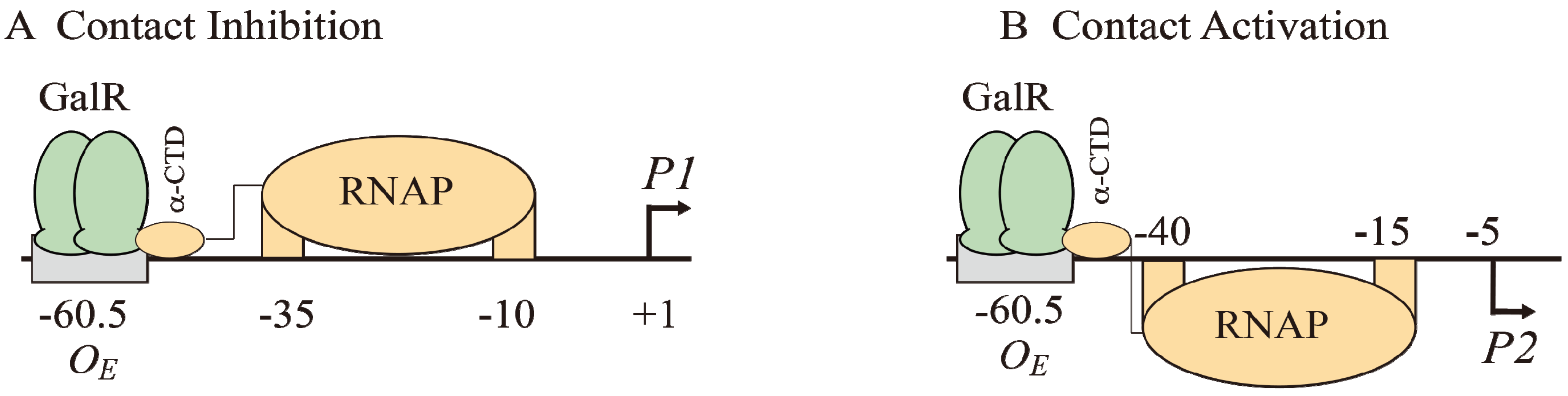1. Introduction
The study of the galactose (
gal) operon, which encodes enzymes for an amphibolic pathway of
d-galactose metabolism, first revealed a plethora of gene regulatory mechanisms by which bacterial genes are regulated: (i) beside substrate induction of a specific catabolic pathway or end-product repression of a specific biosynthetic pathway, accumulation or depletion of metabolic intermediates in the cell globally regulates the expression of a wide variety of genes to compensate for the accumulation or depletion [
1]; (ii) use of more than one promoter to regulate an operon [
2]; (iii) the mechanism of Rho-mediated premature transcription termination [
3]; (iv) gene regulation by a DNA element located within a structural gene [
4]; (v) DNA looping to repress gene transcription [
4]; (vi) the global gene activator, CRP, can also represses a gene [
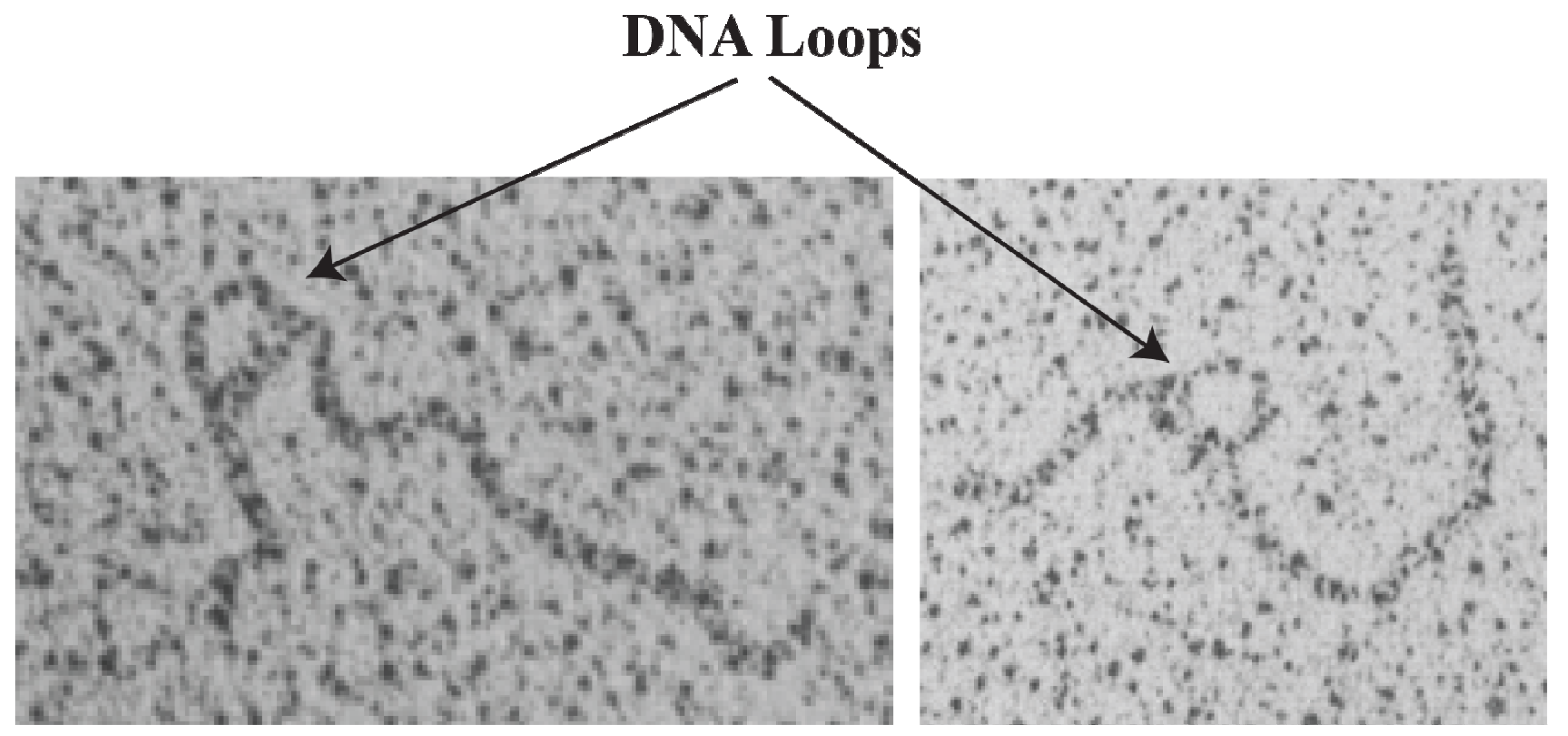
2]; (vii) demonstration of trajectory of DNA loops [
5]; and (viii) phage protein mediated transcription anti-termination in bacterial genes [
6]. Here we review more recent revelations that provide several new aspects of the multiple regulatory pathways by which
gal promoters are regulated at the transcription level.
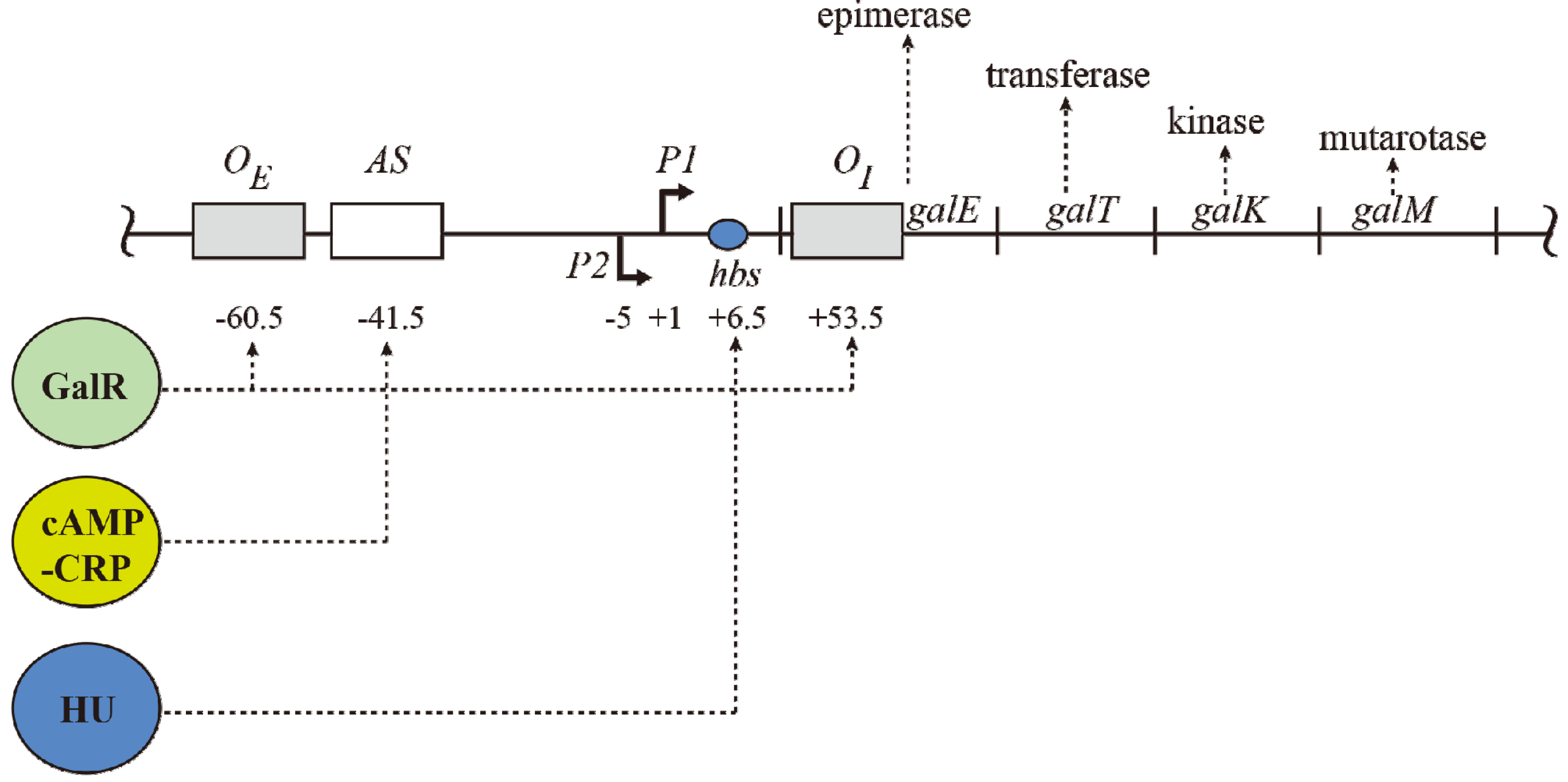
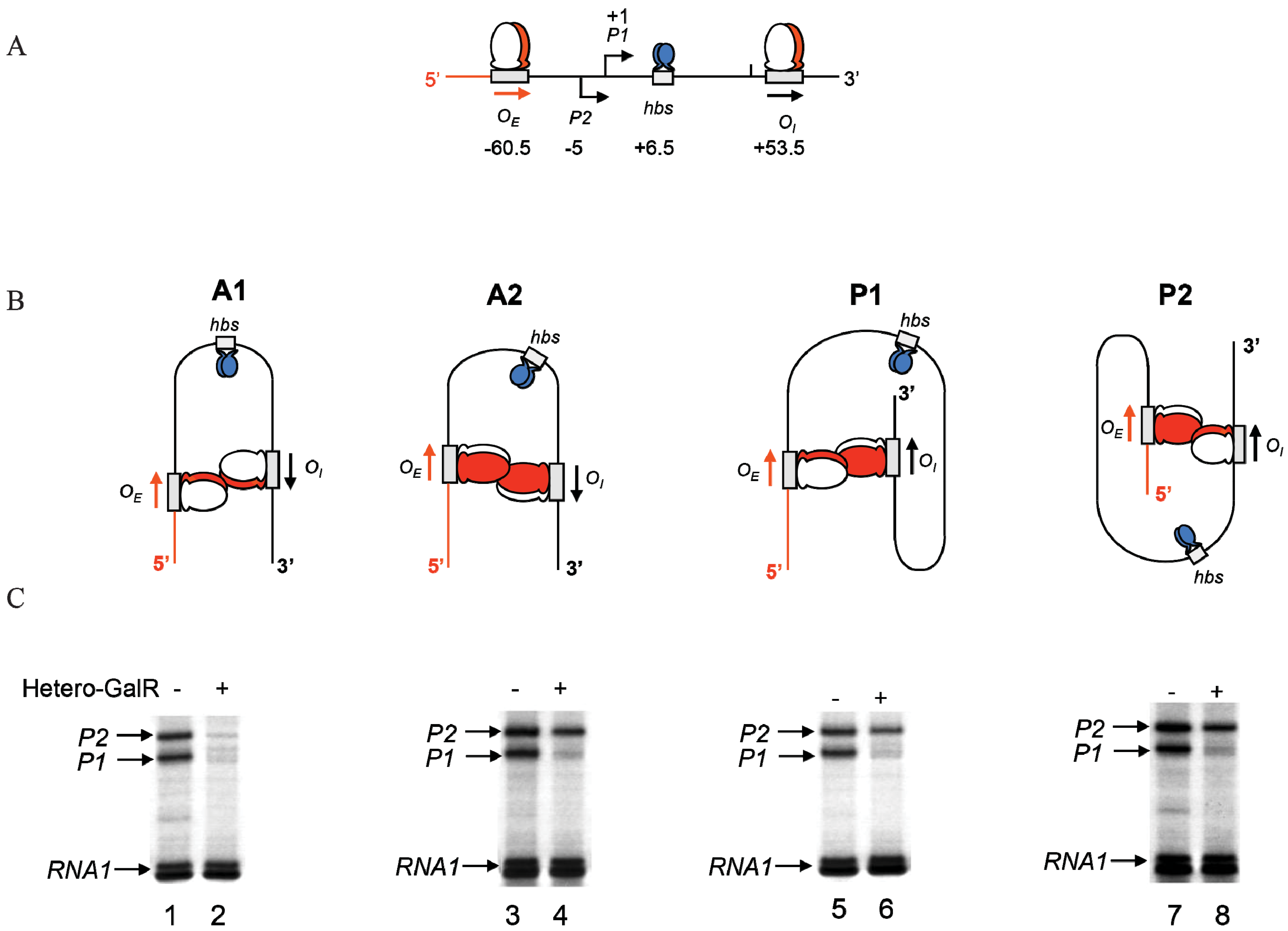
2. The Gal Operon
The
gal operon is transcribed from two overlapping promoters,
P1 and
P2, with transcription start points marked as +1 and −5, respectively (
Figure 1) [
2,
7,
8]. Why two promoters? Each promoter responds to different regulators for coping with physiological needs as enzymes encoded in the
gal operon are needed for both catabolic and anabolic metabolisms. Both promoters are intrinsically expressed at significant levels. Nonetheless,
cAMP and its receptor protein
CRP
complex (CCC) enhances
P1 but represses
P2, whereas GalR represses
P1 and enhances
P2 (details below). CCC acts by binding to a single site
AS and GalR to two operators.
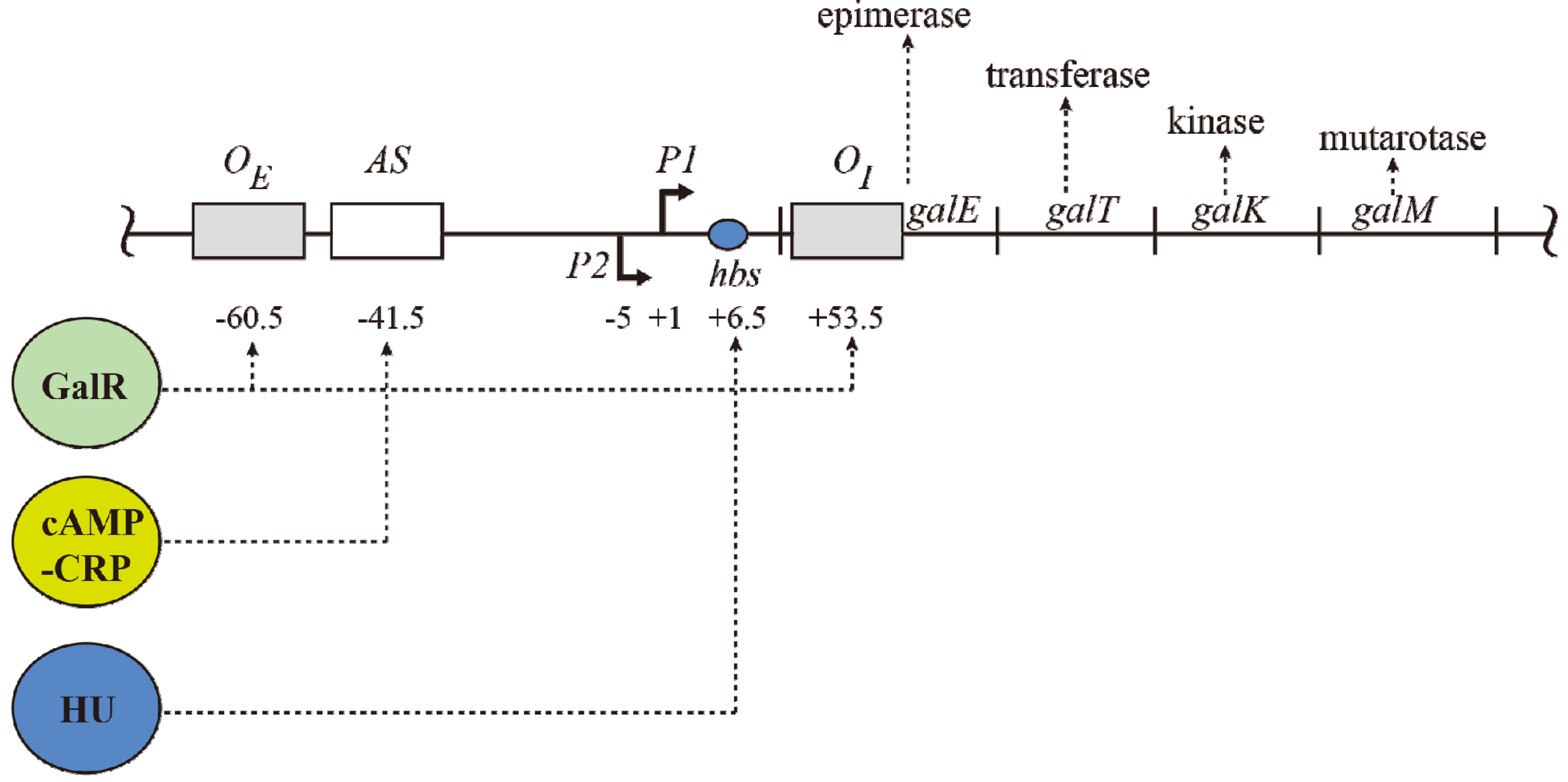
Figure 1.
The gal operon. The gal structural genes, galETKM, encode the enzymes epimerase, transferase, kinase and mutarotase, respectively. The transcription start point (tsp) of P1 is +1 and that of P2 is −5 The numbering system is relative to +1, with numbers downstream of +1 as positive (+) and numbers upstream as negative (−). Operators (OE & OI), hbs: HU binding site, AS: activating site. GalR (green) binds to two operators (OE & OI). HU (blue) binds to the HU binding site (hbs) and cAMP-CRP (yellow) complex binds to the activating site (AS). The map is not drawn to scale.
Figure 1.
The gal operon. The gal structural genes, galETKM, encode the enzymes epimerase, transferase, kinase and mutarotase, respectively. The transcription start point (tsp) of P1 is +1 and that of P2 is −5 The numbering system is relative to +1, with numbers downstream of +1 as positive (+) and numbers upstream as negative (−). Operators (OE & OI), hbs: HU binding site, AS: activating site. GalR (green) binds to two operators (OE & OI). HU (blue) binds to the HU binding site (hbs) and cAMP-CRP (yellow) complex binds to the activating site (AS). The map is not drawn to scale.
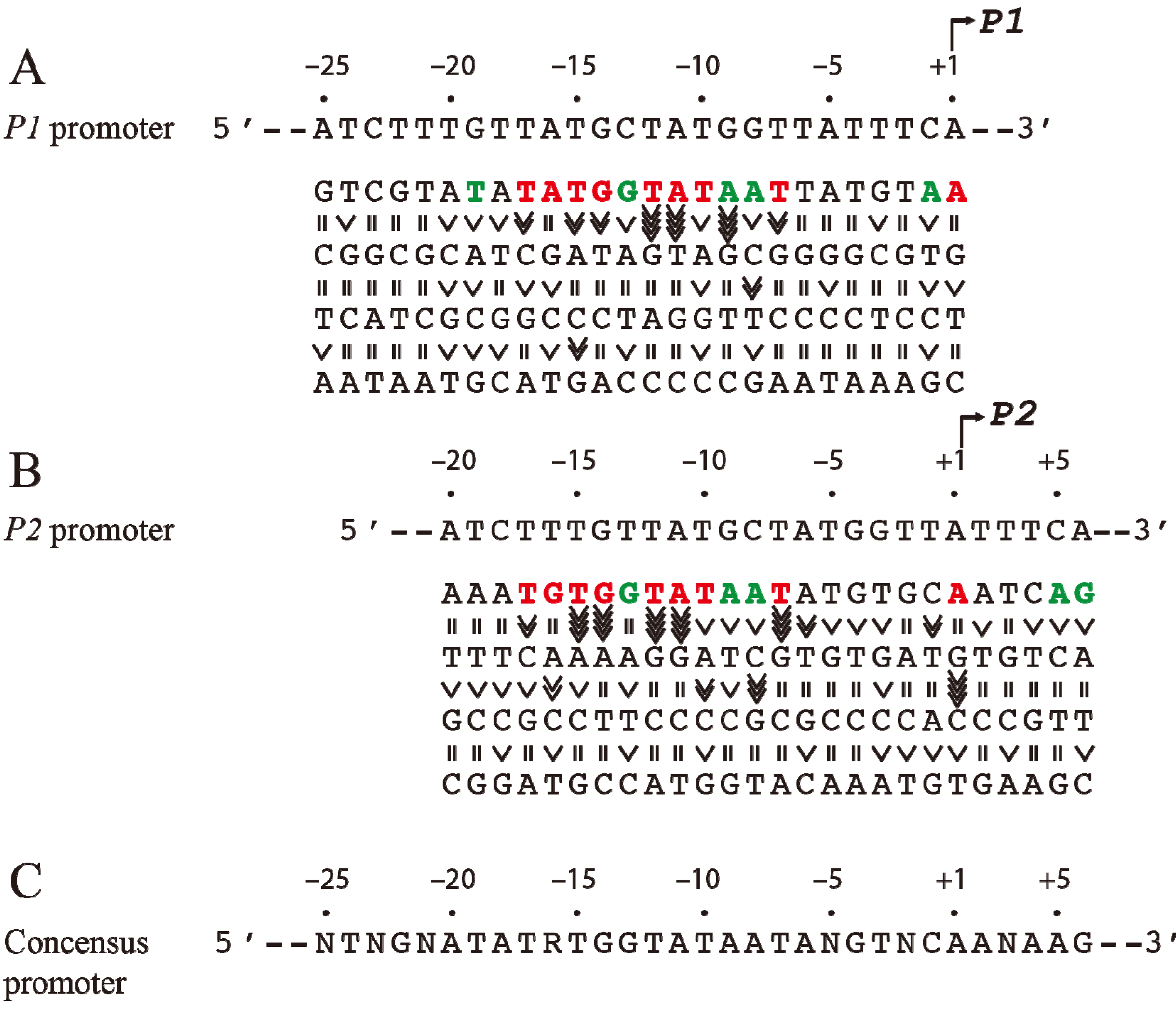
3. Intrinsic Strength of Promoters
Since the
tsps for
P1 and
P2 are separated by 5-bp (half of a helical turn on B-DNA), the two promoters are located on opposite faces of the DNA. The intrinsic strength of each promoter depends on the contribution of several critical base pairs in the promoter region [
9]. Both
P1 and
P2 do not have functional −35 elements but are composed of ex-10 and −10 sequences as authenticated by mutational studies [
10,
11]. In the absence of regulatory protein,
P2 is transcribed 3-fold more efficiently than
P1 [
12]. The intrinsic strength of a promoter was postulated to be dependent on the presence of and the closeness of DNA sequences of the different elements to their consensus forms so that the frequency of occurrence of a base pair at a given position of the element reflects its relative importance in promoter function [
13]. But, the significance of the base pair frequency concept in promoter strength was developed without regard to the context sequence. It is probable that the contribution of a base pair to the promoter strength may depend upon the presence of a specific base pair at another seemingly unrelated position in the promoter. This would not be known by looking for consensus sequences among heterologous promoters. A meaningful approach would be to assess the contribution of a base pair at a given position in the promoter under the context sequence that was kept constant. To study the effects of individual base pairs on the intrinsic strength of the promoters, each base pair in the overlapping
gal promoter region (from −20 to the +5) was mutated systematically to the other three base pairs and the promoter activities were analyzed by an
in vitro transcription assay [
14]. First, it was observed that purines at the non-template strand at the
tsp of
P1 and
P2 are favorable for the initiation of transcription while pyrimidines are unfavorable with a preference for A = G >> C = T at the
tsp (
Figure 2). The
tsp is determined by counting 12 base pairs from the “master base” −11A (see below) located within the −10 element of
P1 and
P2 [
15,
16]. Next, base pairs −7T, −11A, and −12T were found to be critical determinants of promoter activity. Mutating the corresponding −7T, −11A or −12T to another base inactivated
P1 and
P2 [
14]. In addition, base pairs in the ex-10 elements (−15T and −14G) of
P1 and
P2 were also critical for promoter activities as expected from previous results. In summary, the base pair frequency within known consensus elements correlated well with promoter strength. Surprisingly however,
P1 and
P2 promoter strengths increased by substitution of several native base pairs by some others located in the −20 to −16 segment,
i.e., outside the ex-10 and −10 standard elements of both promoters with a consensus sequence of
−20ATATA/G
−16 for the region; no sequence requirement in that segment was predicted before. How this new sequence element influences promoters is unknown. The results of the exhaustive mutational analysis about DNA sequence requirements in
gal promoters are summarized in
Figure 2 [
14].
The steps of closed and open complex formation in
gal promoters were studied by the indirect abortive initiation method [
17]. The mechanism of base pair opening during transcription initiation by RNA polymerase at the
galP1 promoter was directly assayed by 2-aminopurine (2,AP) fluorescence [
18]. The fluorescence of 2,AP is quenched when present in DNA duplex and enhanced when the 2,AP:T base pair is distorted or deformed. The increase of 2,AP fluorescence was used to monitor base pair distortion at several individual positions in the promoter. Base pair distortions during isomerization were observed at every position tested except at −11 in which the substitution created a defective promoter. The isomerization appeared to be a multi-step process. Three distinct hitherto unresolved steps in kinetic terms were observed, where significant fluorescence change occurred: a fast step with a half-life of around 1 s, which is followed by two slower steps occurring with a half-life in the range of minutes at 25 °C. Contrary to commonly held expectations, base pairs at different positions opened by 2,AP assays without any obvious pattern, suggesting that base pair opening is an asynchronous multi-step process. Note that 2,AP was used only at positions where there was an A in the “opening” region of the promoter.
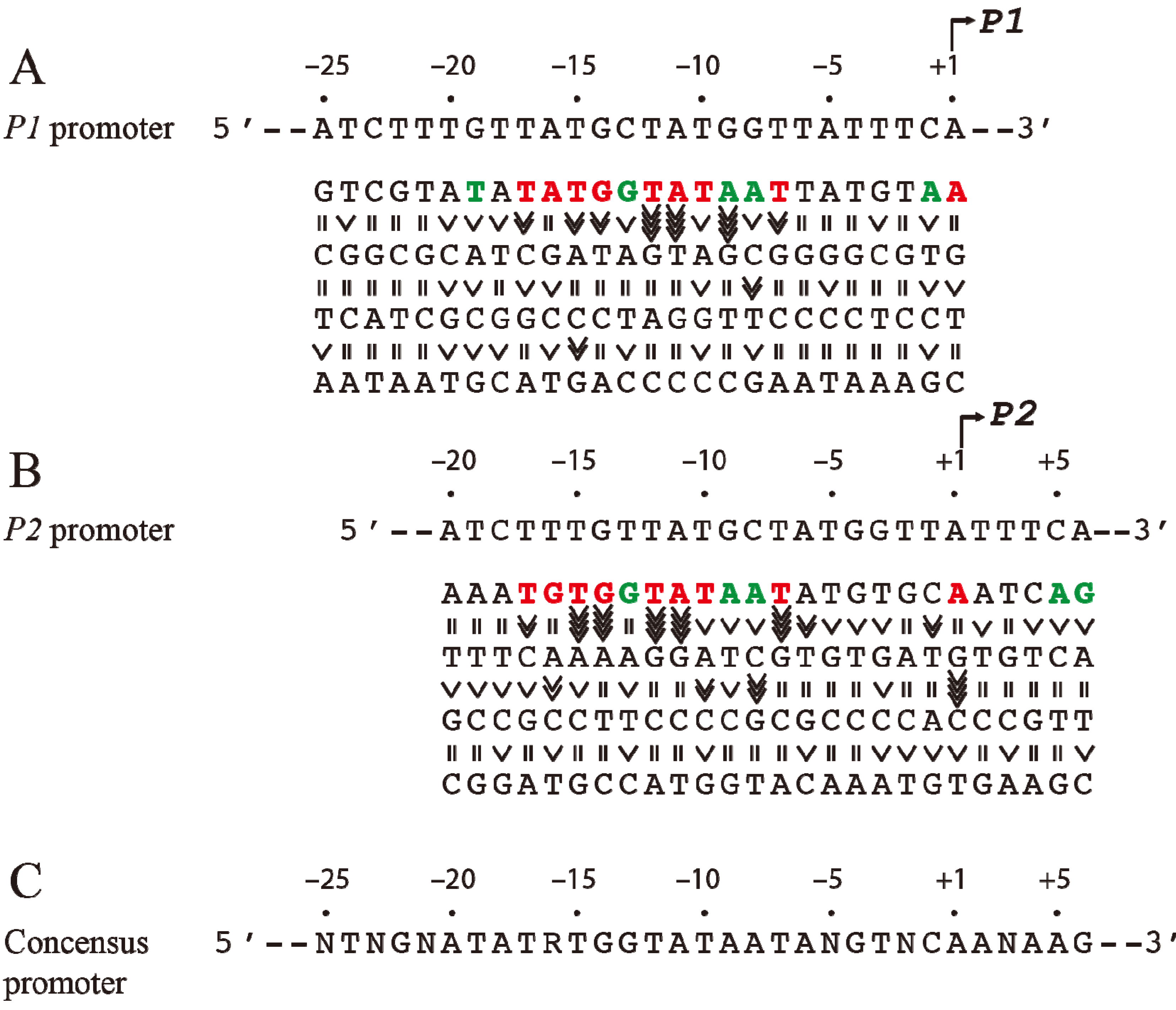
Figure 2.
Base pair requirement in
gal promoters. (
A) The DNA sequence from −25 to +1 of
P1 and a summary of the effect of base pair changes from +1 to −25 on
P1 transcription; (
B) The DNA sequence from −20 to +6 of
P2 and a summary of the effect of base pair changes from −20 to +6 on
P2 transcription; (
C) Consensus promoter region of
P1 and
P2 derived from the results shown in (A) and (B). R = A or G, N = any nucleotide. Base pair is in red if it is unique for promoter function, green if it improves promoter function, and black if it is degenerate. The symbol “>>>>” in vertical shapes represents 4.1-fold or more difference in promoter function from the wild type; “>>>”, 3.1 to 4-fold; “>>”, 2 to 3-fold; “>” less than 2-fold; “=” indicates equal (reproduced with permission from Elsevier, [
14]).
Figure 2.
Base pair requirement in
gal promoters. (
A) The DNA sequence from −25 to +1 of
P1 and a summary of the effect of base pair changes from +1 to −25 on
P1 transcription; (
B) The DNA sequence from −20 to +6 of
P2 and a summary of the effect of base pair changes from −20 to +6 on
P2 transcription; (
C) Consensus promoter region of
P1 and
P2 derived from the results shown in (A) and (B). R = A or G, N = any nucleotide. Base pair is in red if it is unique for promoter function, green if it improves promoter function, and black if it is degenerate. The symbol “>>>>” in vertical shapes represents 4.1-fold or more difference in promoter function from the wild type; “>>>”, 3.1 to 4-fold; “>>”, 2 to 3-fold; “>” less than 2-fold; “=” indicates equal (reproduced with permission from Elsevier, [
14]).
The −11A within the −10 box is termed the “master control switch” because DNA melting and DNA strand opening first occur at −11 during isomerization from the closed to the open complex followed by opening at subsequent positions (−11 to +3) [
15,
19,
20]. A mutant −11A does not allow base pairs at other positions to open whereas the reverse is not the case. Crystal structure studies showed that −7T and 11A flip out into hydrophobic pockets in an open complex [
21,
22]. This explains why any mutation in −7 and −11 positions results in the loss of
gal promoter activities [
14,
15,
19]. It also explains why −7T and −11A bases are highly conserved in the −10 element of promoters and play important roles during the formation of the open complex. It has been proposed that during isomerization, strand opening occurs from −11 to +3 to form a single-stranded DNA bubble, while −12T remains as part of the upstream double-stranded DNA bound to RNAP [
14].
4. Role of CCC
The
gal operon includes a 16-bp activating site (
AS) located at −40.5 that binds the regulator CCC for activating
P1 and repressing
P2 (
Figure 3A) [
8,
23,
24,
25]. A typical result of CCC action at the
gal promoters is shown in
Figure 3B. The overlapping of the
AS at −40.5 with the −35 element of
P1 is a feature of CCC-regulated Class II promoters. In contrast, in Class I promoters, the
AS is located upstream (−61.5) to the promoter region for RNA polymerase (RNAP).
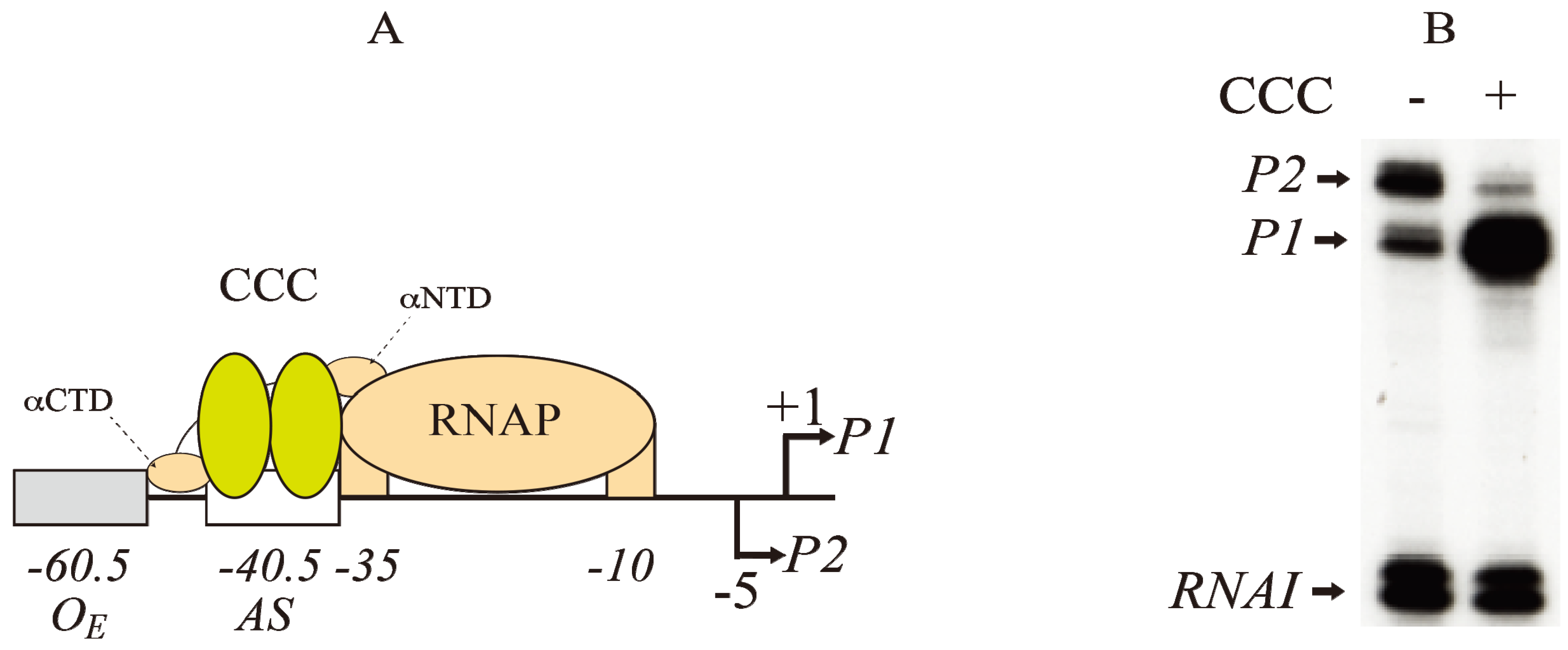
Figure 3.
Regulation of
gal promoters by CRP complex (CCC). (
A) Model of interactions between CCC (yellow) at the activating site (−40.5) and RNAP (brown) at the −10 and −35 elements of
P1 (+1).
P2 is located at −5 The αNTD and αCTD of RNAP contact both subunits of Class II promoters at CCC as shown in
Figure 4 (adapted from [
25]); (
B) RNAs made typically from
P1 and
P2 promoters in the absence (−) and presence (+) of CCC as analyzed by gel electrophoresis. The concentrations of cAMP and CRP are 100 μM and 50 nM, respectively.
RNAI is a control RNA in the plasmid (reproduced with permission from Elsevier [
14]).
Figure 3.
Regulation of
gal promoters by CRP complex (CCC). (
A) Model of interactions between CCC (yellow) at the activating site (−40.5) and RNAP (brown) at the −10 and −35 elements of
P1 (+1).
P2 is located at −5 The αNTD and αCTD of RNAP contact both subunits of Class II promoters at CCC as shown in
Figure 4 (adapted from [
25]); (
B) RNAs made typically from
P1 and
P2 promoters in the absence (−) and presence (+) of CCC as analyzed by gel electrophoresis. The concentrations of cAMP and CRP are 100 μM and 50 nM, respectively.
RNAI is a control RNA in the plasmid (reproduced with permission from Elsevier [
14]).
CCC represses
P2 by decreasing open complex formation of RNAP. In contrast, CCC activates
P1 by increasing both closed complex formation and isomerization from the closed complex to the open complex of RNAP [
17]. The
AS of CCC is located on the same face as RNAP at
P1 and on the opposite face of RNAP at
P2. By binding to
AS, CCC switches transcription initiation from
P2 to
P1. The activation of
P1 by CCC is also dependent on the superhelical density of the DNA. The maximal
P1 activity (12-fold) was observed at a superhelical density of −0.051, but the activity decreases at both higher and lower densities on a plasmid of 3528 bp [
26]. In the absence of CCC,
P2 activity is maximal (2-fold) also at a superhelical density of −0.051.
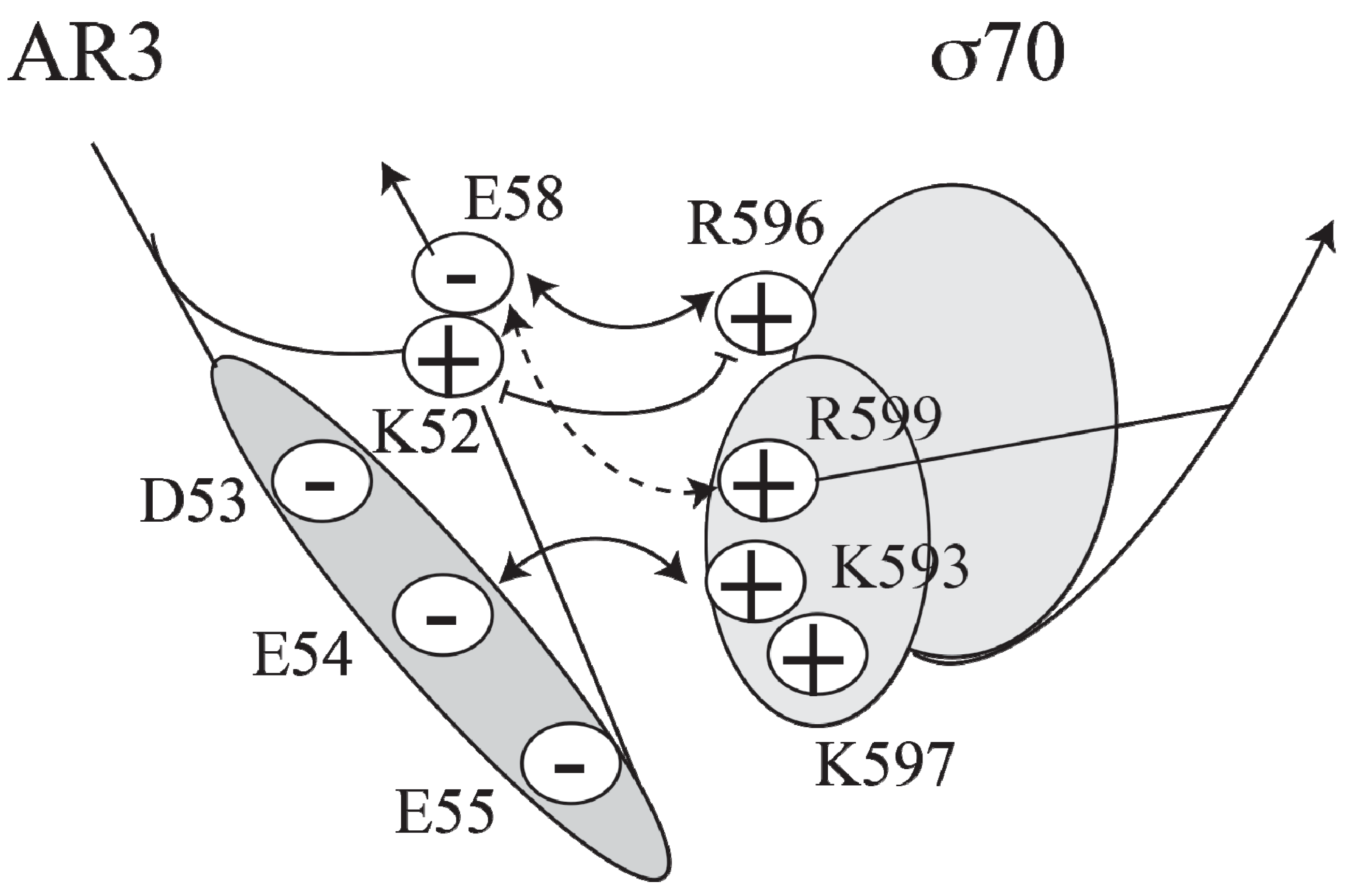
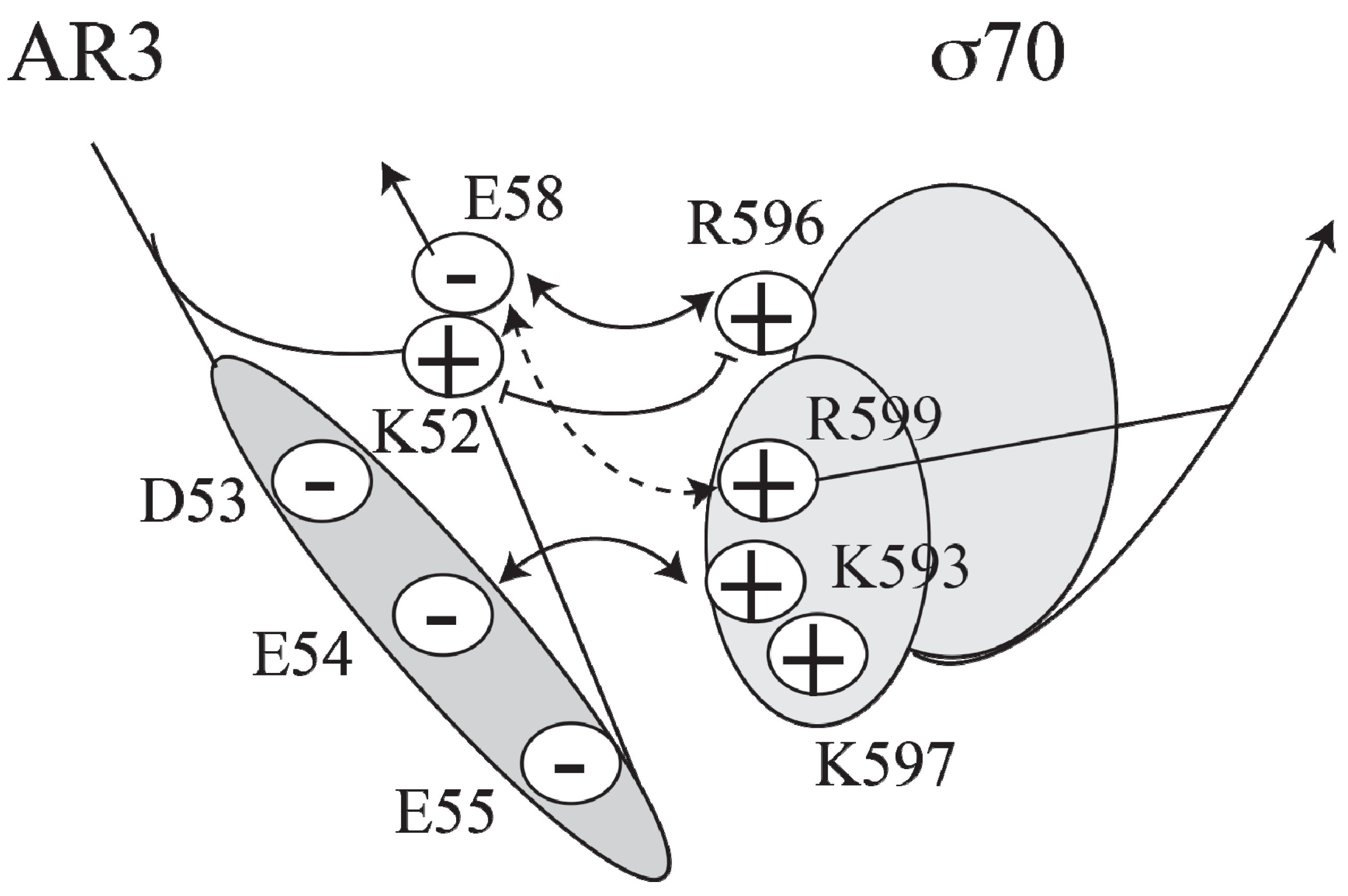
Figure 4.
Model of the interactions between AR3 and sigma 70 (adapted from [
27]).
Figure 4.
Model of the interactions between AR3 and sigma 70 (adapted from [
27]).
5. CCC and RNAP Interactions
Cooperative binding between CCC and RNAP was demonstrated using DNase I protection assays [
28]. Amino acids involved in the interaction of CCC with RNAP that produced cooperative binding are confined to three activating regions (AR1, AR2 and AR3) of CRP [
29,
30,
31,
32,
33,
34,
35,
36,
37]. The alpha carboxyl-terminal domain (αCTD, residues 249–329) of RNAP interacts with CRP at AR1 (residues 156–164), the alpha amino-terminal domain (αNTD, residues 8–235) interacts with CRP at AR2 (residues 1, 9, 21, 96 and 101), and the σ70 subunit of RNAP interacts with CRP at AR3 (residues 52–58) [
25,
29,
34,
38]. The crystal structure of CCC-αCTD-DNA complex has been determined [
36].
CCC induces transcription at
P1, a Class II promoter, by making three different activatory contacts with different surfaces of holo RNA polymerase [
34]. One of the contacts is located in the downstream subunit of the CRP dimer at the
AS site and has been predicted to interact with region 4 of the RNAP σ70 subunit [
27,
39]. A cluster of negatively charged residues (D53, E54, E55 and E58) in AR3 of CRP interacts with a cluster of positively charged residues (K593, K597, R599 and R596) in σ70 (
Figure 4).
RNAP predominantly forms a binary complex at the
P2 promoter in the absence of CCC and a ternary complex at the
P1 promoter in the presence of CCC. Very high concentrations of heparin are able to dissociate CRP from the
P1 ternary complex without changing the properties of the complex. Thus, CCC is not required for the maintenance of the RNAP complex and plays no role in the subsequent steps in
P1 transcription as was true for several other promoters [
40], suggesting that interaction between CCC and RNAP is needed only transiently for the activation of transcription.
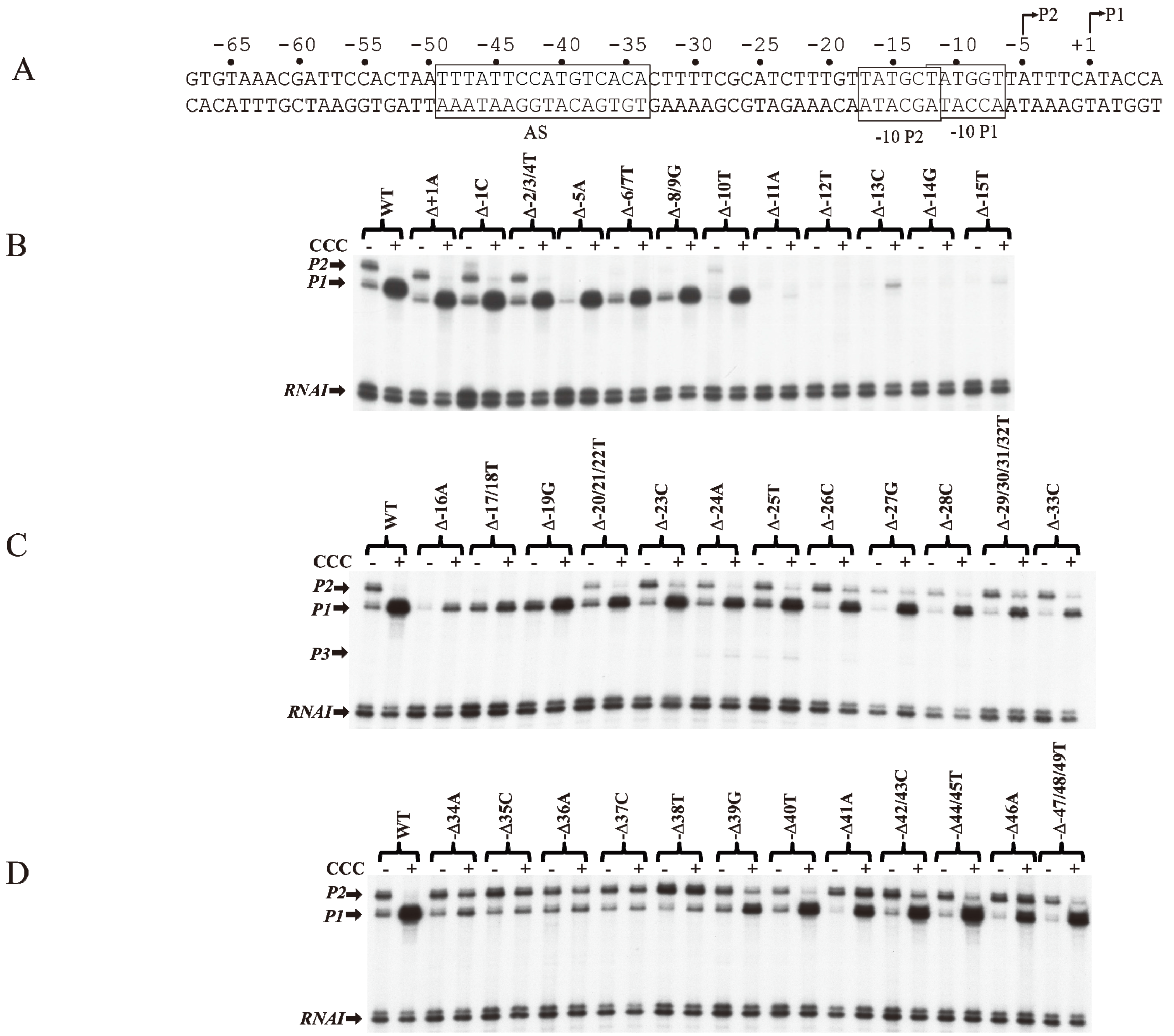
6. CCC Action on Templates with Single bp Deletions
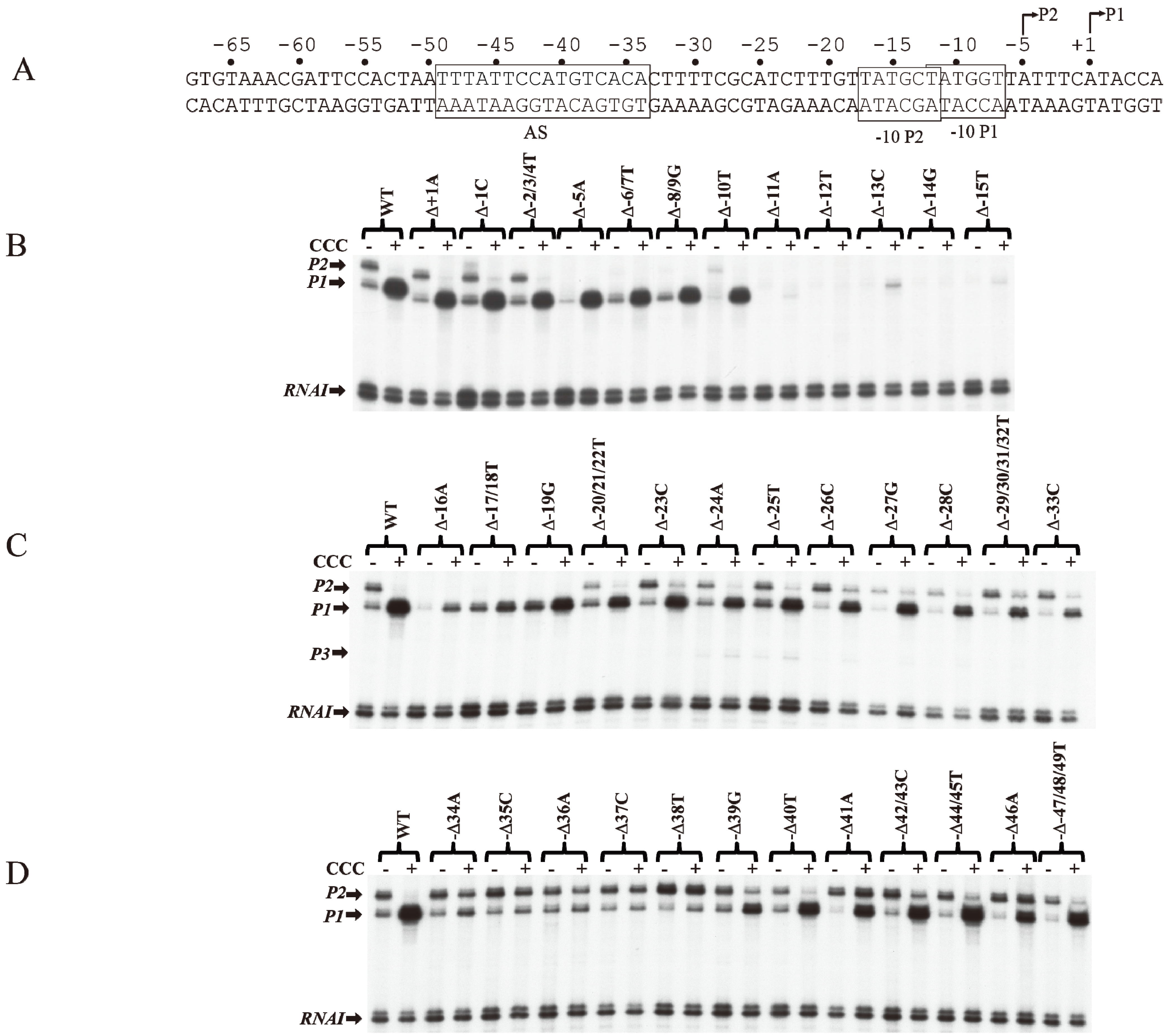
The role of individual base pairs from −49 to +1 on CCC action was investigated by systematically deleting each base pair and monitoring the effect of CCC on
P1 activation and
P2 repression (
Figure 5A). Deletion of one base pair from positions +1A to −10T (Δ+1A to Δ−10T) does not affect the activation of
P1 or the repression of
P2 by CCC (
Figure 5B). The deletion of 1-bp shifted the next adenine from +3 in WT to +2 in the deletion templates (Δ+1A to Δ−10T), allowing the
tsp of
P1 to initiate at the new +2A [
16].
P2 with
tsp of −5A was inactivated with single bp deletion from Δ−5A to Δ−8/9G, because, no adenine or guanine is available from −4 to −2 to initiate
P2. Single bp deletion of −11A or −12T inactivated both promoters. Interestingly, the
tsp of
P1 initiated at the new +2A on Δ−11A as observed by the faint transcript band in the presence of CCC. In Δ−12T,
P1 initiates at the WT +1A, suggesting that if the distance from −11T to +1 (12-nt) is shortened, RNAP will choose the next downstream purine to initiate transcription.
The −12T position is the first base of the −10 element of
P1 and the last base of the −10 element of
P2. It is not surprising that the intrinsic transcription of both
P1 and
P2 was inactivated, and CCC failed to activate
P1. The −13C to −17T sequence contains the ex-10 of
P1 (
−15TG
−14) and the −10 element of
P2 (
−17TATGCT
−12). Sigma region 2.5 of RNAP recognizes the ex-10 motif of promoters [
41,
42,
43,
44]. Detailed analyses of the ex-10 showed the importance of the ex-10 element in transcription regulation [
43]. Deletions of −16A and −17T/−18T result in approximately 5- and 2-fold activation of
P1 by CCC, respectively (
Figure 5C).
P2 was inactivated from −5A to −19G because its
tsp, ex-10 and −10 elements (
−20TGTTATGCT
−12) are altered. From −20T to −33C, the regulation of
P1 and
P2 is restored.
CCC is known to protect the
AS region in the
gal DNA from −50 to −25 bp by DNase I protection assays [
10,
31,
45,
46,
47].
AS contained a non-consensus (NC)
half-site and a consensus (C)
half-site separated by a 6-bp spacer [
48,
49,
50,
51]. The
AS extends from −49 to −34. Thirteen mutations each of which inhibits CCC action are located in the consensus half-site from −38 to −34 (
) proximal to the promoters [
23,
52]. When −34A is deleted, there is only marginal activation of
P1 and repression of
P2 (
Figure 5D). There was no noticeable change in
P1 or
P2 levels in the absence or presence of CCC when a base pair in the consensus half-site is deleted. These results suggest that CCC fails to bind to
AS when a base pair is deleted in the consensus half-site. The basal level of
P2 was increased by 2-fold in Δ−38T. Perhaps a stronger −35 element of
P2 is created with Δ−38T. The activation of
P1 by CCC was restored with single base pair deletions upstream of the consensus half-site from Δ−39G to Δ−47/48/49T.
P1 was activated only 4-fold in Δ−39G. These results suggest that the 6-bp spacer between the consensus and non-consensus half-sites do not affect CCC binding. These also suggest that mutations of the non-consensus half-site do not affect the activation of
P1 by CCC. Interestingly, Δ−41A and Δ−46A are the only two mutations in
P1, which were activated 9-fold in Δ−41A and 8-fold in Δ−46A by CCC. However, in both Δ−41A and Δ−46A templates, CCC activated
P2 marginally. Busby and colleagues showed that the consensus half-site is inactivated by three substitution mutations, p35 (−35 CG to GC), p37 (−37CG to AT) and p38 (−38 TA to AT) [
23,
52]. They also showed that
AS, unlike in WT, is not protected by CCC in p35, p37 and p38 mutants [
10,
46]. EMSA shows no stable complexes of CCC binding to a 144-bp DNA fragment containing p35, p37, or p38 mutations [
46].
In summary, (i) the distance between −11 and +1 determines the start point selection of P1 and P2. If a purine is not available at +1, RNAP selects the next downstream purine within 12–13 bp from −11A; (ii) the −7T, −11A and −12T are critical bases of the −10 elements of P1 and P2 for promoter function. Any deletion or substitution of these bases prevents intrinsic transcription. CCC restores transcription from P1 in −7T and −12T, but not in −11A; (iii) both base pairs in the ex-10 elements (−15TG−14) are critical in both P1 and P2 because deleting or substituting one of them inactivates both promoters; (iv) any base pair deletion in the spacer region from −20 to −33 does not affect the activation and repression of P1 and P2 by CCC, respectively; (v) CCC fails to activate P1 or repress P2 when any base pair in the consensus half-site (−34 to −38) of AS is deleted; (vi) any base pair deletion except −41 and −46A in the non-consensus half-sites does not affect the regulation of the promoters by CCC. The conclusion from the results of single base pair deletions about the role of base pairs in the promoters are mostly the same as from the results of single base pair substitutions in the gal promoters.
Figure 5.
Effect of base pair deletions on in vitro transcription of gal promoters. (A) Sequence of gal DNA from −68 to +6 with tsp of P2 (−5) and P1 (+1). The −10 element of P1 and −10 element of P2 are boxed. The CCC site (AS) is also boxed; (B) mRNAs made from P1 and P2 from WT and mutant templates (Δ−1 to Δ−15) (reproduced with permission from John Wiley and Sons); (C) mRNAs made from P1 and P2 from WT and mutant templates (Δ−16 to Δ−33); (D) mRNAs made from P1 and P2 from templates with WT and mutant templates (Δ−34 to Δ−49).
Figure 5.
Effect of base pair deletions on in vitro transcription of gal promoters. (A) Sequence of gal DNA from −68 to +6 with tsp of P2 (−5) and P1 (+1). The −10 element of P1 and −10 element of P2 are boxed. The CCC site (AS) is also boxed; (B) mRNAs made from P1 and P2 from WT and mutant templates (Δ−1 to Δ−15) (reproduced with permission from John Wiley and Sons); (C) mRNAs made from P1 and P2 from WT and mutant templates (Δ−16 to Δ−33); (D) mRNAs made from P1 and P2 from templates with WT and mutant templates (Δ−34 to Δ−49).
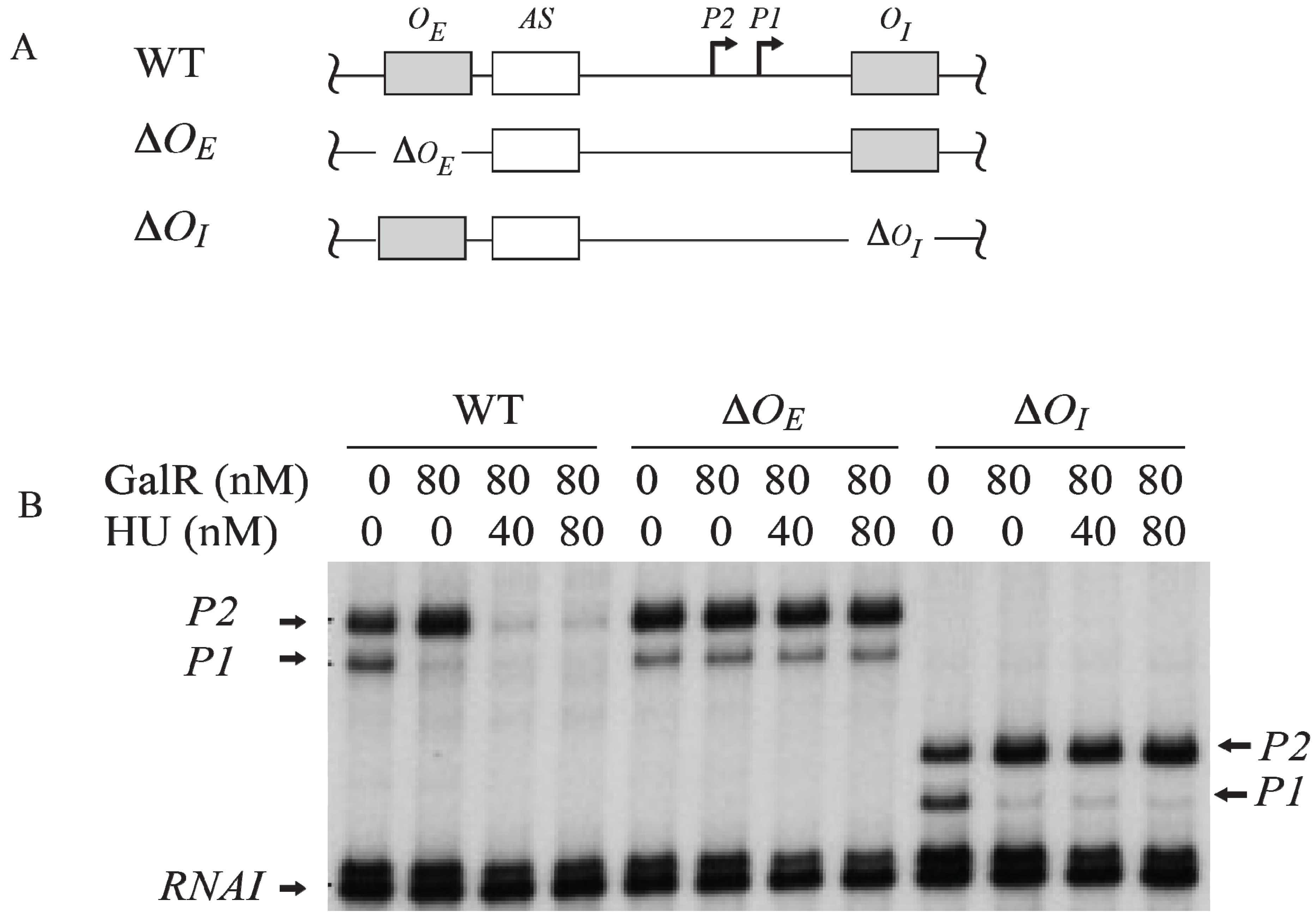
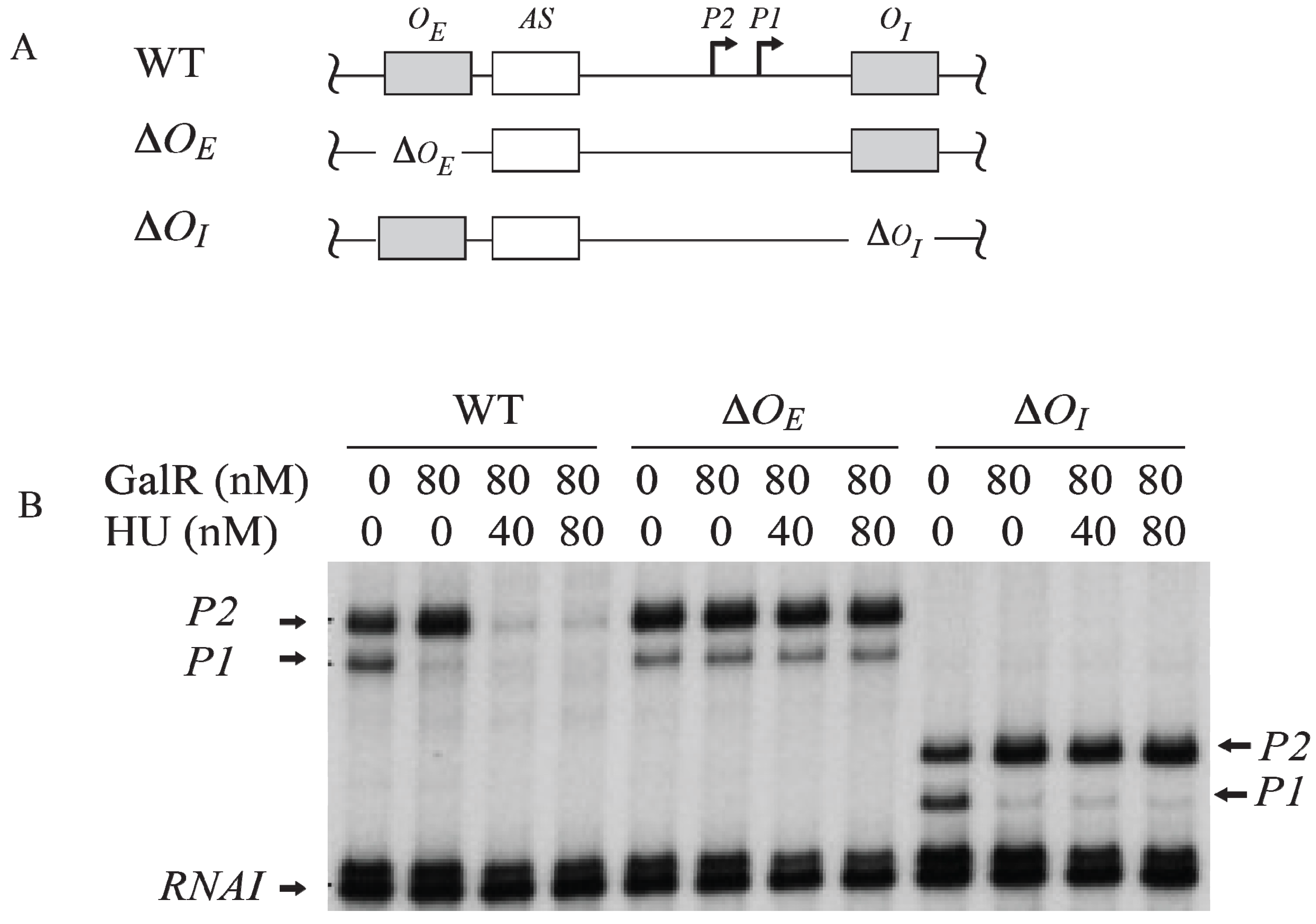
7. Regulation by GalR-OE Complex
To investigate the role of each operator in contact inhibition of
P1, and contact activation of
P2,
OE or
OI was subjected to mutational analysis (
Figure 6A). The result shows that the GalR-
OE complex formation is sufficient for the repression of
P1 (
Figure 6B) [
53,
54,
55,
56]. When
OE was deleted, there was no inhibition of
P1, no activation of
P2. When
OI was deleted, GalR-
OE complex still repressed
P1 and activated
P2. When
OI was deleted, the length of the transcripts from
P1 and
P2 was reduced by 16-nt since
OI is located downstream of both promoters. There is no change in the length of the transcripts from
P1 and
P2 when
OE was deleted because the
tsps of the promoters are located downstream of the
OE.
Figure 6.
Role of the operators in the transcription regulation of
gal promoters. (
A) Templates showing Δ
OE and Δ
OI deletions; (
B) mRNAs made from
P1 and
P2 on
OE and
OI, Δ
OE and
OI, and
OE and Δ
OI templates in the presence of GalR (80 nM) and HU (40 nM, 80 nM) (adapted from [
54]).
Figure 6.
Role of the operators in the transcription regulation of
gal promoters. (
A) Templates showing Δ
OE and Δ
OI deletions; (
B) mRNAs made from
P1 and
P2 on
OE and
OI, Δ
OE and
OI, and
OE and Δ
OI templates in the presence of GalR (80 nM) and HU (40 nM, 80 nM) (adapted from [
54]).
The operon consists of two GalR binding sites (16-bp operators),
OE (external operator, located at position −60.5) and
OI (internal operator within
galE, located at +53.5) [
2,
4,
57]. The
galE gene starts at an ATG (methionine code) located at position starting at +27. The operators are located 113-bp (~11 DNA helical turns) from each other (center to center distance) (
Figure 1). GalR binds to each operator as a dimer.
The binding of GalR to
OE represses
P1 and activates
P2 (
Figure 6B and
Figure 7A). GalR bound to
OE is located on the same DNA face as RNAP bound to
P1 (
Figure 7A), but on the opposite DNA face as RNAP bound to
P2 (
Figure 7B). GalR represses
P1 by inhibiting the rate determining open complex formation through RNAP contacts [
58]. This mode of repression is termed “contact inhibition” (
Figure 7A). While
P1 is repressed by GalR-
OE complex,
P2 is activated by GalR-
OE complex by a direct contact between GalR and RNAP, “contact activation” (
Figure 6B and
Figure 7B). GalR-
OE enhances open complex formation at
P2 presumably in the same way CCC does in
P1 [
12,
53,
59]. In
P1, GalR energetically traps RNAP at an intermediary complex [
53]. GalR mutants (
nc, for negative control) that bind to
OE and do not repress
P1 but represses
P2 have been isolated. These mutations presumably define the contact points of GalR to which RNA polymerase binds while occupying
P1 and need to be characterized further [
60]. The contact points of RNAP for GalR are unknown.
Figure 7.
Models of GalR-RNAP contacts. (A) Contact inhibition of P1: GalR (light green) at OE is interacting with the α-CTD of RNAP (brown) at the −10 and −35 elements of P1; (B) Contact activation of P2 with the α-CTD of RNAP at the −10 and −35 elements of P2.
Figure 7.
Models of GalR-RNAP contacts. (A) Contact inhibition of P1: GalR (light green) at OE is interacting with the α-CTD of RNAP (brown) at the −10 and −35 elements of P1; (B) Contact activation of P2 with the α-CTD of RNAP at the −10 and −35 elements of P2.
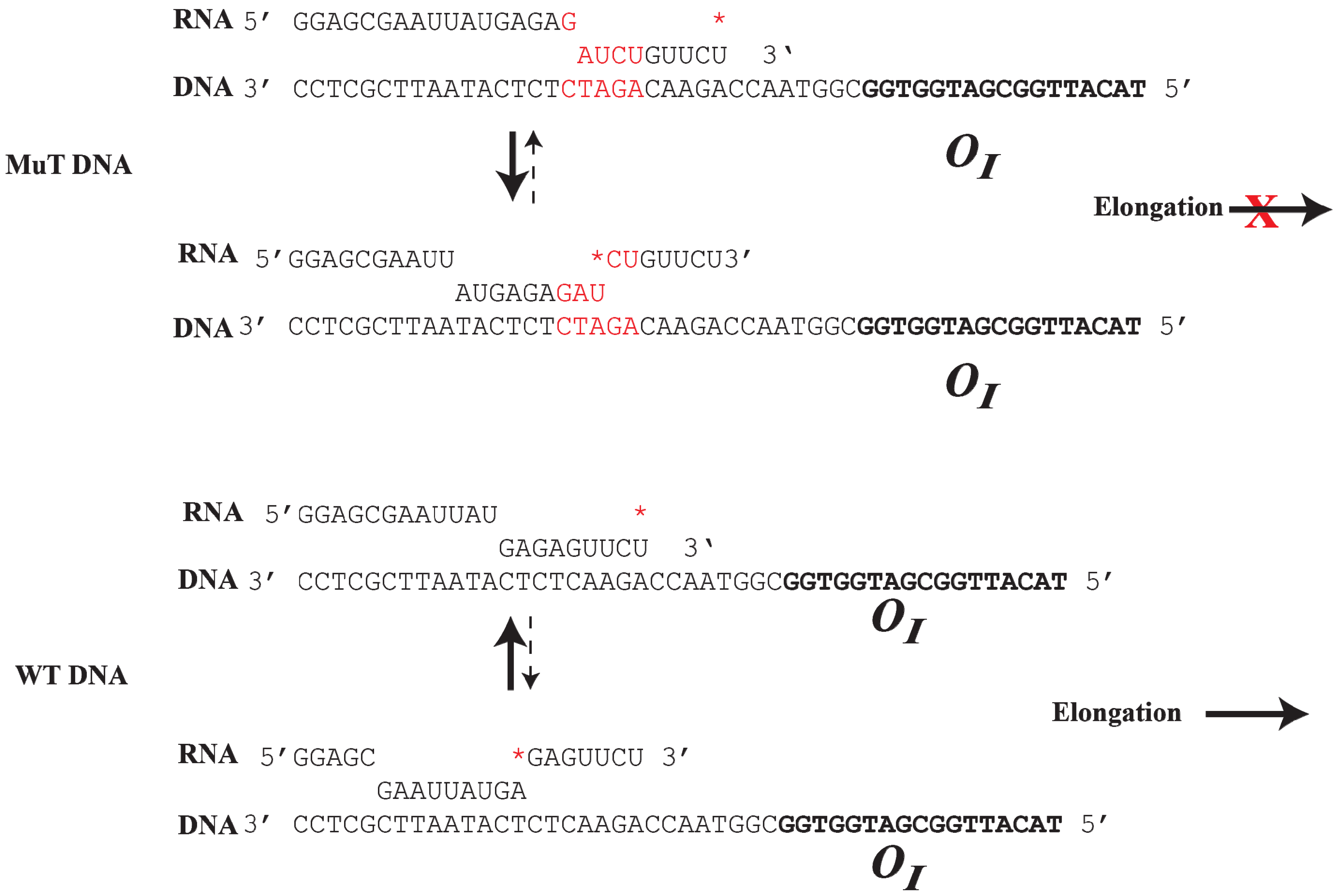
8. Roadblock of RNAP by GalR-OI Complex
The
OI operator is located at position +53.5, which is in the path of elongating RNAP complex transcribing from
P1 and
P2. The question is whether GalR-
OI complex can inhibit or block RNAP elongation [
61]. Unexpectedly, transcription from the
gal promoters under
in vitro conditions overrides the expected physical block created by the presence of the GalR bound to
OI (
Figure 8). It has been shown that although a stretch of pyrimidine residues (UUCU) in the RNA/DNA hybrid located immediately upstream of
OI weakens the RNA/DNA hybrid and favors RNA polymerase pausing and backtracking after encountering the roadblock, a stretch of purines (GAGAG) in the RNA present immediately upstream of the pause sequence in the hybrid acts as an anti-pause element by stabilizing the RNA/DNA duplex and preventing further backtracking. This facilitates forward translocation of RNAP, including overriding of the DNA-bound GalR barrier at
OI [
61]. Consequently, when the GAGAG sequence is separated from the pyrimidine sequence by a 5-bp DNA insertion, RNAP backtracking is favored from a weak hybrid to a more stable hybrid (
Figure 8). The roadblock of RNAP by GalR-
OI complex in the template with the 5-bp insertion was rescued by the transcription elongation factor, GreB, but not GreA. GreB and GreA cleave backtracked RNA in the catalytic center of RNAP to create a new 3'-end of the RNA, which can then be elongated [
62,
63,
64,
65]. As expected, the roadblock is also rescued by
d-galactose, which dissembles the GalR-
OI complex, allowing RNAP to continue transcription [
61]. The ability of a native DNA sequence to override roadblocks in transcription elongation in the
gal operon uncovers a previously unknown way of regulating transcription.
Figure 8.
Role of DNA sequence in RNAP elongation or backtracking. (
Top): The mutant DNA contains an insertion of 5-bp (GATCT, red color), which creates a weak RNA:DNA hybrid at the 3' end of the RNA. RNAP prefers to backtrack to a more stable RNA:DNA hybrid from a weak hydrid, preventing elongation; (
Bottom): In WTDNA, a strong 9-bp RNA:DNA hybrid is formed at the
gal pause site upstream from
OI. RNAP prefers to elongate instead of backtracking by 7-bp to a weak RNA:DNA hybrid. The
* indicates the 3' end of the RNA (reproduced with permission from Elsevier [
61]).
Figure 8.
Role of DNA sequence in RNAP elongation or backtracking. (
Top): The mutant DNA contains an insertion of 5-bp (GATCT, red color), which creates a weak RNA:DNA hybrid at the 3' end of the RNA. RNAP prefers to backtrack to a more stable RNA:DNA hybrid from a weak hydrid, preventing elongation; (
Bottom): In WTDNA, a strong 9-bp RNA:DNA hybrid is formed at the
gal pause site upstream from
OI. RNAP prefers to elongate instead of backtracking by 7-bp to a weak RNA:DNA hybrid. The
* indicates the 3' end of the RNA (reproduced with permission from Elsevier [
61]).
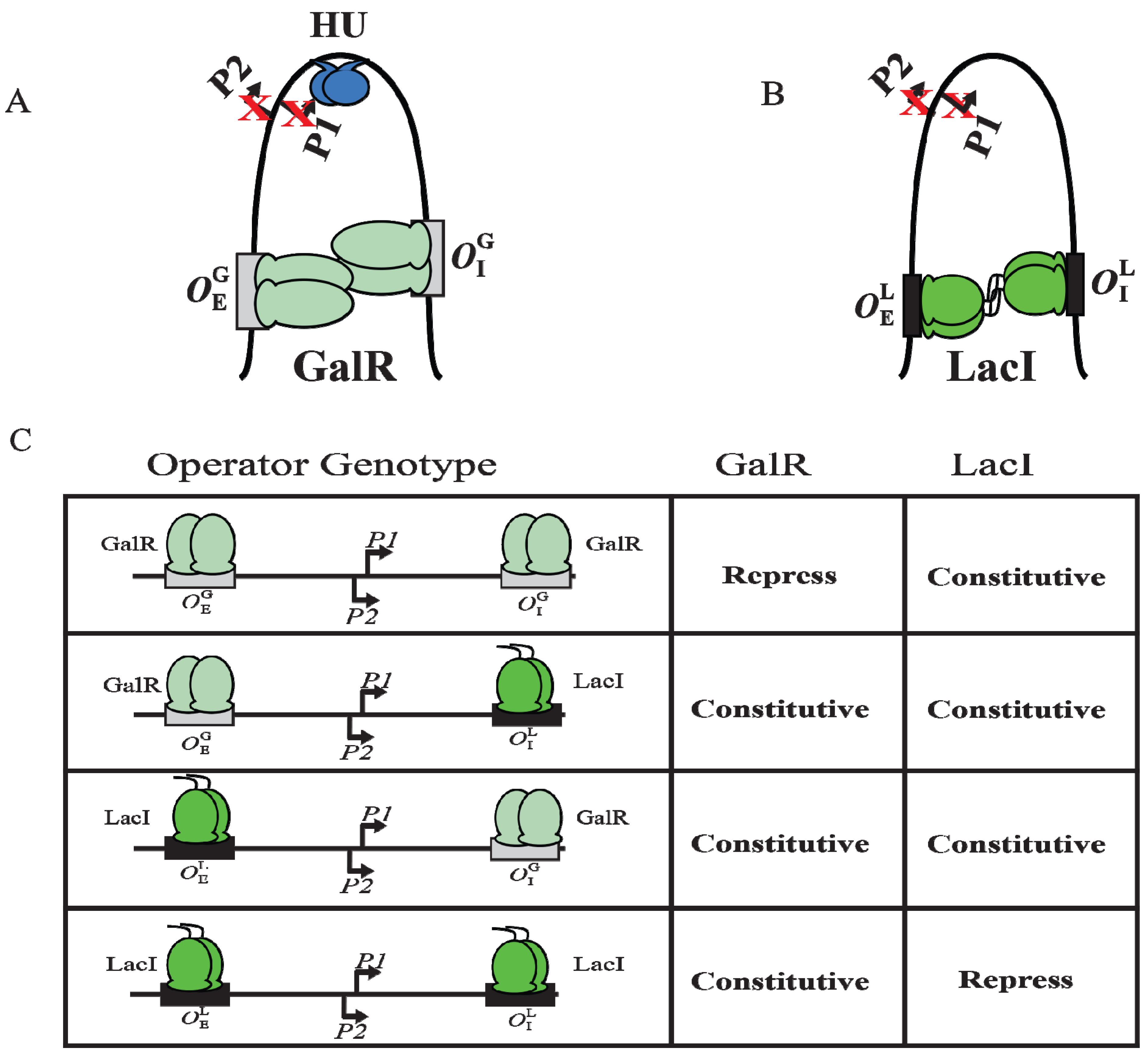
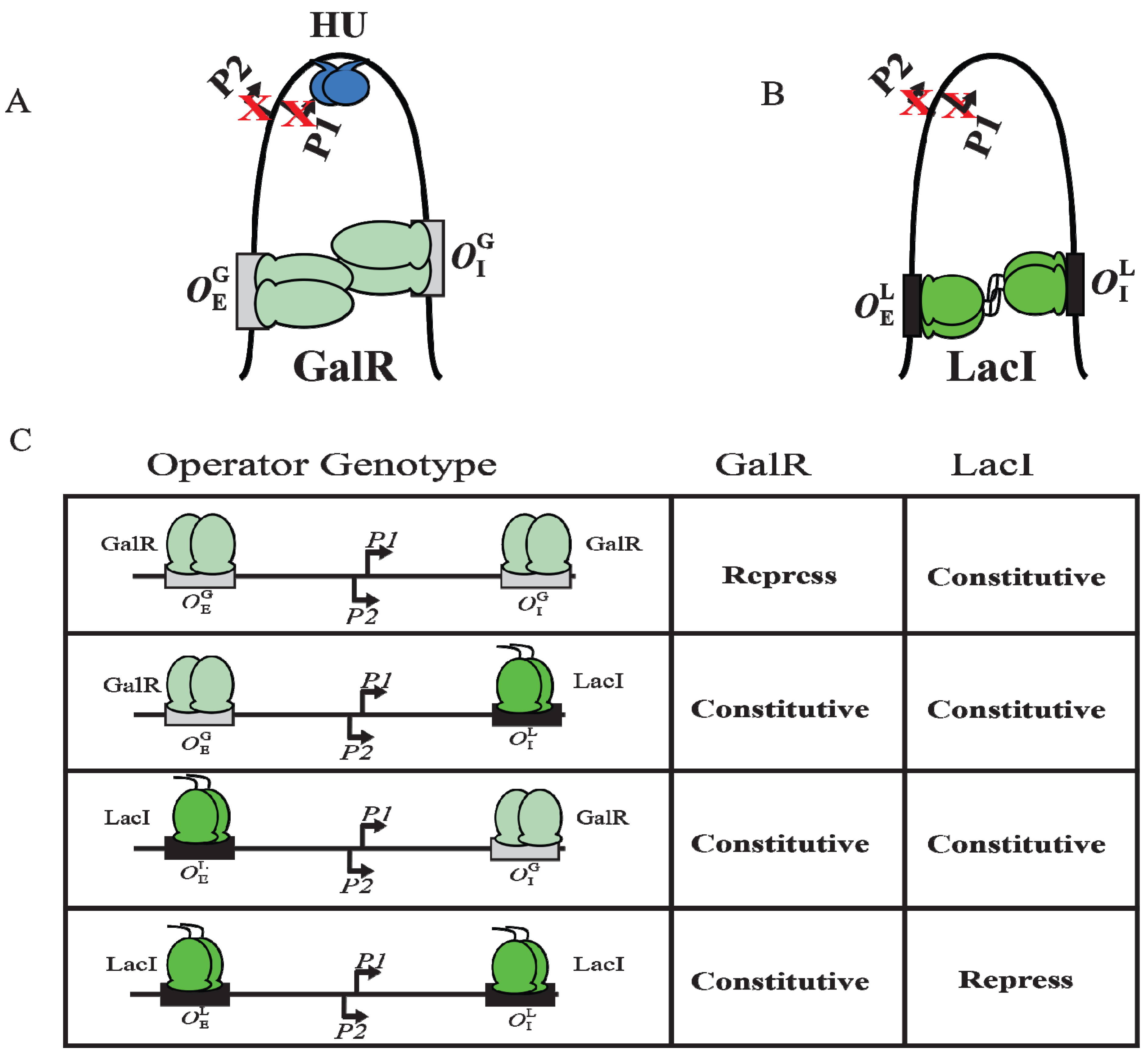
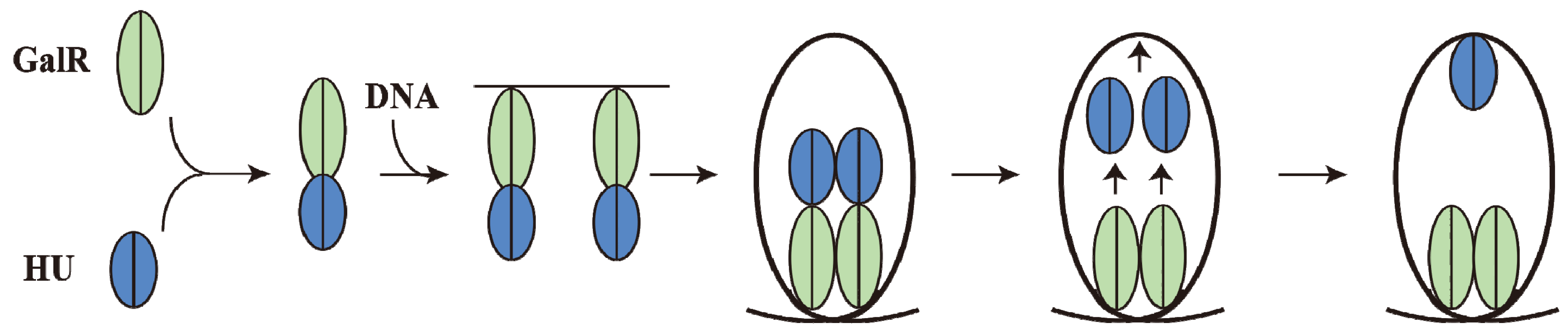
9. DNA Looping
Although GalR binding to a specific operator (
OE or
OI) has different regulatory outcomes, simultaneous binding of GalR to both operators (in the presence of HU; see later) represses both
P1 and
P2 (
Figure 6B). It was proposed that GalR bound to the distally located operators interact with each other forming a loop of the intervening DNA that contains the promoters (
Figure 9A). To test this model, a set of bipartite operators was constructed by converting
gal operators to
lac operators in various combinations (
,
,
,
) and
gal repression was studied
in vivo [
66]. GalR and LacI are part of the GalR-LacI family, in which members show 60% homology in sequence [
67]. Simultaneous repression of both promoters occurred only with homologous operators (
or
) in the presence of the cognate repressor (
Figure 9A–C) [
66]. GalR does not recognize
lac operators and LacI does not recognize
gal operators. These results suggest that the occupation of both operators by heterologous proteins was not sufficient for complete repression of the promoters. It was also inferred that protein-protein interactions occur between homologous proteins bound to cognate operators to form DNA loop and being about repression.
Figure 9.
DNA looping by GalR and LacI. (
A) Repressome formation by GalR-
OE and GalR-
OI interactions with HU (blue) and supercoiled DNA. DNA looping repressed both
P1 and
P2; (
B) DNA looping by LacI binding to
lac operators; (
C) The
in vivo level of galactokinase, a product of the
gal operon is reported as repressed or constitutive in the presence of GalR and LacI on
,
,
and
templates. GalR is in light green and LacI is in dark green. The
gal operators are in grey rectangular boxes, while the
lac operators are in black rectangular boxes (adapted from [
66]).
Figure 9.
DNA looping by GalR and LacI. (
A) Repressome formation by GalR-
OE and GalR-
OI interactions with HU (blue) and supercoiled DNA. DNA looping repressed both
P1 and
P2; (
B) DNA looping by LacI binding to
lac operators; (
C) The
in vivo level of galactokinase, a product of the
gal operon is reported as repressed or constitutive in the presence of GalR and LacI on
,
,
and
templates. GalR is in light green and LacI is in dark green. The
gal operators are in grey rectangular boxes, while the
lac operators are in black rectangular boxes (adapted from [
66]).
10. Mechanism of Repression by DNA Looping
Although an interaction between GalR-
OE and GalR-
OI complexes to generate a DNA loop was predicted from the
in vivo results,
in vitro experiments could not demonstrate DNA looping in
gal DNA in the presence of GalR [
68].
In vitro, DNA looping additionally needs the presence of the histone-like protein HU and supercoiled DNA (see below). HU assists the GalR-
OE complex and the GalR-
OI complex in stabilizing a higher-order complex structure, resulting in a DNA loop (see below). The histone binding site (
hbs) is located in the apex of the loop where the DNA is bent by HU binding, forming the higher-order structure and repressing both
P2 and
P1 (
Figure 6B) [
68]. The higher-order structure is termed “repressosome” (
Figure 9A).
P2 is repressed only by the repressosome. Incidentally, repression of both
P1 and
P2 at the same time by GalR-HU mediated DNA looping overrides the GalR-
OE and CCC-AS mediated DNA looping differential regulation of
P1 and
P2. Mechanistically, synergistic binding of GalR to distal sites forms 113 bp DNA loop which is a topologically closed domain containing the two promoters [
56]. A closed DNA loop of 11 helical turns, which is in-flexible to torsional changes, disables the promoters either by resisting DNA unwinding needed for open complex formation or by impeding the processive DNA contacts by an RNA polymerase in flux during transcription initiation. Interaction between two proteins bound to different sites on DNA modulating the activity of the intervening segment toward other proteins by allostery may be a common mechanism of regulation in DNA-multiprotein complexes.
As mentioned the
P1 promoter of
gal contains only ex-10 and −10 DNA elements and no −35 element. Thus, recognition of
P1 does not require specific contacts between RNA polymerase and its −35 element region. To investigate whether specific recognition of the −35 element would affect the regulation of
P1 by GalR, variants of
P1 in which the −35 element was restored were constructed and their regulation by DNA looping were studied by
in vitro transcription assays [
69]. The results showed that the GalR-mediated DNA loop is less efficient in repressing P1 transcription when RNA polymerase binds to the −10 and −35 elements concomitantly. The most likely explanation of RNA polymerase binding to −35 element inhibiting DNA looping is that RNA polymerase binding to −35 element is known to create a bend in the DNA at an improper position inhibits DNA loop formation.
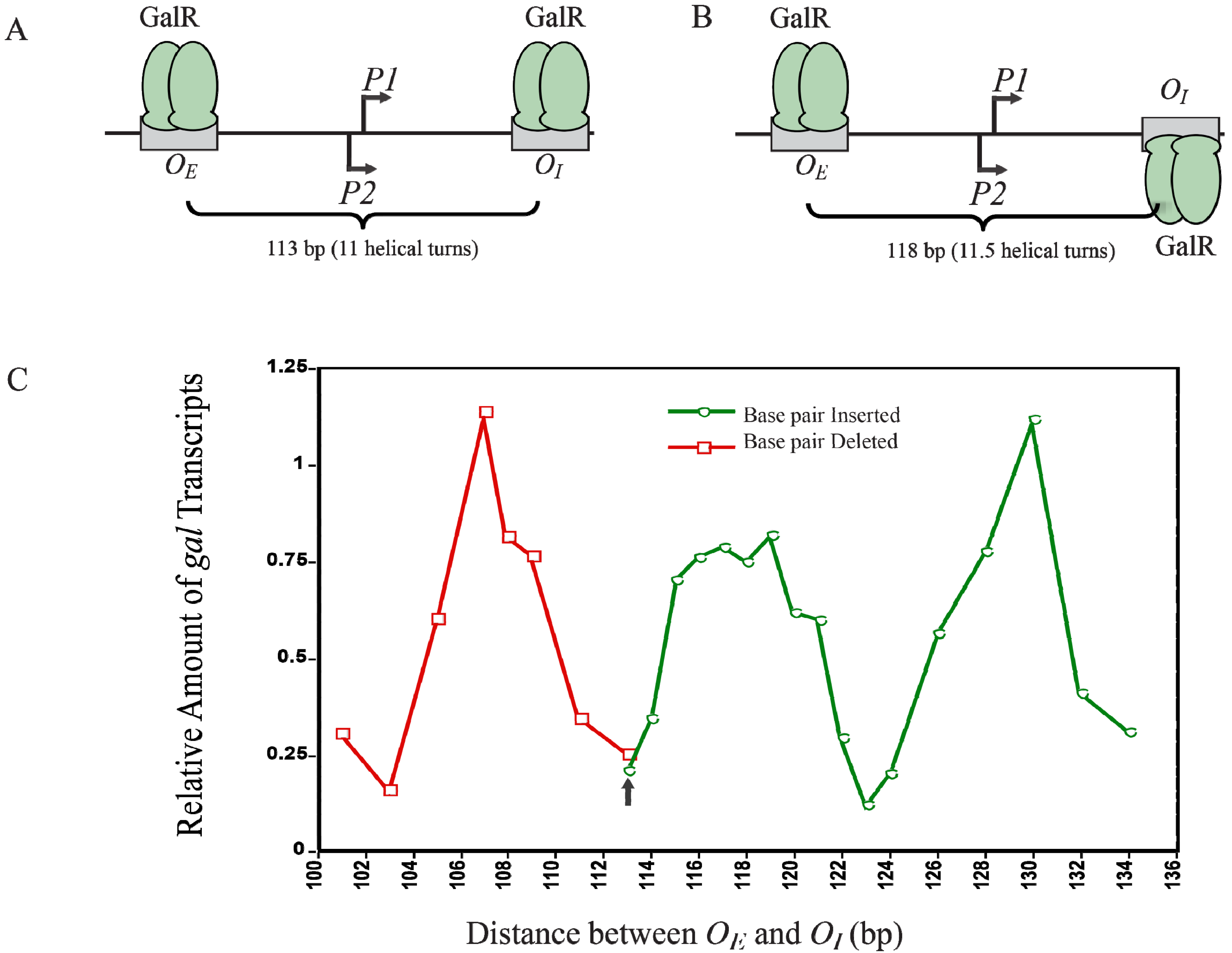
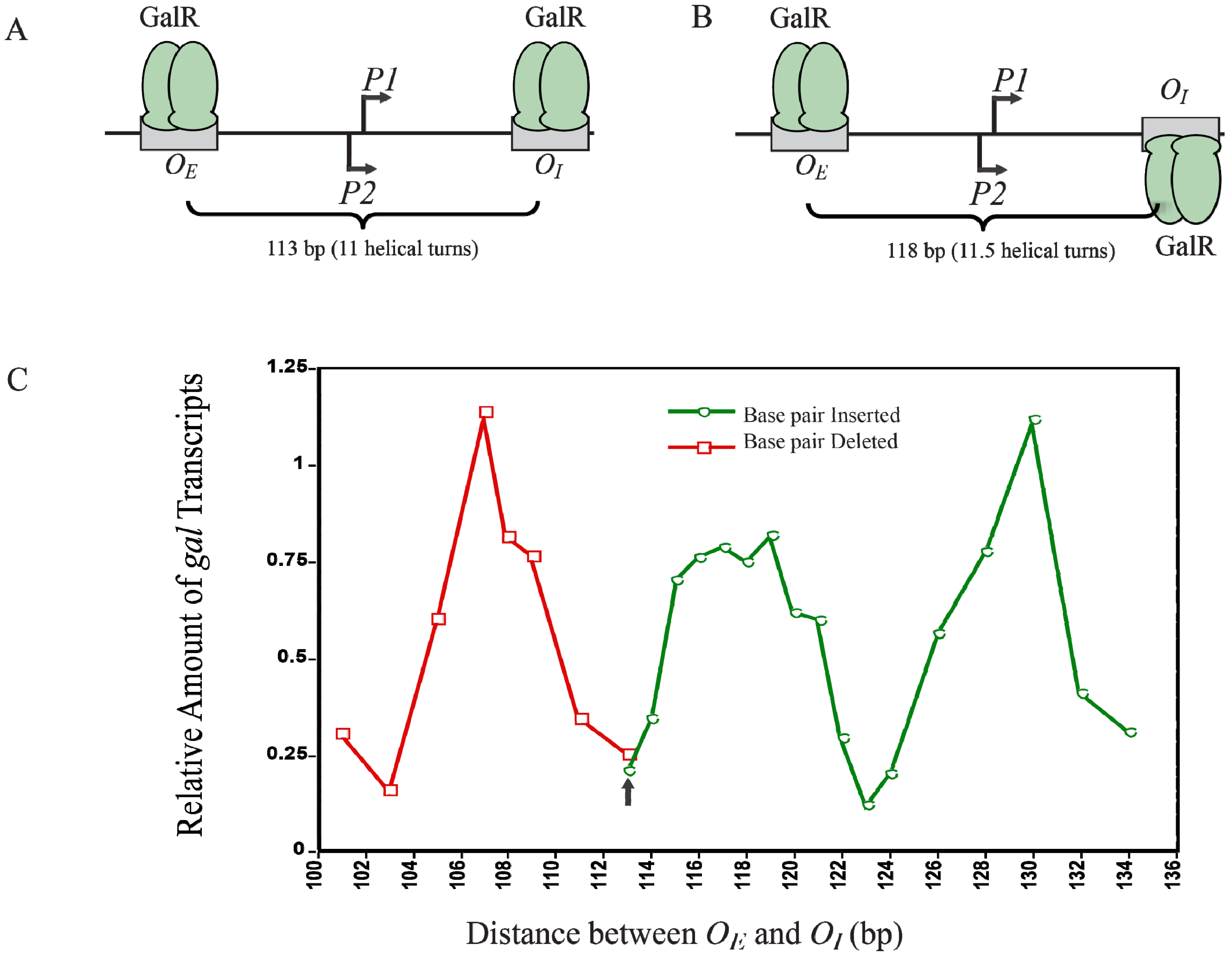
12. Helical Arrangement of Operators
The centers of the operators
OE and
OI are separated by 11 DNA helical turns, thus making the location of two operator-bound GalR on the same face of DNA. This arrangement energetically allows an interaction between two DNA bound GalR dimers, and consequent DNA looping and transcription repression, (
Figure 12A). To investigate the dependence of transcription repression on the relative helical turns of the location of
OE and
OI, the helical arrangement of
OE and
OI was changed by either deleting 2- to 12-bp within positions −50 to −38 to decrease the number of helical turns or by inserting 1- to 21-bp between positions +32 and +33 to increase the helical turns [
73].
Figure 12C shows that the optimal repression of
gal RNA synthesis is achieved at a net distance of 103-, 113-, 123-, and 133-bp, corresponding to 10, 11, 12 and 13 full helical turns, respectively. However, when a 5-bp segment was deleted (108-bp distance) or 5- and 15-bp segments were inserted (118- and 128-bp distance respectively),
gal repression was lifted as judged by the high expression of RNAs even in the presence of GalR, HU and supercoiled DNA presumably making the GalR-
OE and GalR-
OI complexes now located on the opposite face of the DNA more difficult to make GalR-GalR contact for DNA looping (
Figure 12B). Moreover, the loop size between
OE and
OI can be increased up to a total of 19 helical turns to maintain loping-mediated repression [
72]. Above 19 helical turns, the repression was reduced, perhaps because a bound RNAP is able to overcome DNA torsional stress and form open complex.
Figure 12.
Looping-mediated repression of
gal transcription is dependent on the helical distance between operators. (
A) GalR-
OE and GalR-
OI are located on the same face of the DNA at a distance of 113-bp (11 helical turns); (
B) GalR-
OE and GalR-
OI are located on the opposite face of DNA at a distance of 118-bp; (
C) Relative amount of transcription
vs. distance between the two operators in base pair in the presence of GalR and HU. The results of deleted base pairs (12 bp) are in red and the results of inserted base pairs are in green. The arrow indicates the WT distance between operators (adapted from [
73]).
Figure 12.
Looping-mediated repression of
gal transcription is dependent on the helical distance between operators. (
A) GalR-
OE and GalR-
OI are located on the same face of the DNA at a distance of 113-bp (11 helical turns); (
B) GalR-
OE and GalR-
OI are located on the opposite face of DNA at a distance of 118-bp; (
C) Relative amount of transcription
vs. distance between the two operators in base pair in the presence of GalR and HU. The results of deleted base pairs (12 bp) are in red and the results of inserted base pairs are in green. The arrow indicates the WT distance between operators (adapted from [
73]).
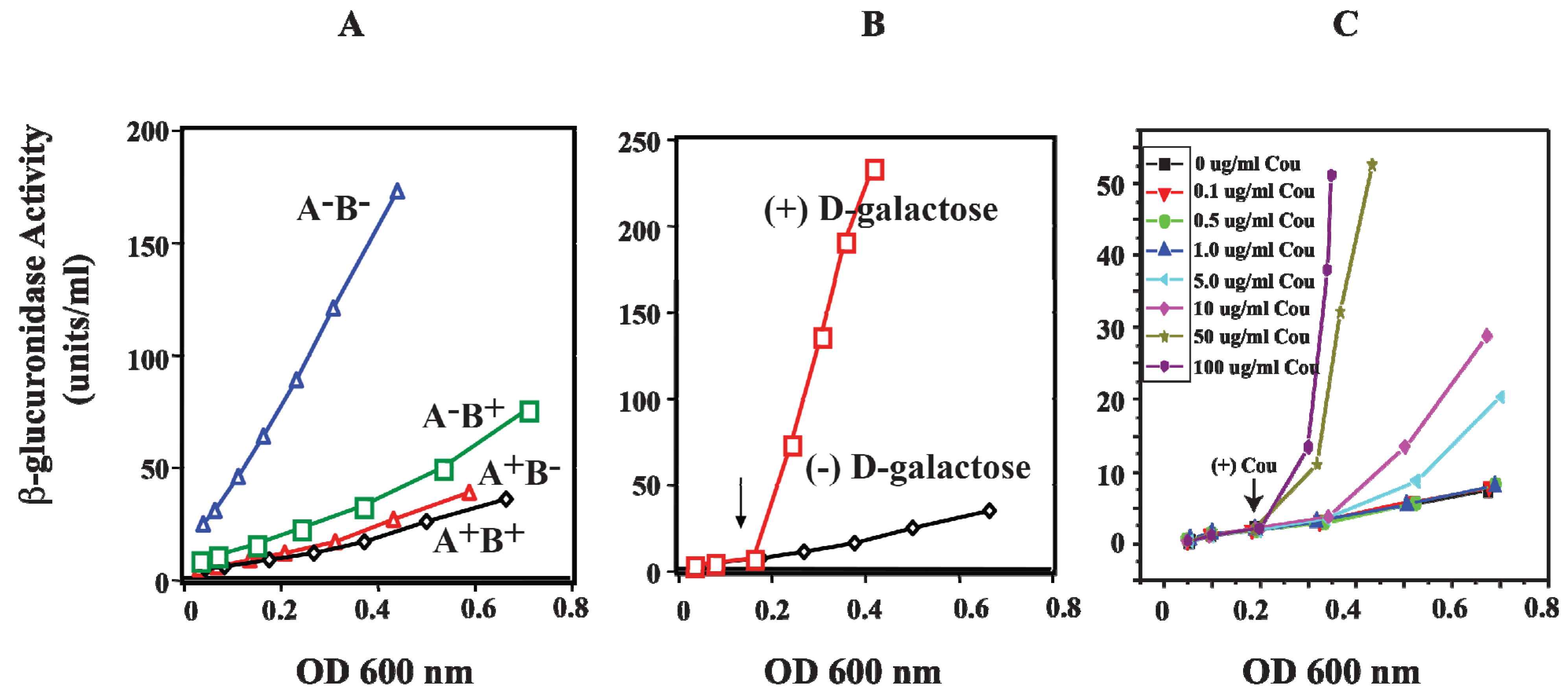
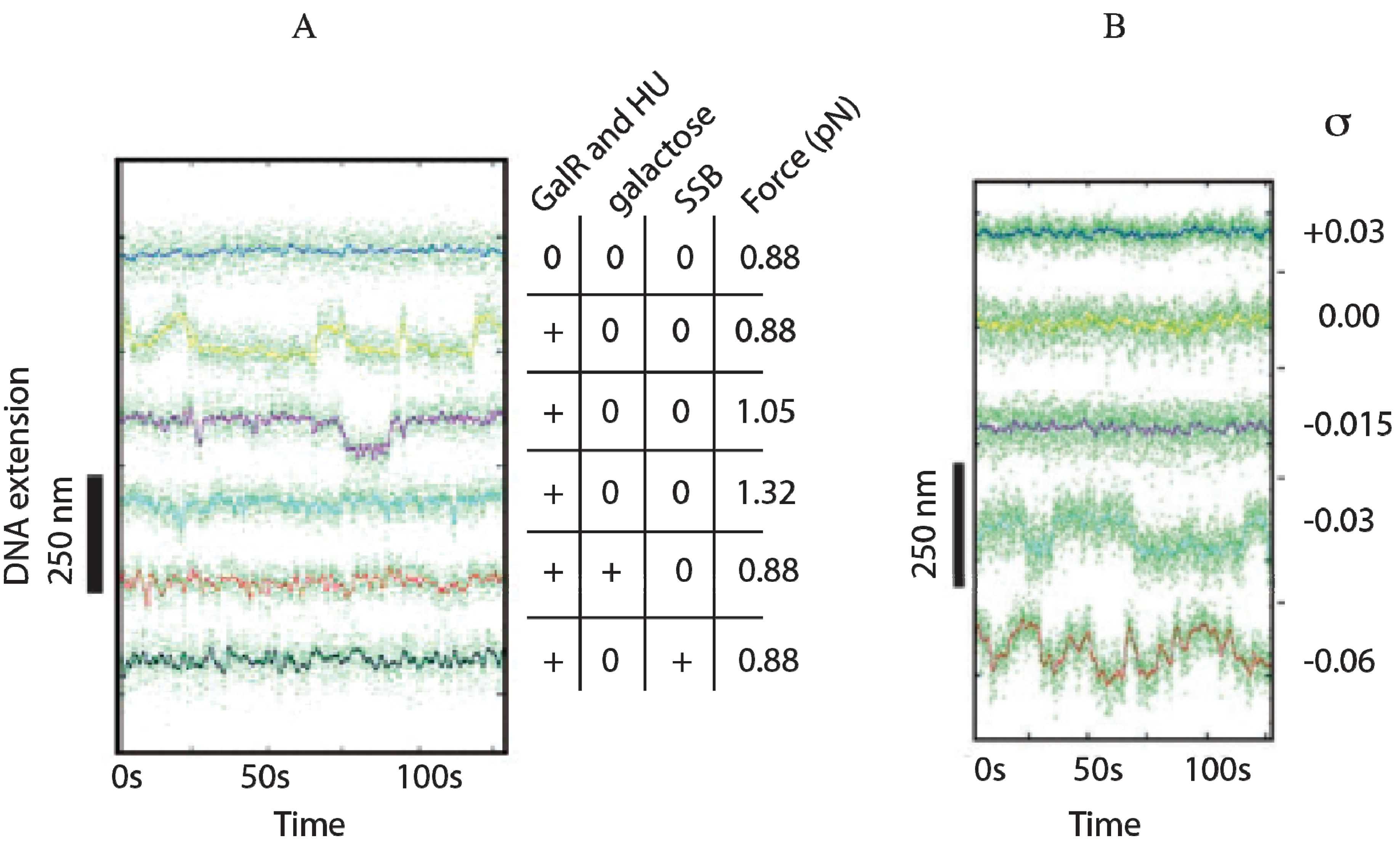
13. Role of HU in DNA Looping
HU is a small basic protein consisting of a heterodimer of α and β subunits, and binds to a 9-bp DNA [
74,
75]. The
hupA gene codes for Huα (~9 kDa) and the
hupB gene codes for Huβ (~9 kDa) [
76,
77]. The involvement of HU in DNA looping and the repression of the
gal operon were investigated
in vivo by monitoring the activity of β-glucuronidase from
gusA fused to the
P2 promoter. The
hupA and
hupB genes were deleted (Δ
hupA::cm
R and Δ
hupB::km
R) to generate
hupA
+B
−,
hupA
−B
+, and
hupA
−B
− strains [
78]. In wild-type strain (
hupA
+B
+), the β-glucuronidase activity of
P2 was repressed when both genes are present (
Figure 13A). In the presence of the
hupA gene (
hupA
+B
−),
P2 is strongly repressed as in wild-type cells, suggesting that
hupA is sufficient to achieve complete repression of
P2 by DNA looping. When
hupA is inactivated (
hupA
−B
+), the repression of
P2 is slightly weaker than that for
hupA
+B
+ and
hupA
+B
−. The derepression of β-glucuronidase activity of
P2 was completely constitutive only in the
hupA
−B
− strain. The
P2 activity of
hupA
−B
− is comparable to that of
P2 activity in the presence of
d-galactose, an inducer of the
gal operon (
Figure 13B) [
78].
Figure 13.
Effect of HU and
d-galactose on
P2 in vivo. (
A) β-glucuronidase activity as a reporter of the
P2 promoter in WT (
hupA
+B
+) and mutants (
hupA
+B
−,
hupA
−B
+ and
hupA
−B
−) strains; (
B) β-glucuronidase activity from
P2 in WT (
hupA
+B
+) strain in the absence and presence of
d-galactose; (
C) β-glucuronidase activity from
P2 in WT (
hupA
+B
+) strain in the presence of various coumermycin concentrations. The arrow indicates where
d-galactose or coumermycin was added. In each panel, the x-axis shows cell OD (reproduced with permission from John Wiley and Sons Ltd. (Hoboken, NJ, USA) [
78]).
Figure 13.
Effect of HU and
d-galactose on
P2 in vivo. (
A) β-glucuronidase activity as a reporter of the
P2 promoter in WT (
hupA
+B
+) and mutants (
hupA
+B
−,
hupA
−B
+ and
hupA
−B
−) strains; (
B) β-glucuronidase activity from
P2 in WT (
hupA
+B
+) strain in the absence and presence of
d-galactose; (
C) β-glucuronidase activity from
P2 in WT (
hupA
+B
+) strain in the presence of various coumermycin concentrations. The arrow indicates where
d-galactose or coumermycin was added. In each panel, the x-axis shows cell OD (reproduced with permission from John Wiley and Sons Ltd. (Hoboken, NJ, USA) [
78]).
15. Piggybacking HU
GalR mediated DNA looping requires binding of HU to an architecturally critical position on DNA (
hbs) to facilitate the GalR-GalR interaction. It has been shown that GalR piggybacks HU to the critical position on the DNA through a specific GalR-HU interaction [
79]. The thermodynamic parameters of some of the required interactions, GalR-
OE, GalR-GalR, HU-GalR, and HU-GalR-
OE, were studied by analytical ultracentrifugation, fluorescence anisotropy, and fluorescence resonance energy transfer [
80]. The physiological significance of several of these interactions was confirmed by the finding that a mutant HU, which is unable to help looping
in vivo and
in vitro, failed to show the HU-GalR interaction. The results helped to construct a pathway of DNA looping (
Figure 14). Structure-based genetic analysis indicated that the two DNA-bound GalR dimers interact directly and form a stacked tetramer in assembling a transient loop [
81]. The loop is stabilized by HU leaving GalR and binding to the architecturally critical position on the DNA. The GalR-HU contact is likely transient and absent in the final loop structure. A sequence-independent DNA-binding protein being recruited to an architectural site on DNA through a specific association with a regulatory protein may be a common mode for assembly of complex nucleoprotein structures [
80].
Figure 14.
Pathway of Repressosome formation. GalR (light green) and HU (blue) first bind together, then bind to the DNA, resulting in DNA loop formation with HU dissociating from GalR and binding to the apex of the DNA stabilizing the loop involving GalR-GalR interactions (adapted from [
80]).
Figure 14.
Pathway of Repressosome formation. GalR (light green) and HU (blue) first bind together, then bind to the DNA, resulting in DNA loop formation with HU dissociating from GalR and binding to the apex of the DNA stabilizing the loop involving GalR-GalR interactions (adapted from [
80]).
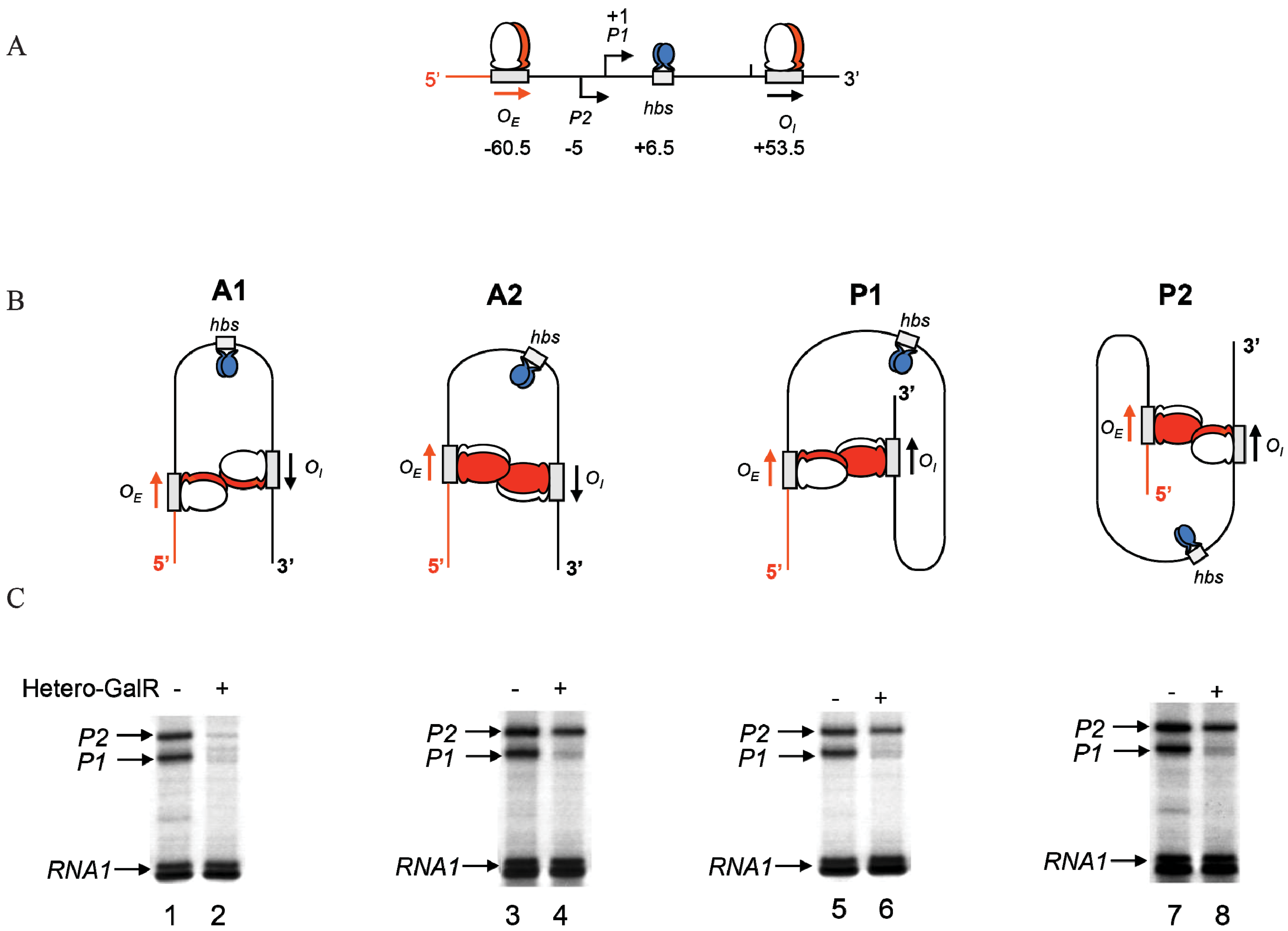
16. DNA Loop Trajectory
In the scheme of DNA looping as shown in
Figure 14, the alignment of the operators in the DNA loop could be in either parallel (P) or antiparallel (A) mode (
Figure 15). Feasibilities of these trajectories were tested by
in vitro transcription repression assays, first by isolating GalR mutants with altered operator specificity and then by constructing proper operator sequences to allow formation of mutant GalR heterodimers bound to specific hybrid operators in such a way as to give rise to only one of the two putative trajectories (parallel (P) or antiparallel (A)) [
5]. A1 loop is formed when the 3'-end of
OE is facing the 3'-end of
OI in a head-to-head (→ ←) orientation. The A2 loop is formed when the 3'-end of
OE is facing away from the 3'-end of
OI in a tail-to-tail (← →) orientation. In P1, the 3'-end of
OE is located in the same direction as the 3'-end of
OI in a head-to-tail (← ←) configuration, while in P2, the 3'-end of
OE is located in the opposite direction as the 3'-end of
OI in a tail-to-head (→ →) configuration. Results show that
OE and
OI adopt a mutual antiparallel orientation in an under-twisted DNA loop, consistent with the energetically optimal structural model. In this structure the center of the HU-binding site is located at the apex of the DNA loop (
Figure 15).
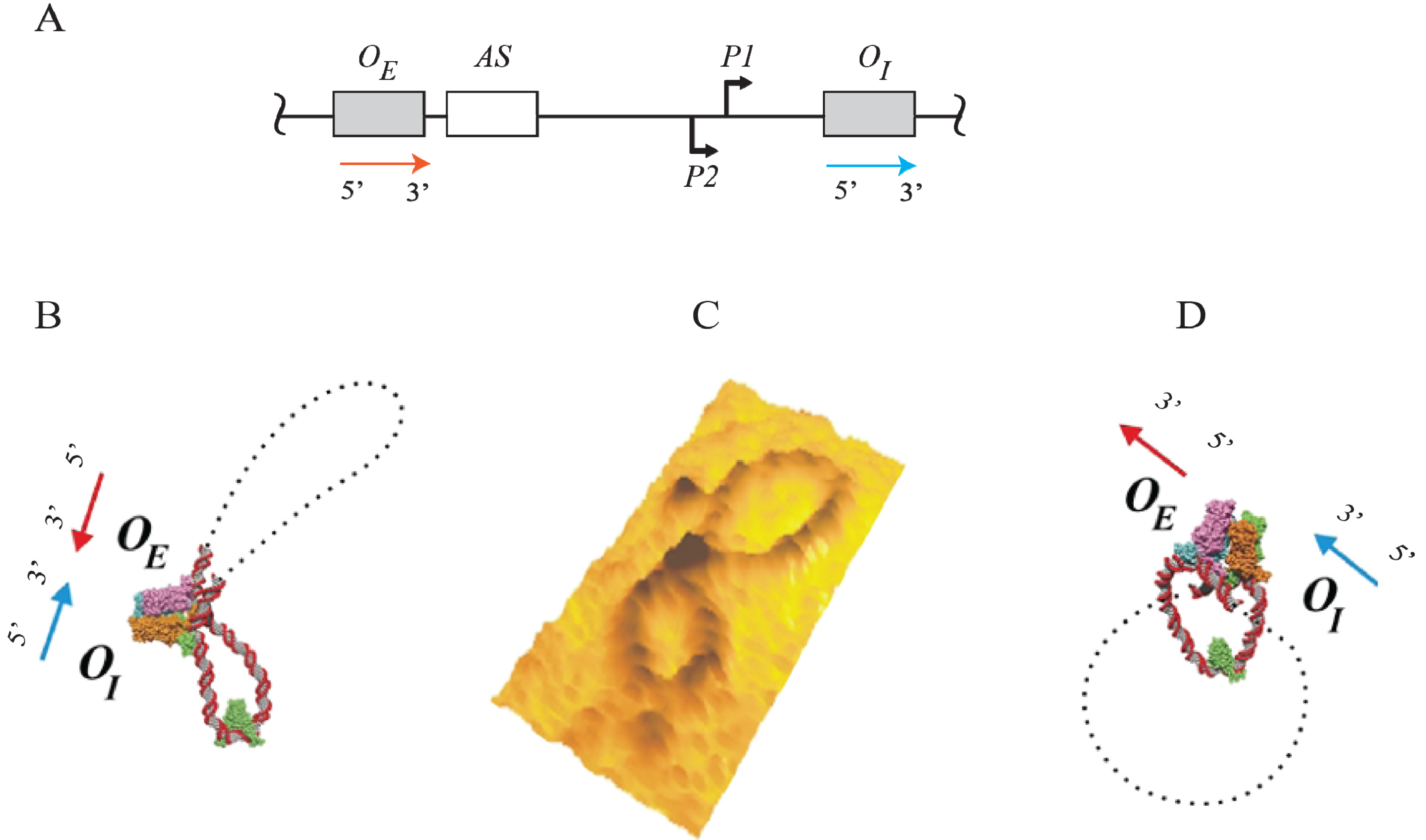
Figure 15.
Trajectory of DNA loops formed by GalR and HU; (
A) The
gal regulatory region containing promoters (
P1 and
P2), operators (
OE and
OI), HU binding site (
hbs). The arrows at
OE and
OI are shown in the direction 5' to 3'; (
B) Two trajectories of DNA loops, “antiparallel” (A), and “parallel” (P); (
C) mRNAs made from
P1 and
P2 in antiparallel or parallel configurations (reproduced with permission from Cold Spring Harbor Laboratory Press, [
5]).
Figure 15.
Trajectory of DNA loops formed by GalR and HU; (
A) The
gal regulatory region containing promoters (
P1 and
P2), operators (
OE and
OI), HU binding site (
hbs). The arrows at
OE and
OI are shown in the direction 5' to 3'; (
B) Two trajectories of DNA loops, “antiparallel” (A), and “parallel” (P); (
C) mRNAs made from
P1 and
P2 in antiparallel or parallel configurations (reproduced with permission from Cold Spring Harbor Laboratory Press, [
5]).
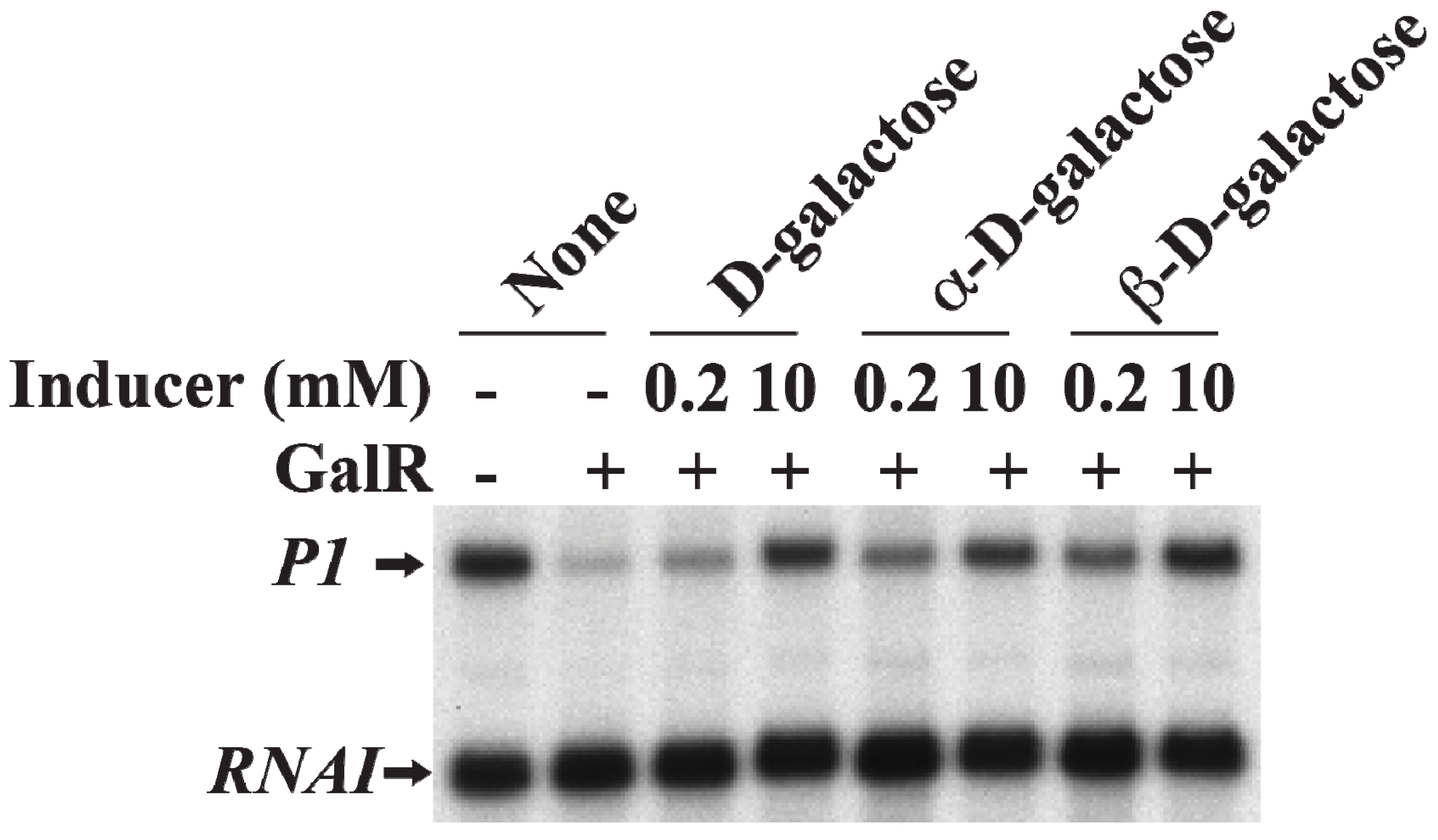
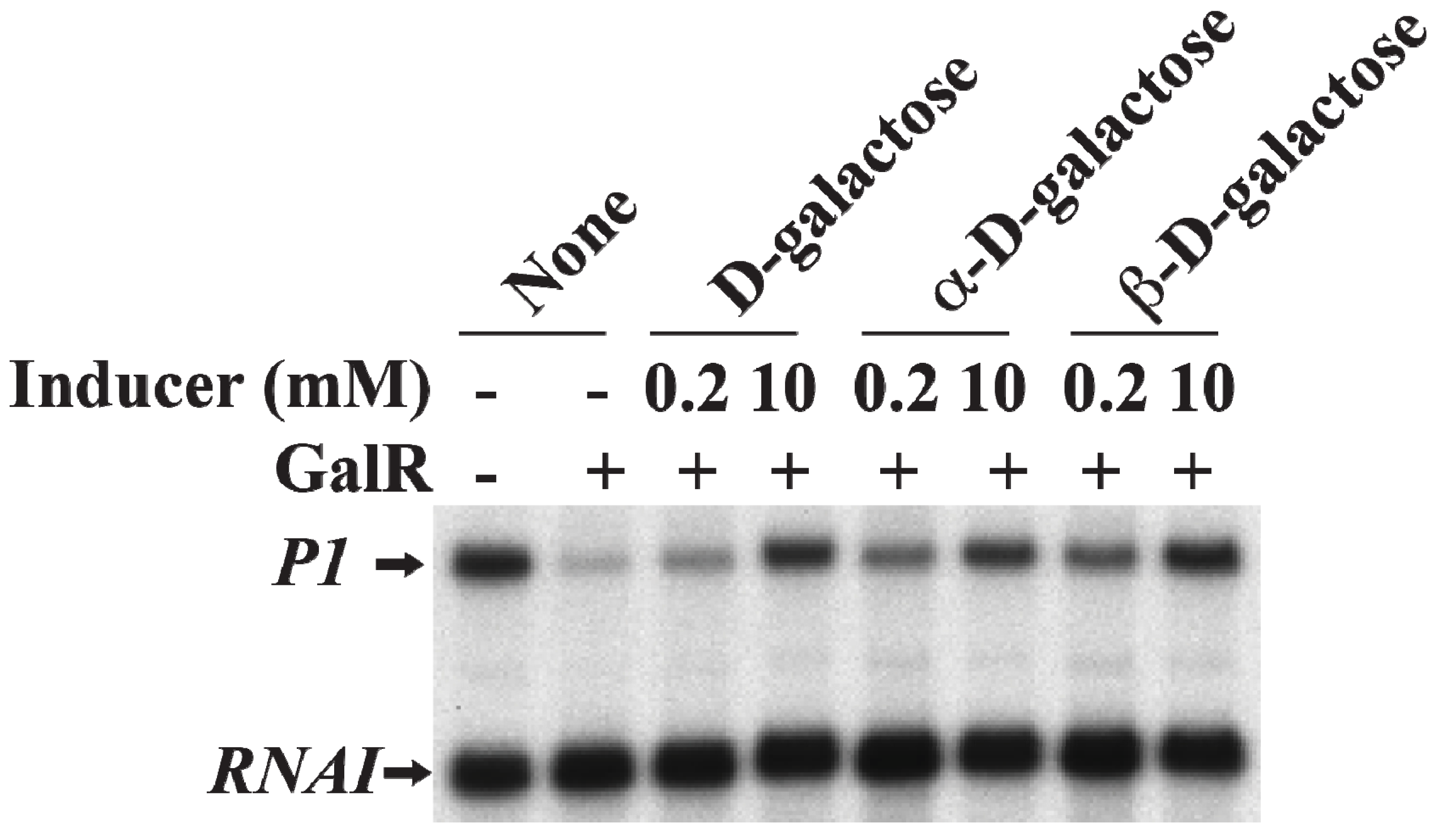
19. Induction of Gal Operon by d-Galactose
d-Galactose, an inducer of GalR, acts by inactivating GalR.
d-galactose binding to GalR results in an allosteric change in the protein, which cannot contact RNAP or bind to DNA anymore. This neutralizes any regulatory effect of GalR.
d-galactose is a mixture of both α-anomer and β-anomer [
86]. Purified α-anomer and β-anomer were used to investigate whether the α-anomer or β-anomer or both can inactivate GalR for
P1 transcription from the repressed state of the promoter
in vitro. The result showed that both α-anomer and β-anomer act as inducer by lifting the repression of
P1 transcription without DNA looping (
Figure 17).
Figure 17.
Derepression of
P1 by
d-galactose. mRNAs made from
P1 in the presence of GalR (80 nM) and
d-galactose, α-
d-galactose or β-
d-galactose 0.2 and 10 nM) (adapted from [
86]).
Figure 17.
Derepression of
P1 by
d-galactose. mRNAs made from
P1 in the presence of GalR (80 nM) and
d-galactose, α-
d-galactose or β-
d-galactose 0.2 and 10 nM) (adapted from [
86]).
How does
d-galactose disrupt the repressosome structure? In case of DNA looping mediated repression of
P2, the presence of
d-galactose first breaks up GalR-GalR tetramers into individual operator-bound dimers in which state
P1 is still largely repressed and
P2 is derepressed [
87]. Next,
d-galactose helps to dissociate GalR from GalR-
OI complex, and finally, GalR dissociates from GalR-
OE complex to disassemble the remaining complex. This also confirms that GalR-
OE complex is more stable than GalR-
OI complex [
87].
20. Conclusions
The gal operon of E. coli plays an important role in cellular metabolism by encoding enzymes that catalyze conversion of d-galactose to energy sources as well as to anabolic substrates. The operon is transcribed from two overlapping promoters, P1 and P2. The importance of individual base pairs at various positions in the −49 to +1 segment in the gal promoters for transcription and its regulation were discerned by substitutions, deletions or insertions of base pairs. First, a 12-bp distance from the master base pair (−11) is a determinant of transcription start point (+1), which is preferably a purine. Second, two of the standard RNAP recognition elements, ex-10 and −10, are responsible for determining the strength of the two gal promoters. Genetic analysis of the two overlapping promoters also identified that the DNA sequence of the segment −20 to −16, which is outside the boundaries of the previously defined promoter elements, contribute to promoter strength in both P1 and P2.
Regulatory proteins, CCC and GalR, regulate the two promoters coordinately and differentially depending on the cellular conditions. (i) CCC binds to the AS element at position −41.5 and activates P1 and represses P2 both at the step of open complex formation; (ii) GalR acts by binding to two operators, OE and OI. The GalR-OE complex inhibits P1 and stimulates P2 by contacting the αCTD of RNAP by preventing open complex formation at P1 and enhancing open complex formation at P2; (iii) Interestingly, the GalR-OI complex does not create a road-block to any elongating RNAP from P1 and P2 because of the presence of anti-pause sequence in the immediate upstream area that occludes pause; (iv) In the presence of the histone-like protein, HU, and supercoiled DNA as template, interactions of the two operator-bound GalR, results in the formation of a DNA loop (repressosome). The trajectory of DNA in the loop is antiparallel, as revealed by biochemical experiments and AFM observations. Genetic analysis of GalR identified the protein interface between GalR-OE and GalR-OI at the looped state. Looping needs the binding of HU to the apex of the looped DNA that stabilizes the repressosome complex. GalR interacts with HU and piggybacks the latter to its binding site. In the final structure, there is no GalR-HU contact. The helical arrangement between GalR-OE and GalR-OI is important for facilitating DNA looping. When they are located on the same face of the DNA looping is favorable; when not on the same face, energetics prevents DNA looping.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
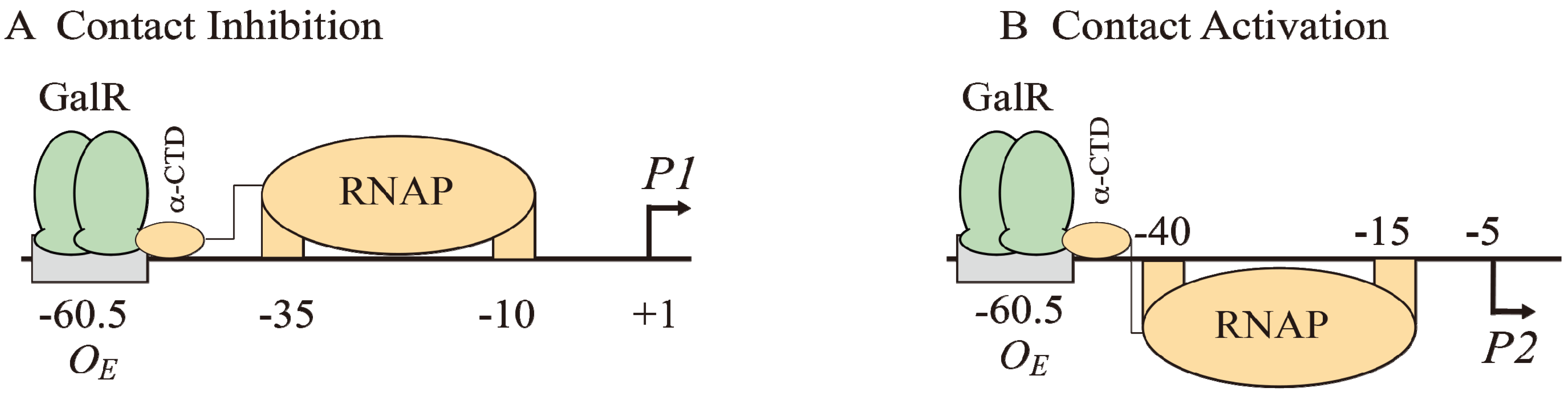{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}