woolfson, D.N. Rational design and application of responsive α-helical peptide hydrogels. Nat. Mater. 2009, 8, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.C.; Nettesheim, F.; Pochan, D.J.; Schneider, J.P.; Wagner, N.J. Fast dynamics of semiflexible chain networks of self-assembled peptides. Biomacromolecules 2009, 10, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, C.J.; Nilsson, B.L. Review self-assembly of amphipathic β-sheet peptides: Insights and applications. Biopolymers 2012, 98, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.G.; Holmes, T.C.; Dipersio, C.M.; Hynes, R.O.; Su, X.; Rich, A. Self-complementary oligopeptide matrices support mammalian-cell attachment. Biomaterials 1995, 16, 1385–1393. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Elbaum-Garfinkle, S.; Langdon, E.M.; Taylor, N.; Occhipinti, P.; Bridges, A.A.; Brangwynne, C.P.; Gladfelter, A.S. RNA controls PolyQ protein phase transitions. Mol. Cell 2015, 60, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Corradini, M.G.; Weiss, R.G.; Raghavan, S.R.; Rogers, M.A. To gel or not to gel: Correlating molecular gelation with solvent parameters. Chem. Soc. Rev. 2015, 44, 6035–6058. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Caran, K.L. Anatomy of a gel. Amino acid derivatives that rigidify water at submillimolar concentrations. J. Am. Chem. Soc. 2000, 122, 11679–11691. [Google Scholar] [CrossRef]
- Lieleg, O.; Ribbeck, K. Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 2011, 21, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Zámecník, J.; Vargová, L.; Homola, A.; Kodet, R.; Syková, E. Extracellular matrix glycoproteins and diffusion barriers in human astrocytic tumours. Neuropathol. Appl. Neurobiol. 2004, 30, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Jin, S.; Verkman, A.S. Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J. 2007, 22, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, Y.S.; Linker, A.; Farquhar, M.G. Increased permeability of the glomerular basement membrane to ferritin after removal of glycosaminoglycans (heparan sulfate) by enzyme digestion. J. Cell Biol. 1980, 86, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 51–59. [Google Scholar]
- Kreuger, J.; Spillmann, D.; Li, J.-P.; Lindahl, U. Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.Q.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.D. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Opinion pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Fawcett, J. The perineuronal net and the control of CNS plasticity. Cell Tissue Res. 2012, 349, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Peters, R. Translocation through the nuclear pore: Kaps pave the way. Bioessays 2009, 31, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Hulsmann, B.B.; Labokha, A.A.; Gorlich, D. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell 2012, 150, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Strawn, L.A.; Shen, T.X.; Shulga, N.; Goldfarb, D.S.; Wente, S.R. Minimal nuclear pore complexes define FG repeat domains essential for transport. Nat. Cell Biol. 2004, 6, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Katikaneni, P.; Skern, T.; Gustin, K.E. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 2008, 82, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Iovine, M.K.; Watkins, J.L.; Wente, S.R. The GLFG repetitive region of the nucleoporin Nup116p interacts with Kap95p, an essential yeast nuclear import factor. J. Cell Biol. 1995, 131, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.; Ribbeck, K.; Akin, D.; Kent, H.M.; Feldherr, C.M.; Gorlich, D.; Stewart, M. Interaction between NTF2 and xFxFG-containing nucleoporins is required to mediate nuclear import of Ran GDP. J. Mol. Biol. 1999, 293, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Chaillan-Huntington, C.; Braslavsky, C.V.; Kuhlmann, J.; Stewart, M. Dissecting the interactions between NTF2, RanGDP, and the nucleoporin xFxFG repeats. J. Biol. Chem. 2000, 275, 5874–5879. [Google Scholar] [CrossRef] [PubMed]
- Ribbeck, K.; Gorlich, D. Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 2001, 20, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E.; Medalia, O.; Zwerger, M. Functional architecture of the nuclear pore complex. Annu. Rev. Biophys. 2012, 41, 557–584. [Google Scholar] [CrossRef] [PubMed]
- Dworetzky, S.I.; Lanford, R.E.; Feldherr, C.M. The effects of variations in the number and sequence of targeting signals on nuclear uptake. J. Cell Biol. 1988, 107, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; Wente, S.R. The nuclear pore complex: A protein machine bridging the nucleus and cytoplasm. Curr. Opin. Cell Biol. 2000, 12, 361–371. [Google Scholar] [CrossRef]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Eisele, N.B.; Frey, S.; Piehler, J.; Gorlich, D.; Richter, R.P. Ultrathin nucleoporin phenylalanine-glycine repeat films and their interaction with nuclear transport receptors. EMBO Rep. 2010, 11, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Gorlich, D. FG/FxFG as well as GLFS repeats form a selective permeability barrier with self-healing properties. EMBO J. 2009, 28, 2554–2567. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Lemke, E.A. Single molecule study of the intrinsically disordered FG-repeat nucleoporin 153. Biophys. J. 2011, 101, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Labokha, A.A.; Fassati, A. Viruses challenge selectivity barrier of nuclear pores. Viruses 2013, 5, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Labokha, A.A.; Gradmann, S.; Frey, S.; Hulsmann, B.B.; Urlaub, H.; Baldus, M.; Gorlich, D. Systematic analysis of barrier-forming FG hydrogels from Xenopus nuclear pore complexes. EMBO J. 2013, 32, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A. Mechanical properties of cytoskeletal polymers. Curr. Opin. Cell Biol. 1991, 3, 4–11. [Google Scholar] [CrossRef]
- Julicher, F.; Kruse, K.; Prost, J.; Joanny, J.F. Active behavior of the cytoskeleton. Phys. Rep. 2007, 449, 3–28. [Google Scholar] [CrossRef]
- Joanny, J.F.; Prost, J. Active gels as a description of the actin-myosin cytoskeleton. HFSP J. 2009, 3, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and function of nuclear bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Courchaine, E.M.; Lu, A.; Neugebauer, K.M. Droplet organelles? EMBO J. 2016, 35, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Handwerger, K.E.; Cordero, J.A.; Gall, J.G. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol. Biol. Cell 2005, 16, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Marko, J.F. The liquid drop nature of nucleoli. Nucleus 2012, 3, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P.; Mitrea, D.M.; Zhu, L.; Richardson, T.M. Coexisting liquid phases underlie nucleolar subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 2009, 10, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stress granules. Curr. Biol. 2009, 19, R397–R398. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Loschi, M.; Desbats, M.A.; Boccaccio, G.L. RNA granules: The good, the bad and the ugly. Cell Signal. 2011, 23, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.M.; Alexander, R.D.; Spiller, M.P.; Beggs, J.D. A role for Q/N-rich aggregation-prone regions in P-body localization. J. Cell Sci. 2008, 121, 2463–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, S.; Richter, R.P.; Goerlich, D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef] [PubMed]
- Arvan, P.; Castle, D. Sorting and storage during secretory granule biogenesis: Looking backward and looking forward. Biochem. J. 1998, 332, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.G.; Reid, J.J.; Daniell, L.W. Intracellular-transport and packaging of prolactin—Quantitative electron-microscope auto-radiographic study of mammotrophs dissociated from rat pituitaries. Endocrinology 1978, 102, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Keeler, C.; Hodsdon, M.E.; Dannies, P.S. Is there structural specificity in the reversible protein aggregates that are stored in secretory granules? J. Mol. Neurosci. 2004, 22, 43–49. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Dannies, P.S. Prolactin and growth hormone aggregates in secretory granules: The need to understand the structure of the aggregate. Endocr. Rev. 2012, 33, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.M.; Castle, J.D. Enhanced glycosylation and sulfation of secretory proteoglycans is coupled to the expression of a basic secretory protein. Mol. Biol. Cell 1998, 9, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Fernandez, J.M. Atomic force microscopy study of the secretory granule lumen. Biophys. J. 1996, 71, 2356–2366. [Google Scholar] [CrossRef]
- Fowler, D.M.; Kelly, J.W. The World of Functional Amyloid. Functional Fold: Amyloid Structures in Nature; Pan Stanford Publishing: Singapore, 2012; pp. 1–14. [Google Scholar]
- Iconomidou, V.A.; Hamodrakas, S.J. Natural protective amyloids. Curr. Protein Pept. Sci. 2008, 9, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Kolter, R. Functional amyloids in bacteria. Int. Microbiol. 2014, 17, 65–73. [Google Scholar] [PubMed]
- Fowler, D.M.; Kelly, J.W. Functional amyloidogenesis and cytotoxicity-insights into biology and pathology. PLoS Biol. 2012, 10, e1001459. [Google Scholar] [CrossRef] [PubMed]
- Wessels, J.G. Hydrophobins: Proteins that change the nature of the fungal surface. Adv. Microb. Physiol. 1997, 38, 1–45. [Google Scholar] [PubMed]
- Wu, C.; Lim, J.Y.; Fuller, G.G.; Cegelski, L. Quantitative analysis of amyloid-integrated biofilms formed by uropathogenic Escherichia coli at the air–liquid interface. Biophys. J. 2012, 103, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Xicohtencatl-Cortes, J.; Hess, S.; Caballero-Olin, G.; Giron, J.A.; Friedman, R.L. Mycobacterium tuberculosis produces pili during human infection. Proc. Natl. Acad. Sci. USA 2007, 104, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Bavdek, A.; Kostanjsek, R.; Antonini, V.; Lakey, J.H.; Serra, M.D.; Gilbert, R.J.C.; Anderluh, G. pH dependence of listeriolysin O aggregation and pore-forming ability. FEBS J. 2012, 279, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Tukel, C.; Wilson, R.P.; Nishimori, J.H.; Pezeshki, M.; Chromy, B.A.; Baumler, A.J. Responses to amyloids of microbial and host origin are mediated through Toll-like receptor 2. Cell Host Microbe 2009, 6, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.; Syed, A.K.; Stephenson, R.E.; Rickard, A.H.; Boles, B.R. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012, 8, e1002744. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Costerton, W.; Cheng, K.-L.; Geesey, G.G.; Ladd, T.L.; Dasgupta, M.; Marrie, T.I. Bacterial biofilms in nature and disease. Ann. Rev. Microbiol. 1987, 41, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Baty, A.M.; Eastburn, C.C.; Techkarnjanaruk, S.; Goodman, A.E.; Geesey, G.G. Spatial and temporal variations in chitinolytic gene expression and bacterial biomass production during chitin degradation. Appl. Environ. Microbiol. 2000, 66, 3574–3585. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Braxton, E.E.; Nistico, L.; Hall-Stoodley, L.; Johnson, S.; Quigley, M.; Post, J.C.; Ehrlich, G.D.; Kathju, S. Direct demonstration of Staphylococcus biofilm in an external ventricular drain in a patient with a history of recurrent ventriculoperitoneal shunt failure. Pediatr. Neurosurg. 2010, 46, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Espeland, E.M.; Wetzel, R.G. Complexation, stabilization, and UV photolysis of extracellular and surface-bound glucosidase and alkaline phosphatase: Implications for biofilm microbiota. Microb. Ecol. 2001, 42, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Le Magrex-Debar, E.; Lemoine, J.; Gellé, M.-P.; Jacquelin, L.-F.; Choisy, C. Evaluation of biohazards in dehydrated biofilms on foodstuff packaging. Int. J. Food Microbiol. 2000, 55, 239–243. [Google Scholar] [CrossRef]
- Teitzel, G.M.; Parsek, M.R. Heavy metal resistance of biofilm and planktonic Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2003, 69, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Fux, C.A.; Costerton, J.W.; Stewart, P.S.; Stoodley, P. Survival strategies of infectious biofilms. Trends Microbiol. 2005, 13, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Klapper, I.; Rupp, C.J.; Cargo, R.; Purvedorj, B.; Stoodley, P. Viscoelastic fluid description of bacterial biofilm material properties. Biotechnol. Bioeng. 2002, 80, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Körstgens, V.; Flemming, H.-C.; Wingender, J.; Borchard, W. Uniaxial compression measurement device for investigation of the mechanical stability of biofilms. J. Microbiol. Methods 2001, 46, 9–17. [Google Scholar] [CrossRef]
- Towler, B.W.; Rupp, C.J.; Cunningham, A.B.; Stoodley, P. Viscoelastic properties of a mixed culture biofilm from rheometer creep analysis. Biofouling 2003, 19, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.W.; He, Y.; Ren, Y.J.; Zerdoum, A.; Libera, M.R.; Sharma, P.K.; van Winkelhoff, A.J.; Neut, D.; Stoodley, P.; van der Mei, H.C.; et al. Viscoelasticity of biofilms and their recalcitrance to mechanical and chemical challenges. FEMS Microbiol. Rev. 2015, 39, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, M.; Komatsuzawa, H.; Sugai, M.; Suginaka, H.; Usui, T. Effect of the growth rate of pseudomonas aeruginosa biofilms on the susceptibility to antimicrobial agents. Chemotherapy 1997, 43, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.B. Hydrophobins: Proteins that self assemble at interfaces. Curr. Opin. Colloid Interface Sci. 2009, 14, 356–363. [Google Scholar] [CrossRef]
- Kwan, A.H.Y.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Permentier, H.P.; Rink, R.; Kruijtzer, J.A.W.; Liskamp, R.M.J.; Wösten, H.A.B.; Poolman, B.; Robillard, G.T. Probing the self-assembly and the accompanying structural changes of hydrophobin SC3 on a hydrophobic surface by mass spectrometry. Biophys. J. 2004, 87, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Aimanianda, V.; Guijarro, J.I.; Sunde, M.; Latgé, J.-P. Hydrophobins—Unique fungal proteins. PLoS Pathog. 2012, 8, e1002700. [Google Scholar] [CrossRef] [PubMed]
- Mackay, J.P.; Matthews, J.M.; Winefield, R.D.; Mackay, L.G.; Haverkamp, R.G.; Templeton, M.D. The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 2001, 9, 83–91. [Google Scholar] [CrossRef]
- Butko, P.; Buford, J.P.; Goodwin, J.S.; Stroud, P.A.; McCormick, C.L.; Cannon, G.C. Spectroscopic evidence for amyloid-like interfacial self-assembly of hydrophobin SC3. Biochem. Biophys. Res. Commun. 2001, 280, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Butko, P.; Goodwin, J.S.; Bufford, J.P.; Stroud, P.; McCormick, C.L.; Cannon, G.C. β-sheet stacking in interfacial self-assembly of hydrophobin SC3. Biophys. J. 2001, 80, 404a. [Google Scholar]
- Wosten, H.A.B.; de Vocht, M.L. Hydrophobins, the fungal coat unravelled. Biochim. Biophys. Acta 2000, 1469, 79–86. [Google Scholar] [CrossRef]
- Zampieri, F.; Wosten, H.A.B.; Scholtmeijer, K. Creating surface properties using a palette of hydrophobins. Materials 2010, 3, 4607–4625. [Google Scholar] [CrossRef] [PubMed]
- Wosten, H.A.B. Hydrophobins: Multipurpose proteins. Annu. Rev. Microbiol. 2001, 55, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Latge, J.P.; Beauvais, A.; Vey, A. Wall synthesis in the entomophthorales and its role in the immune-reaction of infected insects. Dev. Comp. Immunol. 1986, 10, 639. [Google Scholar]
- Valo, H.K.; Laaksonen, P.H.; Peltonen, L.J.; Linder, M.B.; Hirvonen, J.T.; Laaksonen, T.J. Multifunctional hydrophobin: Toward functional coatings for drug nanoparticles. ACS Nano 2010, 4, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Altman, G.H.; Diaz, F.; Jakuba, C.; Calabro, T.; Horan, R.L.; Chen, J.; Lu, H.; Richmond, J.; Kaplan, D.L. Silk-based biomaterials. Biomaterials 2003, 24, 401–416. [Google Scholar] [CrossRef]
- Wenk, E.; Merkle, H.P.; Meinel, L. Silk fibroin as a vehicle for drug delivery applications. J. Control. Release 2011, 150, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.J.; Park, J.Y.; Li, C.M.; Jin, H.J.; Valluzzi, R.; Kaplan, D.L. Structure and properties of silk hydrogels. Biomacromolecules 2004, 5, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.L.; Holland, C.; Porter, D.; Shao, Z.Z.; Vollrath, F. Concentration state dependence of the rheological and structural properties of reconstituted silk. Biomacromolecules 2009, 10, 2724–2728. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.-Y.; Chen, J.-P.; Leu, Y.-L.; Wang, H.-Y. Characterization and evaluation of silk protein hydrogels for drug delivery. Chem. Pharm. Bull. 2006, 54, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, T.; Watanabe, A.; Tsuchiya, T.; Ikoma, R.; Hidaka, M.; Sugihara, M. New oral dosage form for elderly patients. II. Release behavior of benfotiamine from silk fibroin gel. Chem. Pharm. Bull. 1995, 43, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kluge, J.A.; Leisk, G.G.; Kaplan, D.L. Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials 2008, 29, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; Obrien, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Sun, Y.; Huang, H.W. How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys. J. 2012, 102, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced β cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide—pinpointing amino-acid-residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Charge, S.B.P.; Dekoning, E.J.P.; Clark, A. Effect of pH and insulin on fibrillogenesis of islet amyloid polypeptide in vitro. Biochemistry 1995, 34, 14588–14593. [Google Scholar] [CrossRef] [PubMed]
- Bantchev, G.B.; Schwartz, D.K. Surface shear rheology of β-casein layers at the air/solution interface: Formation of a two-dimensional physical gel. Langmuir 2003, 19, 2673–2682. [Google Scholar] [CrossRef]
- Mackie, A.R.; Gunning, A.P.; Ridout, M.J.; Wilde, P.J.; Morris, V.J. Orogenic displacement in mixed β-lactoglobulin/β-casein films at the air/water interface. Langmuir 2001, 17, 6593–6598. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.Q.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNAPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackman, P.; Sarparanta, J.; Lehtinen, S.; Vihola, A.; Evila, A.; Jonson, P.H.; Luque, H.; Kere, J.; Screen, M.; Chinnery, P.F.; et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann. Neurol. 2013, 73, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobagyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. TAR DNA binding protein-43 (RDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Z.; Wei, Y.Y.; Lu, Y.M.; Song, J.X. ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Chauhan, V.S. Self-assembling properties of peptides derived from TDP-43 C-terminal fragment. Langmuir 2014, 30, 3845–3856. [Google Scholar] [CrossRef] [PubMed]
- Daigle, J.G.; Lanson, N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Stabile, S.; Velde, C.V. Endogenous RDP-43, but not FUS, contributes to stress granule assembly via G3BP. Mol. Neurodegener. 2012, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.K.; Chian, R.J.; Boggio, K.J.; Quaresma, A.J.C.; Nickerson, J.A.; Bosco, D.A. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. α-synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hsu, L.J.; Sisk, A.; Xia, Y.; Takeda, A.; Sundsmo, M.; Masliah, E. Human recombinant NACP/α-synuclein is aggregated and fibrillated in vitro: Relevance for lewy body disease. Brain Res. 1998, 799, 301–306. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, J.H.; Hahn, J.S.; Paik, S.R. Granular assembly of α-synuclein leading to the accelerated amyloid fibril formation with shear stress. PLoS ONE 2009, 4, e4177. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Miranker, A.D. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 12341–12346. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Bhak, G.; Lee, S.; Park, J.W.; Cho, S.; Paik, S.R. Amyloid hydrogel derived from curly protein fibrils of α-synuclein. Biomaterials 2010, 31, 5986–5995. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Zhou, K.; Ghosh, D.; Jha, N.N.; Singh, P.K.; Jacob, R.S.; Bernard, C.C.; Finkelstein, D.I.; Forsythe, J.S.; Maji, S.K. Implantable amyloid hydrogels for promoting stem cell differentiation to neurons. NPG Asia Mater. 2016, 8, e304. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer‘s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.S.; Ghosh, D.; Singh, P.K.; Basu, S.K.; Jha, N.N.; Das, S.; Sukul, P.K.; Patil, S.; Sathaye, S.; Kumar, A.; et al. Self-healing hydrogels composed of amyloid nano fibrils for cell culture and stem cell differentiation. Biomaterials 2015, 54, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, L.J. Comparative vibrational spectroscopy of intracellular tau and extracellular collagen I reveals parallels of gelation and fibrillar structure. J. Biol. Chem. 2004, 279, 7395–7404. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; da Rocha, E.L.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Yu, H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol. Neurodegener. 2012, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer‘s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Hebda, J.A.; Miranker, A.D. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: Insights from type II diabetes. Annu. Rev. Biophys. 2009, 38, 125–152. [Google Scholar] [CrossRef] [PubMed]
- Garner, C.C.; Tucker, R.P.; Matus, A. Selective localization of messenger RNA for cytoskeletal protein RNAP2 in dendrites. Nature 1988, 336, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Robins, S.P. Biochemistry and functional significance of collagen cross-linking. Biochem. Soc. Trans. 2007, 35, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; do Amaral, J.B.; Guimaraes, A.; Saraiva, M.J. Up-regulation of the extracellular matrix remodeling genes, biglycan, neutrophil gelatinase-associated lipocalin, and matrix metalloproteinase-9 in familial amyloid polyneuropathy. FASEB J. 2005, 19, 124–126. [Google Scholar] [PubMed]
- Deb, S.; Gottschall, P.E. Increased production of matrix metalloproteinases in enriched astrocyte and mixed hippocampal cultures treated with β-amyloid peptides. J. Neurochem. 1996, 66, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.H.; Trapani, L.M.; Siemeski, A.; MacKellar, D.; Gong, H.Y.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Matsudaira, P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 10889–10894. [Google Scholar] [CrossRef] [PubMed]
- Robbers, L.F.H.J.; Baars, E.N.; Brouwer, W.P.; Beek, A.M.; Hofman, M.B.M.; Niessen, H.W.M.; van Rossum, A.C.; Marcu, C.B. T1 mapping shows increased extracellular matrix size in the myocardium due to amyloid depositions. Circ. Cardiovasc. Imaging 2012, 5, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.; Bokhari, S.; Maurer, M.S. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2595–2597. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, T.; Helmke, S.; Patel, A.R.; Ruberg, F.L.; Packman, J.; Cheung, K.; Grogan, D.; Maurer, M.S. Pressure–volume relationships in patients with transthyretin (ATTR) cardiac amyloidosis secondary to V122I mutations and wild-type transthyretin transthyretin cardiac amyloid study (TRACS). Circ. Heart Fail. 2011, 4, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Breen, K.C.; Bruce, M.; Anderton, B.H. β-amyloid precursor protein mediates neuronal cell–cell and cell–surface adhesion. J. Neurosci. Res. 1991, 28, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Debeer, F.C.; Baltz, M.L.; Holford, S.; Feinstein, A.; Pepys, M.B. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid-P component. J. Exp. Med. 1981, 154, 1134–1149. [Google Scholar] [CrossRef]
- Brandan, E.; Inestrosa, N.C. Extracellular-matrix components and amyloid in neuritic plaques of Alzheimer’s disease. Gen. Pharmacol. 1993, 24, 1063–1068. [Google Scholar] [CrossRef]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate modulus directs neural stem cell behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef] [PubMed]
- Bott, K.; Upton, Z.; Schrobback, K.; Ehrbar, M.; Hubbell, J.A.; Lutolf, M.P.; Rizzi, S.C. The effect of matrix characteristics on fibroblast proliferation in 3D gels. Biomaterials 2010, 31, 8454–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Peyton, S.R.; Putnam, A.J. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell. Physiol. 2005, 204, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Arha, M.; Choudhary, S.; Ashton, R.S.; Bhatia, S.R.; Schaffer, D.V.; Kane, R.S. The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 2009, 30, 4695–4699. [Google Scholar] [CrossRef] [PubMed]
- Matus, A. Actin-based plasticity in dendritic spines. Science 2000, 290, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Dowjat, W.K.; Wisniewski, H.; Wisniewski, T. Alzheimer‘s disease presenilin-1 expression modulates the assembly of neurofilaments. Neuroscience 2001, 103, 1–8. [Google Scholar] [CrossRef]
- Weeds, A.G.; Gooch, J.; Mclaughlin, P.; Maury, C.P.J. Variant plasma gelsolin responsible for familial amyloidosis (Finnish type) has defective actin severing activity. FEBS Lett. 1993, 335, 119–123. [Google Scholar] [CrossRef]
- Haltia, M.; Prelli, F.; Ghiso, J.; Kiuru, S.; Somer, H.; Palo, J.; Frangione, B. Amyloid protein in familial amyloidosis (Finnish type) is homologous to gelsolin, an actin-binding protein. Biochem. Biophys. Res. Commun. 1990, 167, 927–932. [Google Scholar] [CrossRef]
- Heredia, L.; Helguera, P.; de Olmos, S.; Kedikian, G.; Vigo, F.S.; LaFerla, F.; Staufenbiel, M.; de Olmos, J.; Busciglio, J.; Caceres, A.; et al. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid β-induced degeneration: A potential mechanism of neuronal dystrophy in Alzheimer‘s disease. J. Neurosci. 2006, 26, 6533–6542. [Google Scholar] [CrossRef] [PubMed]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [PubMed]
- Hiruma, H.; Katakura, T.; Takahashi, S.; Ichikawa, T.; Kawakami, T. Glutamate and amyloid β-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J. Neurosci. 2003, 23, 8967–8977. [Google Scholar] [PubMed]
- Song, C.; Perides, G.; Wang, D.; Liu, Y.F. β-amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J. Neurochem. 2002, 83, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Woodard, D.; Bell, D.; Tipton, D.; Durrance, S.; Cole, L.; Li, B.; Xu, S.H. Gel formation in protein amyloid aggregation: A physical mechanism for cytotoxicity. PLoS ONE 2014, 9, e104152. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, H.; Popovic, S.; Taya, M. Pores and diffusion characteristics of porous gels. Polymer 2000, 41, 7201–7207. [Google Scholar] [CrossRef]
- Masaro, L.; Zhu, X.X. Physical models of diffusion for polymer solutions, gels and solids. Prog. Polym. Sci. 1999, 24, 731–775. [Google Scholar] [CrossRef]
- Verkman, A.S. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 2002, 27, 27–33. [Google Scholar] [CrossRef]
- Adalbert, R.; Coleman, M.P. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2013, 39, 90–108. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jean, L.; Foley, A.C.; Vaux, D.J.T. The Physiological and Pathological Implications of the Formation of Hydrogels, with a Specific Focus on Amyloid Polypeptides. Biomolecules 2017, 7, 70. https://doi.org/10.3390/biom7040070
Jean L, Foley AC, Vaux DJT. The Physiological and Pathological Implications of the Formation of Hydrogels, with a Specific Focus on Amyloid Polypeptides. Biomolecules. 2017; 7(4):70. https://doi.org/10.3390/biom7040070
Chicago/Turabian StyleJean, Létitia, Alex C. Foley, and David J. T. Vaux. 2017. "The Physiological and Pathological Implications of the Formation of Hydrogels, with a Specific Focus on Amyloid Polypeptides" Biomolecules 7, no. 4: 70. https://doi.org/10.3390/biom7040070