
Functional Amyloid Protection in the Eye Lens: Retention of α-Crystallin Molecular Chaperone Activity after Modification into Amyloid Fibrils

Abstract
:1. Introduction
2. Results
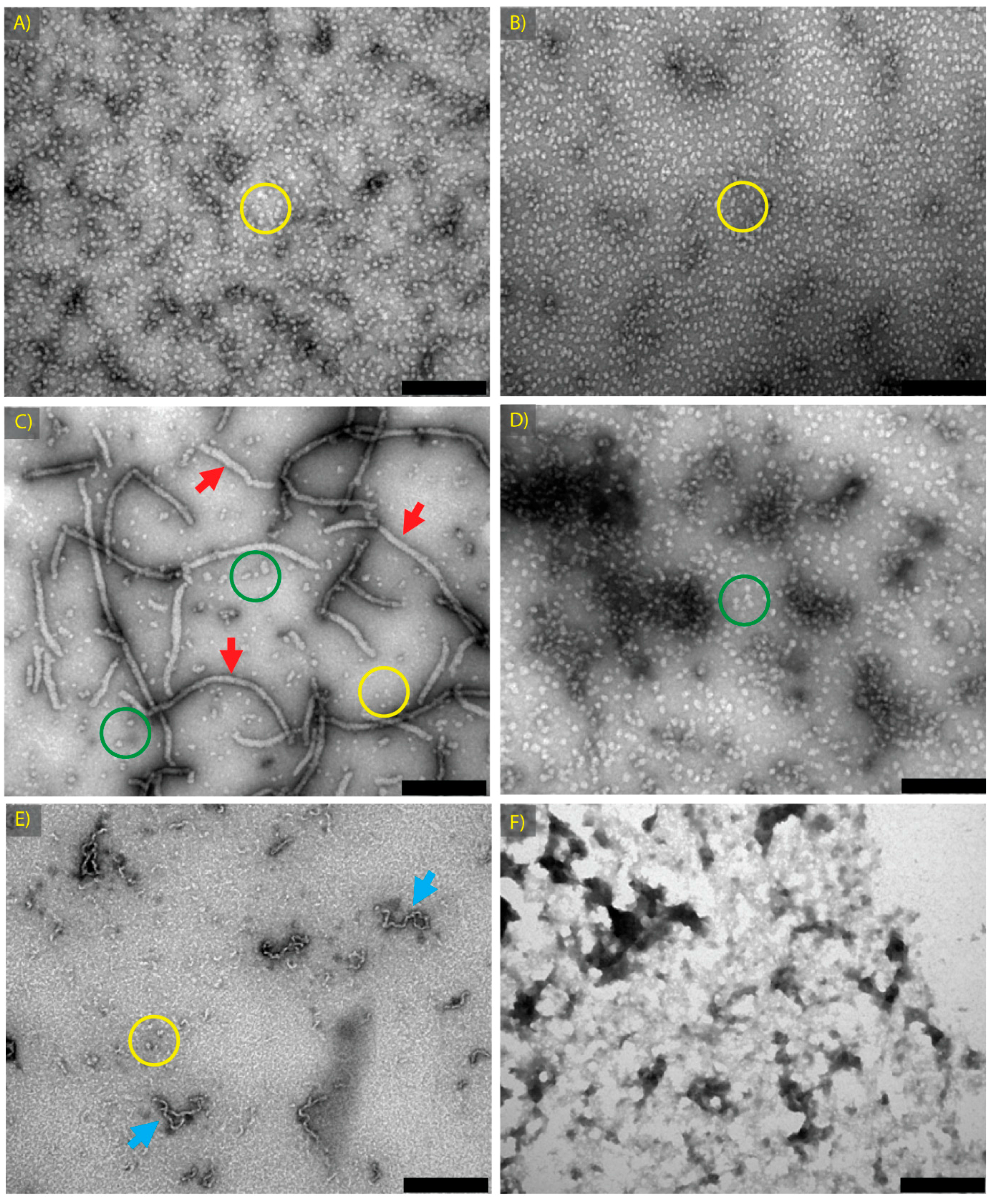
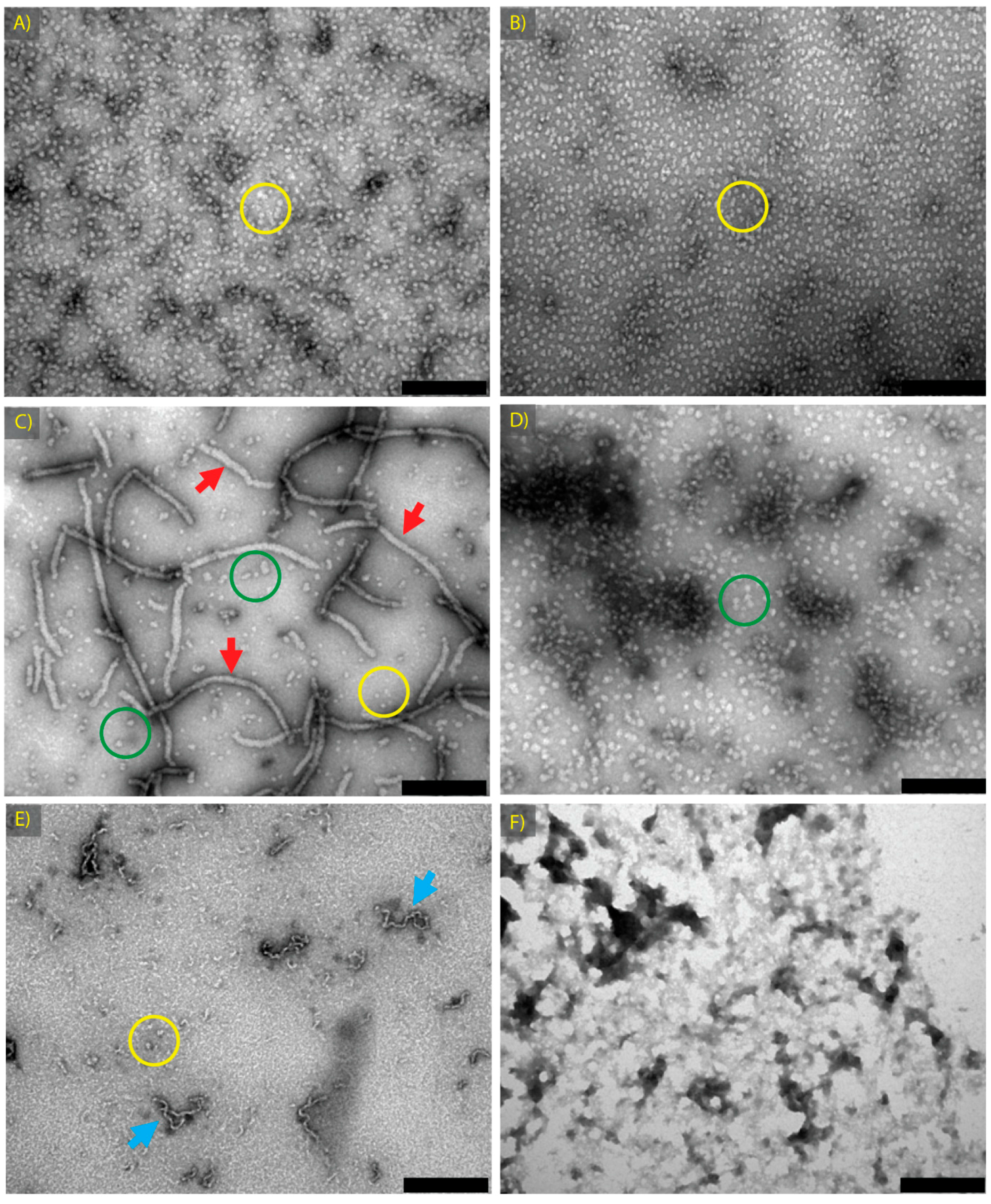
2.1. α-Crystallin Amyloid Fibril Formation
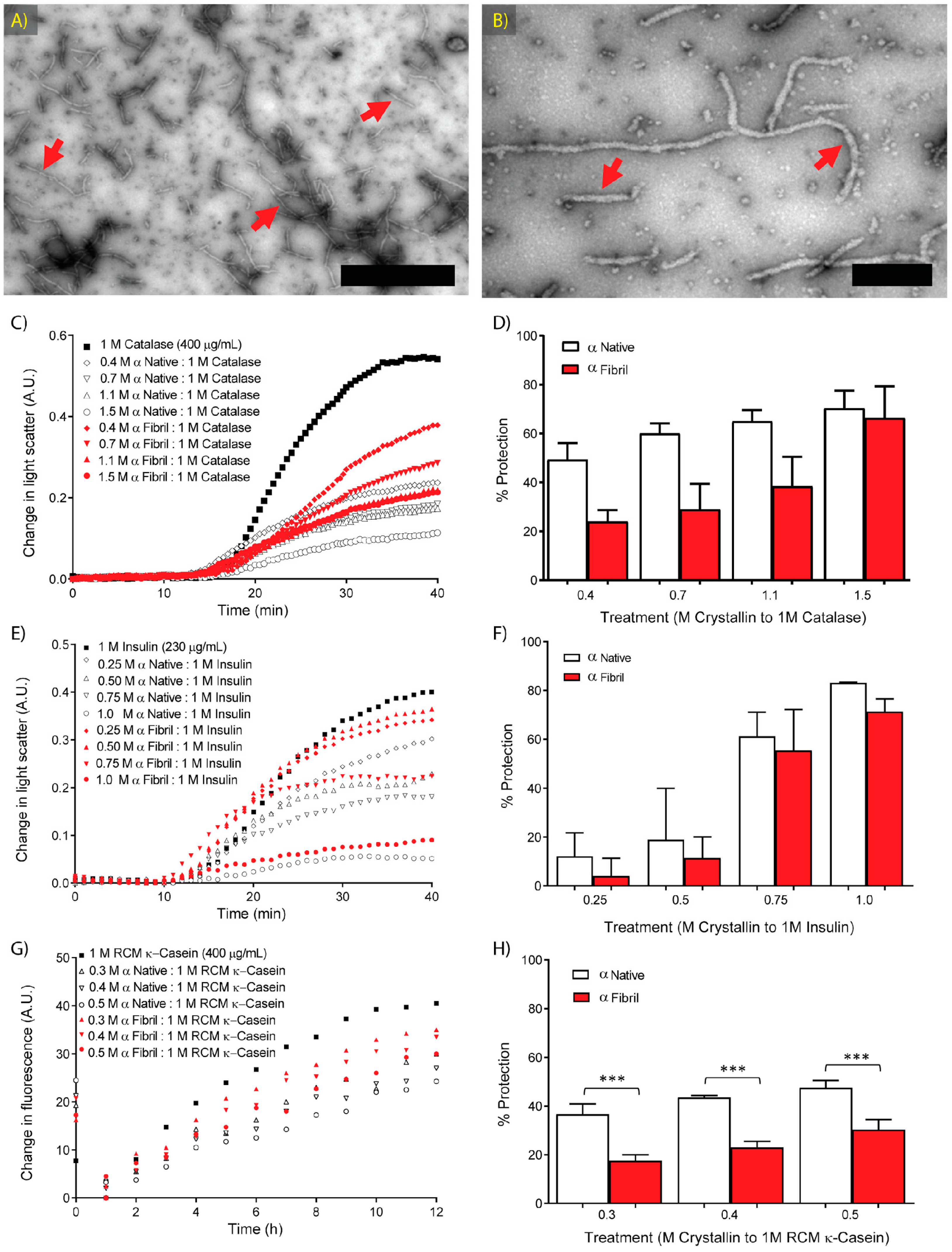
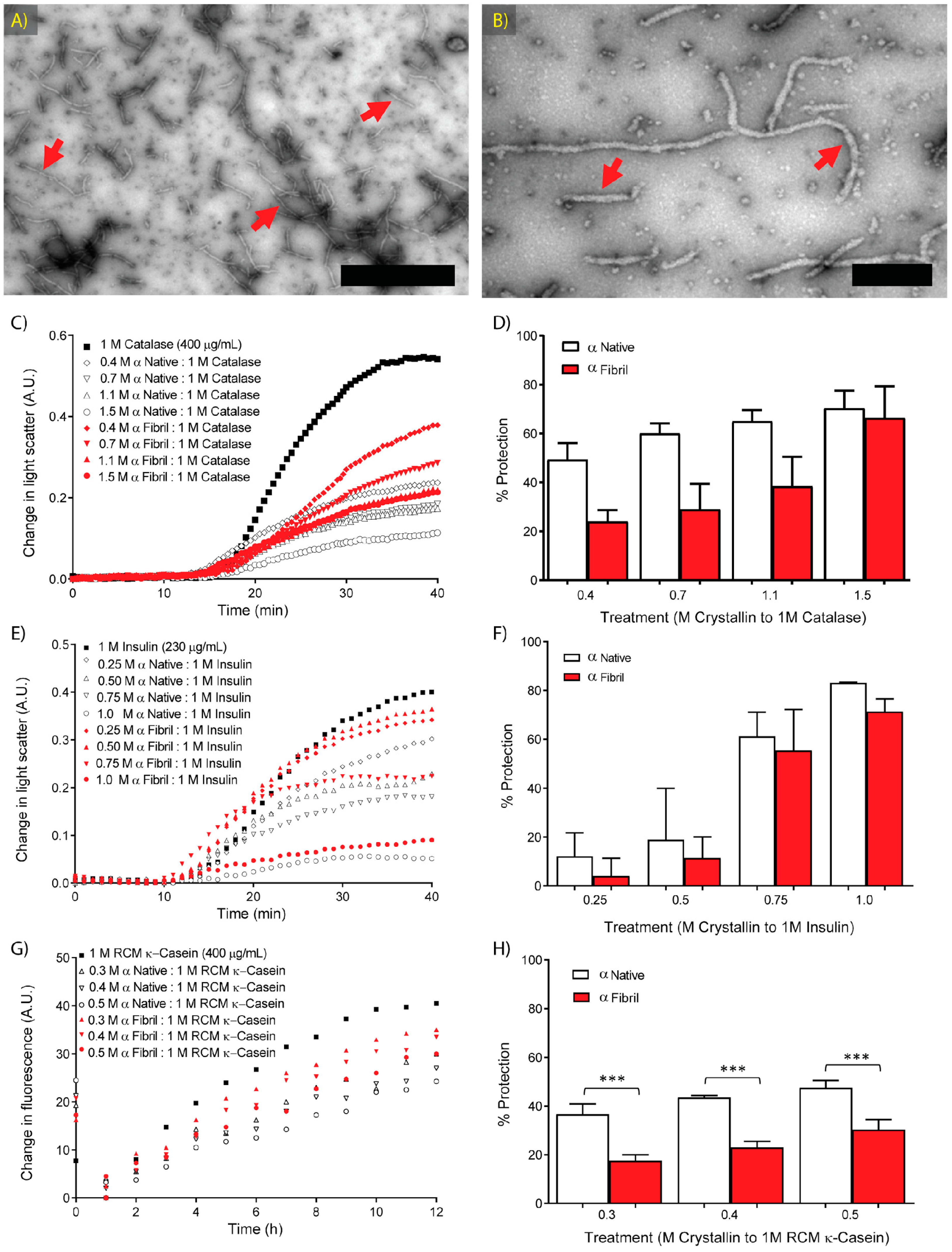
2.2. Molecular Chaperone Activity of Native and Fibrillar α-Crystallin against Amorphous Aggregation
2.3. Molecular Chaperone Activity of Native and Fibrillar α-Crystallin against κ-Casein Fibrillar Aggregation
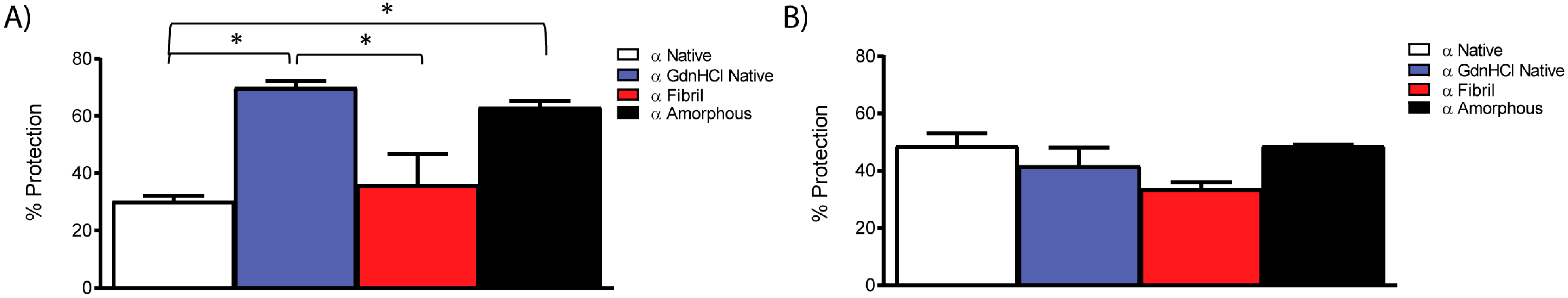
2.4. Molecular Chaperone Activity of Native and Fibrillar α-Crystallin as Compared to Non-Chaperone Based Amorphous and Fibrillar Aggregates
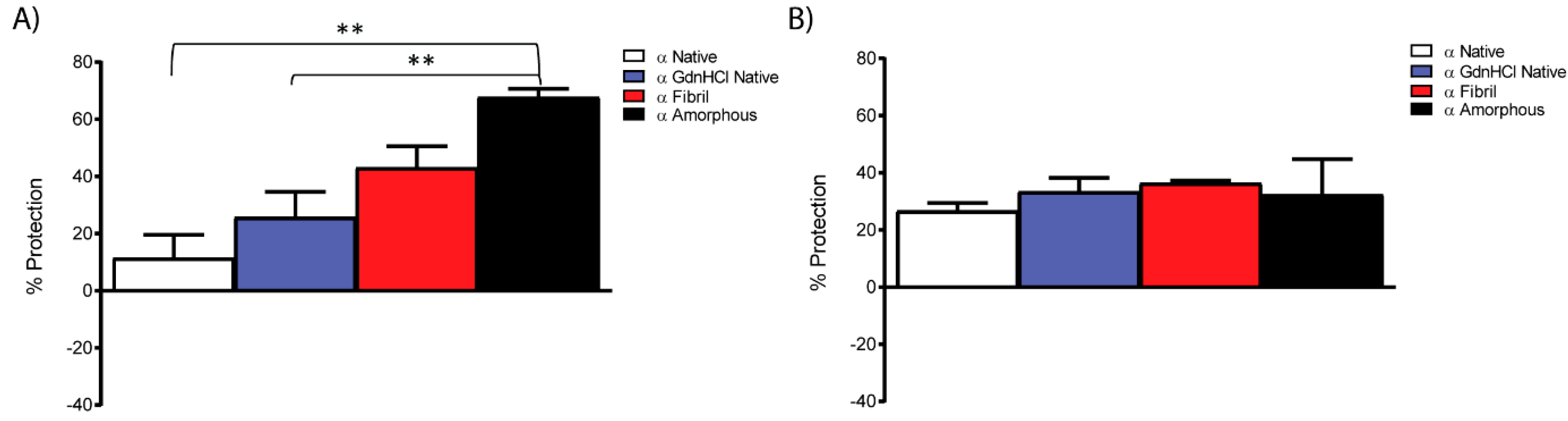
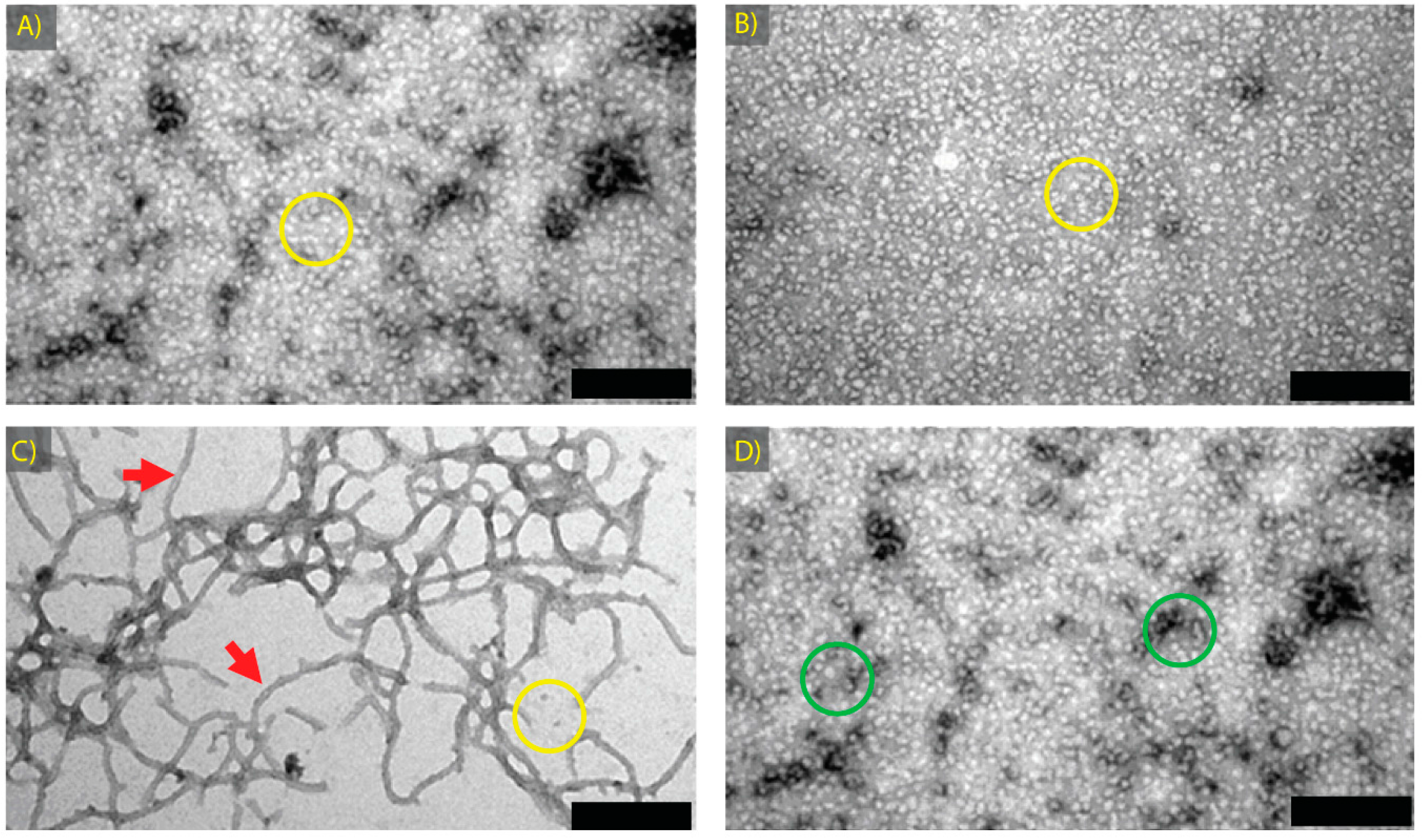
2.5. Chaperone Activity of αB-Crystallin Fibrils and Other Species
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Bovine Crystallin Protein Extraction
4.2.2. Bovine Crystallin Separation
4.2.3. Expression and Purification of Human αB-Crystallin
4.2.4. Aggregate Formation for Use as Molecular Chaperones
Refolded Aggregate Formation for Use as Molecular Chaperones
Amorphous Aggregate Formation for Use as Molecular Chaperones
Amyloid Fibril Formation by α-Crystallin Using GdnHCl
4.2.5. Monitoring Protein Aggregation
Induction of Amorphous Aggregation
Light Scattering Assessment of Thermally Induced Amorphous Aggregation
Light Scattering Assessment of Disulfide-Bond Reduction Induced Amorphous Aggregation
4.2.6. Characterisation of Amyloid Fibril Formation
Amyloid Fibril Formation by RCM κ-Casein
Thioflavin T Assay of Amyloid Fibril Formation
Transmission Electron Microscopy
Removal of GdnHCl from α-Crystallin Samples for Use as Molecular Chaperones
Quantification of Molecular Chaperone Activity
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bloemendal, H.; de Jong, W.; Jaenicke, R.; Lubsen, N.H.; Slingsby, C.; Tardieu, A. Ageing and vision: Structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 2004, 86, 407–485. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.J. Biophysical chemistry of the ageing eye lens. Biophys. Rev. 2015, 7, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Augusteyn, R.C. On the growth and internal structure of the human lens. Exp. Eye Res. 2010, 90, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Meehan, S.; Berry, Y.; Luisi, B.; Dobson, C.M.; Carver, J.A.; MacPhee, C.E. Amyloid fibril formation by lens crystallin proteins and its implications for cataract formation. J. Biol. Chem. 2004, 279, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Bassnett, S. Lens organelle degradation. Exp. Eye Res. 2002, 74, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abgar, S.; Backmann, J.; Aerts, T.; Vanhoudt, J.; Clauwaert, J. The structural differences between bovine lens αA- and αB-crystallin. Eur. J. Biochem. 2000, 267, 5916–5925. [Google Scholar] [PubMed]
- Horwitz, J.; Huang, Q.; Ding, L. The native oligomeric organization of α-crystallin, is it necessary for its chaperone function? Exp. Eye Res. 2004, 79, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Aquilina, J.A.; Benesch, J.L.; Bateman, O.A.; Slingsby, C.; Robinson, C.V. Polydispersity of a mammalian chaperone: Mass spectrometry reveals the population of oligomers in αB-crystallin. Proc. Natl. Acad. Sci. USA 2003, 100, 10611–10616. [Google Scholar] [CrossRef] [PubMed]
- Ecroyd, H.; Carver, J.A. Crystallin proteins and amyloid fibrils. Cell. Mol. Life Sci. 2009, 66, 62–81. [Google Scholar] [CrossRef] [PubMed]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Groenen, P.J.; Merck, K.B.; Jong, W.W.; Bloemendal, H. Structure and modifications of the junior chaperone α-crystallin. Eur. J. Biochem. 1994, 225, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1830–1843. [Google Scholar] [CrossRef] [PubMed]
- Augusteyn, R.C. α-Crystallin: A review of its structure and function. Clin. Exp. Optom. 2004, 87, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.A.; Grosas, A.B.; Ecroyd, H.; Quinlan, R.A. The functional roles of the unstructured N-and C-terminal regions in αB-crystallin and other mammalian small heat-shock proteins. Cell Stress Chaperones 2017, 22, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Kapoor, M.; Sinha, S.; Reddy, G.B. Insights into hydrophobicity and the chaperone-like function of αA- and αB-crystallins: An isothermal titration calorimetric study. J. Biol. Chem. 2005, 280, 21726–21730. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Kumar, R.S.; Kumar, G.S.; Quinn, P.T. Synthesis and characterization of a peptide identified as a functional element in αA-crystallin. J. Biol. Chem. 2000, 275, 3767–3771. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ghosh, J.G.; Clark, J.I.; Jiang, S. Studies of αB crystallin subunit dynamics by surface plasmon resonance. Anal. Biochem. 2006, 350, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, J.; Padmanabha Udupa, E.G.; Wang, J.; Sharma, K.K. Mini-αB-crystallin: A functional element of αB-crystallin with chaperone-like activity. Biochemistry 2006, 45, 3069–3076. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive sequences in the molecular chaperone, human αB crystallin modulate the fibrillation of amyloidogenic proteins. Int. J. Biochem. Cell Biol. 2008, 40, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.I. Functional sequences in human αB crystallin. Biochim. Biophys. Acta BBA Gen. Subj. 2016, 1860, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Mainz, A.; Peschek, J.; Stavropoulou, M.; Back, K.C.; Bardiaux, B.; Asami, S.; Prade, E.; Peters, C.; Weinkauf, S.; Buchner, J. The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 2015, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Rao, C.M. Differential temperature-dependent chaperone-like activity of αA- and αB-crystallin homoaggregates. J. Biol. Chem. 1999, 274, 34773–34778. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.B.; Das, K.P.; Petrash, J.M.; Surewicz, W.K. Temperature-dependent chaperone activity and structural properties of human αA- and αB-crystallins. J. Biol. Chem. 2000, 275, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Das, B.K.; Liang, J.J. Detection and characterization of α-crystallin intermediate with maximal chaperone-like activity. Biochem. Biophys. Res. Commun. 1997, 236, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.A.; Lindner, R.A. NMR spectroscopy of α-crystallin. Insights into the structure, interactions and chaperone action of small heat-shock proteins. Int. J. Biol. Macromol. 1998, 22, 197–209. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Burgio, M.R.; Kim, C.J.; Dow, C.C.; Koretz, J.F. Correlation between the chaperone-like activity and aggregate size of α-crystallin with increasing temperature. Biochem. Biophys. Res. Commun. 2000, 268, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Das, K.P. Unfolding and refolding of bovine α-crystallin in urea and its chaperone activity. Protein J. 2007, 26, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Ortwerth, B.J. Effect of cross-linking on the chaperone-like function of α crystallin. Exp. Eye Res. 1995, 61, 413–421. [Google Scholar] [CrossRef]
- Augusteyn, R.C. Dissociation is not required for α-crystallin’s chaperone function. Exp. Eye Res. 2004, 79, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Fink, A.L. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 2004, 1698, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein aggregation and its consequences for human disease. Protein Pept. Lett. 2006, 13, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Vendruscolo, M. Theoretical approaches to protein aggregation. Protein Pept. Lett. 2006, 13, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.L. Amyloid fibrils: From disease to design. New biomaterial applications for self-assembling cross-β fibrils. Aust. J. Chem. 2007, 60, 333–342. [Google Scholar] [CrossRef]
- Stefani, M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. [Google Scholar] [CrossRef] [PubMed]
- Shewmaker, F.; McGlinchey, R.P.; Wickner, R.B. Structural insights into functional and pathological amyloid. J. Biol. Chem. 2011, 286, 16533–16540. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.; Kwan, A.H.; Sunde, M. Functional amyloid: Widespread in Nature, diverse in purpose. Essays Biochem. 2014, 56, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Mezzenga, R. Amyloid fibrils as building blocks for natural and artificial functional materials. Adv. Mater. 2016, 28, 6546–6561. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.A.; Nicholls, K.A.; Aquilina, J.A.; Truscott, R.J. Age-related changes in bovine α-crystallin and high-molecular-weight protein. Exp. Eye Res. 1996, 63, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.-H.; Azari, P.; Himmel, M.E.; Squire, P.G. Isolation and physical characterization of bovine lens crystallins. Int. J. Pept. Protein Res. 1979, 13, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Meehan, S.; Knowles, T.P.; Baldwin, A.J.; Smith, J.F.; Squires, A.M.; Clements, P.; Treweek, T.M.; Ecroyd, H.; Tartaglia, G.G.; Vendruscolo, M. Characterisation of amyloid fibril formation by small heat-shock chaperone proteins human αA-, αB- and R120G αB-crystallins. J. Mol. Biol. 2007, 372, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Ecroyd, H.; Meehan, S.; Horwitz, J.; Aquilina, J.A.; Benesch, J.L.B.; Robinson, C.V.; MacPhee, C.; Carver, J.A. Mimicking phosphorylation of αB-crystallin affects its chaperone activity. Biochem. J. 2007, 401, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J.; Huang, Q.L.; Linlin, D.; Bova, M.P. Lens α-crystallin: Chaperone-like properties. Methods Enzymol. 1998, 290, 365–383. [Google Scholar] [PubMed]
- Farrell, H.M.; Cooke, P.H.; Wickham, E.D.; Piotrowski, E.G.; Hoagland, P.D. Environmental influences on bovine κ-casein: Reduction and conversion to fibrillar (amyloid) structures. J. Protein Chem. 2003, 22, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Thorn, D.C.; Meehan, S.; Sunde, M.; Rekas, A.; Gras, S.L.; MacPhee, C.E.; Dobson, C.M.; Wilson, M.R.; Carver, J.A. Amyloid fibril formation by bovine milk κ-casein and its inhibition by the molecular chaperones αS-and β-casein. Biochemistry 2005, 44, 17027–17036. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.A.; Duggan, P.J.; Ecroyd, H.; Liu, Y.; Meyer, A.G.; Tranberg, C.E. Carboxymethylated-κ-casein: A convenient tool for the identification of polyphenolic inhibitors of amyloid fibril formation. Bioorg. Med. Chem. 2010, 18, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Haley, D.A.; Bova, M.P.; Huang, Q.-L.; Mchaourab, H.S.; Stewart, P.L. Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies. J. Mol. Biol. 2000, 298, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Meehan, S.; The University of Adelaide, Adelaide, South Australia, Australia. Personal communication, 2009.
- Raman, B.; Rao, C.M. Chaperone-like activity and temperature-induced structural changes of α-crystallin. J. Biol. Chem. 1997, 272, 23559–23564. [Google Scholar] [CrossRef] [PubMed]
- Spinozzi, F.; Mariani, P.; Rustichelli, F.; Amenitsch, H.; Bennardini, F.; Mura, G.M.; Coi, A.; Ganadu, M.L. Temperature dependence of chaperone-like activity and oligomeric state of αB-crystallin. Biochim. Biophys. Acta 2006, 1764, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Ganadu, M.L.; Aru, M.; Mura, G.M.; Coi, A.; Mlynarz, P.; Kozlowski, H. Effects of divalent metal ions on the αB-crystallin chaperone-like activity: Spectroscopic evidence for a complex between copper (II) and protein. J. Inorg. Biochem. 2004, 98, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Raman, B.; Rao, K.S.; Ramakrishna, T.; Rao, C.M. Arginine hydrochloride enhances the dynamics of subunit assembly and the chaperone-like activity of α-crystallin. Mol. Vis. 2005, 11, 249–255. [Google Scholar] [PubMed]
- Ecroyd, H.; Carver, J.A. The effect of small molecules in modulating the chaperone activity of αB-crystallin against ordered and disordered protein aggregation. FEBS J. 2008, 275, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Bakthisaran, R.; Akula, K.K.; Tangirala, R.; Rao, C.M. Phosphorylation of αB-crystallin: Role in stress, aging and patho-physiological conditions. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Ban, T.; Sakai, M.; Pasta, S.Y.; Ramakrishna, T.; Naiki, H.; Goto, Y.; Rao, C.M. αB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid β-peptide and β2-microglobulin. Biochem. J. 2005, 392, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Garvey, M.; Griesser, S.; Griesser, H.J.; Thierry, B.; Nussio, M.R.; Shapter, J.G.; Ecroyd, H.; Giorgetti, S.; Bellotti, V.; Gerrard, J.A.; et al. Enhanced molecular chaperone activity of a small heat-shock protein following covalent immobilization onto a solid-phase support. Biopolymers 2011, 95, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2005, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.L.; Tickler, A.K.; Squires, A.M.; Devlin, G.L.; Horton, M.A.; Dobson, C.M.; MacPhee, C.E. Functionalised amyloid fibrils for roles in cell adhesion. Biomaterials 2008, 29, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Raju, M.; Santhoshkumar, P.; Xie, L.; Sharma, K.K. Addition of αA-crystallin sequence 164–173 to a mini-chaperone DFVIFLDVKHFSPEDLT alters the conformation but not the chaperone-like activity. Biochemistry 2014, 53, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Hains, P.G.; Truscott, R.J.W. Post-translational modifications in the nuclear region of young, aged, and cataract human lenses. J. Proteome Res. 2007, 6, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Farahbakhsh, Z.T.; Huang, Q.-L.; Ding, L.-L.; Altenbach, C.; Steinhoff, H.-J.; Horwitz, J.; Hubbell, W.L. Interaction of α-crystallin with spin-labeled peptides. Biochemistry 1995, 34, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, G.K.A.; Benesch, J.L.P. Dynamical structure of αB-crystallin. Prog. Biophys. Mol. Biol. 2014, 115, 11–20. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Protein | Incubation Conditions | |||||
|---|---|---|---|---|---|---|---|
| Buffer | pH | Temp. (°C) | Time (h) | Morphology | Figure | ||
| α Native | α-Crystallin | 0.1 M sodium phosphate | 7.4 | 4 | 2 | Spherical aggregates ~15 nm in diameter | 2A |
| α GdnHCl Native | α-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 4 | 2 | Spherical aggregates ~15 nm in diameter | 2B |
| α Fibril | α-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 60 | 2 | Long fibrils, (20 nm–1 μm in length), plus short fibrils and/or spherical aggregates | 2C |
| α Amorphous | α-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 90 | 2 | Larger spherical aggregates ~30 nm in diameter, singly and in clumps | 2D |
| βH Fibril | βH-Crystallin | H2O with 10% TFE | 2 | 60 | 18 | Short, curly fibrils, 20 nm to 200 nm in length | 2E |
| ADH Amorphous | ADH | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 90 | 2 | Large non-fibrillar protein clumps | 2F |
| αB Native | αB-Crystallin | 0.1 M sodium phosphate | 7.4 | 4 | 2 | Spherical aggregates ~15 nm in diameter | 4A |
| αB GdnHCl Native | αB-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 4 | 2 | Spherical aggregates ~15 nm in diameter | 4B |
| αB Fibril | αB-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 60 | 2 | Long fibrils, (>1 μm in length) | 4C |
| αB Amorphous | αB-Crystallin | 1 M GdnHCl, 0.1 M sodium phosphate | 7.4 | 90 | 2 | Larger spherical aggregates ~30 nm in diameter, singly or in row-like clumps | 4D |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garvey, M.; Ecroyd, H.; Ray, N.J.; Gerrard, J.A.; Carver, J.A. Functional Amyloid Protection in the Eye Lens: Retention of α-Crystallin Molecular Chaperone Activity after Modification into Amyloid Fibrils. Biomolecules 2017, 7, 67. https://doi.org/10.3390/biom7030067
Garvey M, Ecroyd H, Ray NJ, Gerrard JA, Carver JA. Functional Amyloid Protection in the Eye Lens: Retention of α-Crystallin Molecular Chaperone Activity after Modification into Amyloid Fibrils. Biomolecules. 2017; 7(3):67. https://doi.org/10.3390/biom7030067
Chicago/Turabian StyleGarvey, Megan, Heath Ecroyd, Nicholas J. Ray, Juliet A. Gerrard, and John A. Carver. 2017. "Functional Amyloid Protection in the Eye Lens: Retention of α-Crystallin Molecular Chaperone Activity after Modification into Amyloid Fibrils" Biomolecules 7, no. 3: 67. https://doi.org/10.3390/biom7030067