Evolutionary Implications of Metal Binding Features in Different Species’ Prion Protein: An Inorganic Point of View


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Prion Protein Biological Role
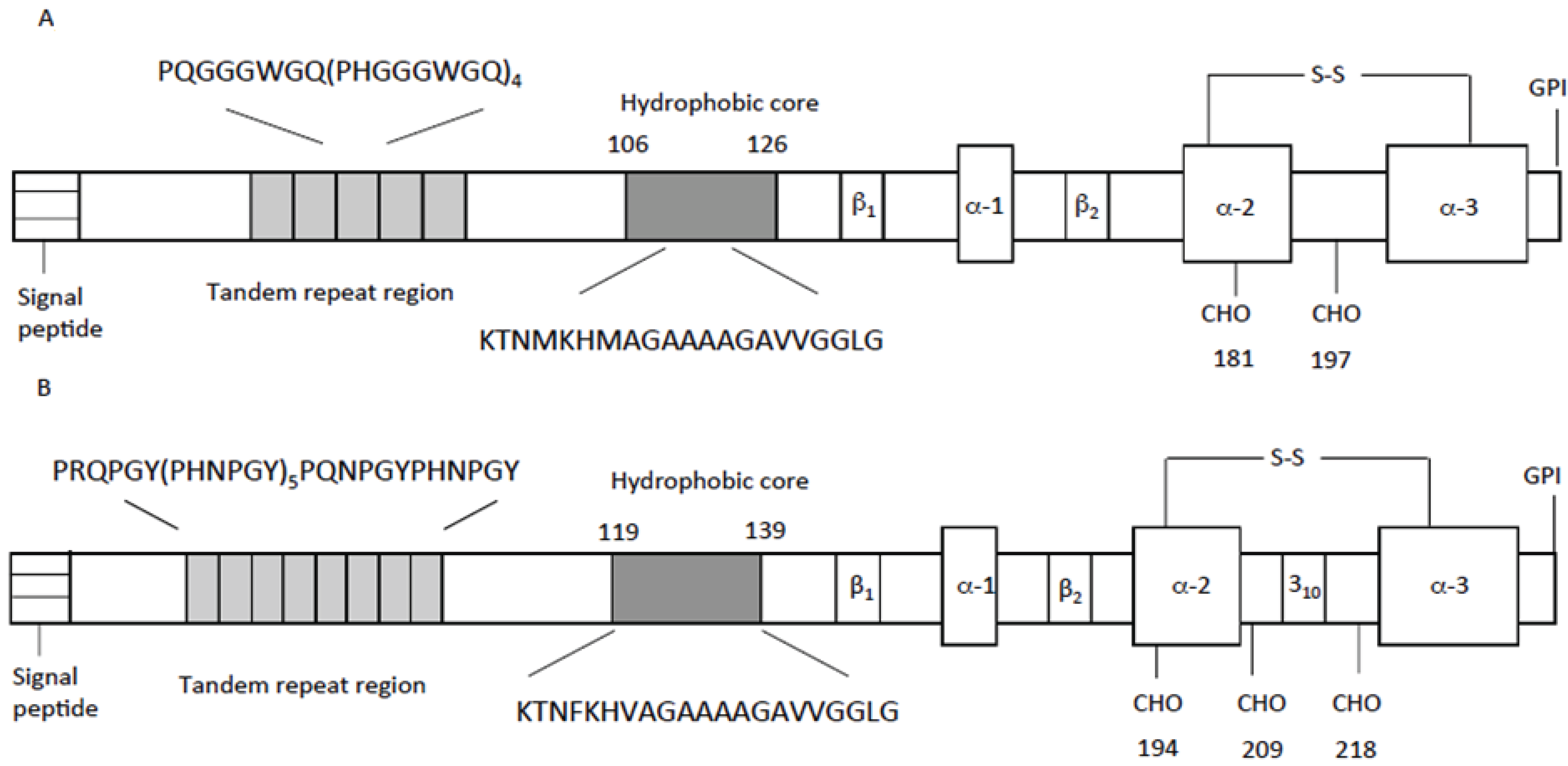
3. Prion Protein Features

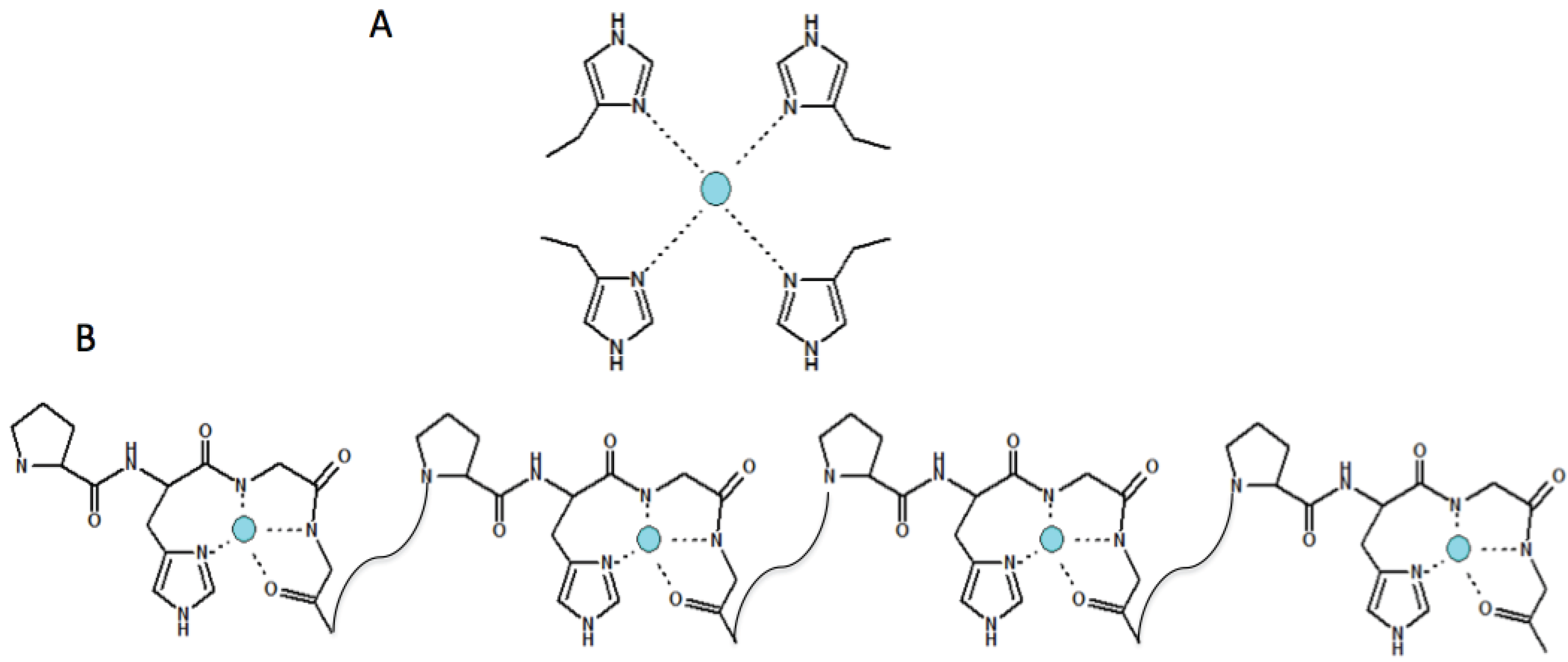
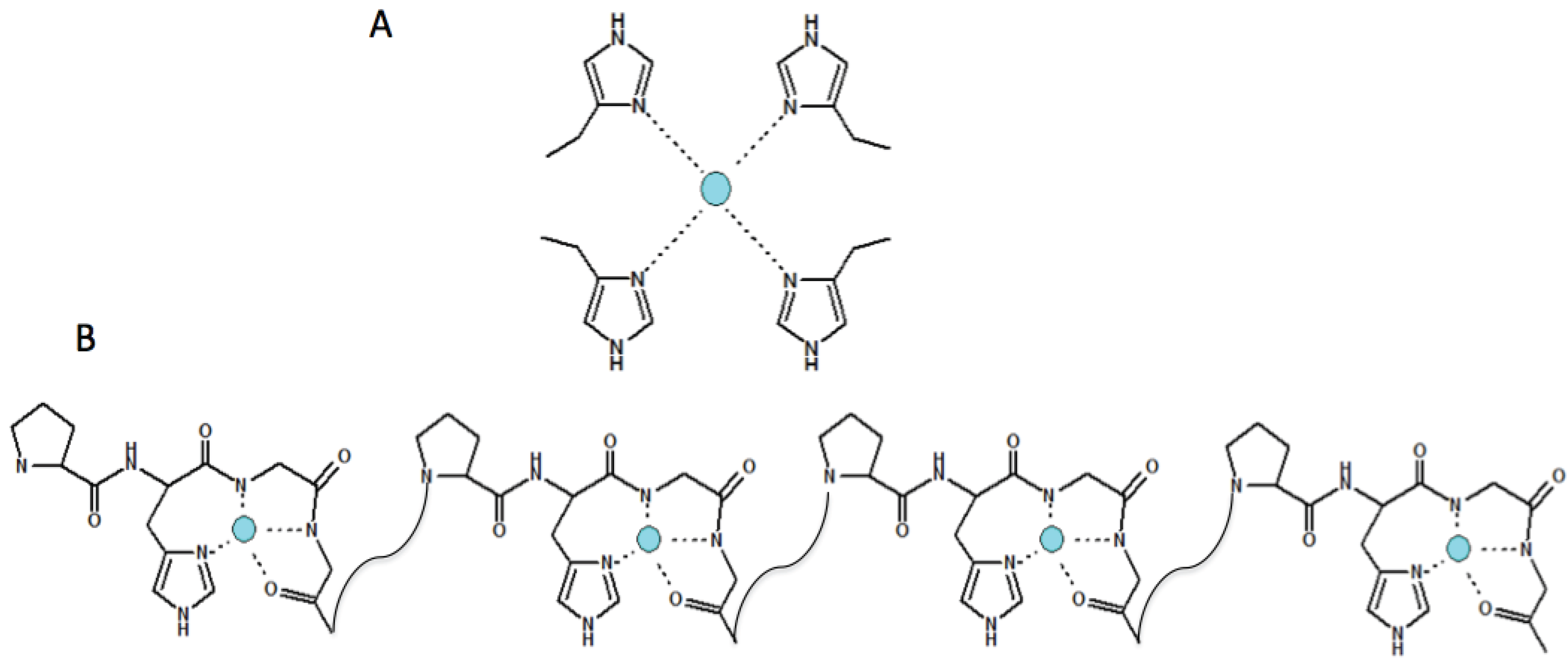
4. Copper(II) Coordination Features within the N-Terminal Domain of Evolutionarily Different Prion Proteins
4.1. Mammals
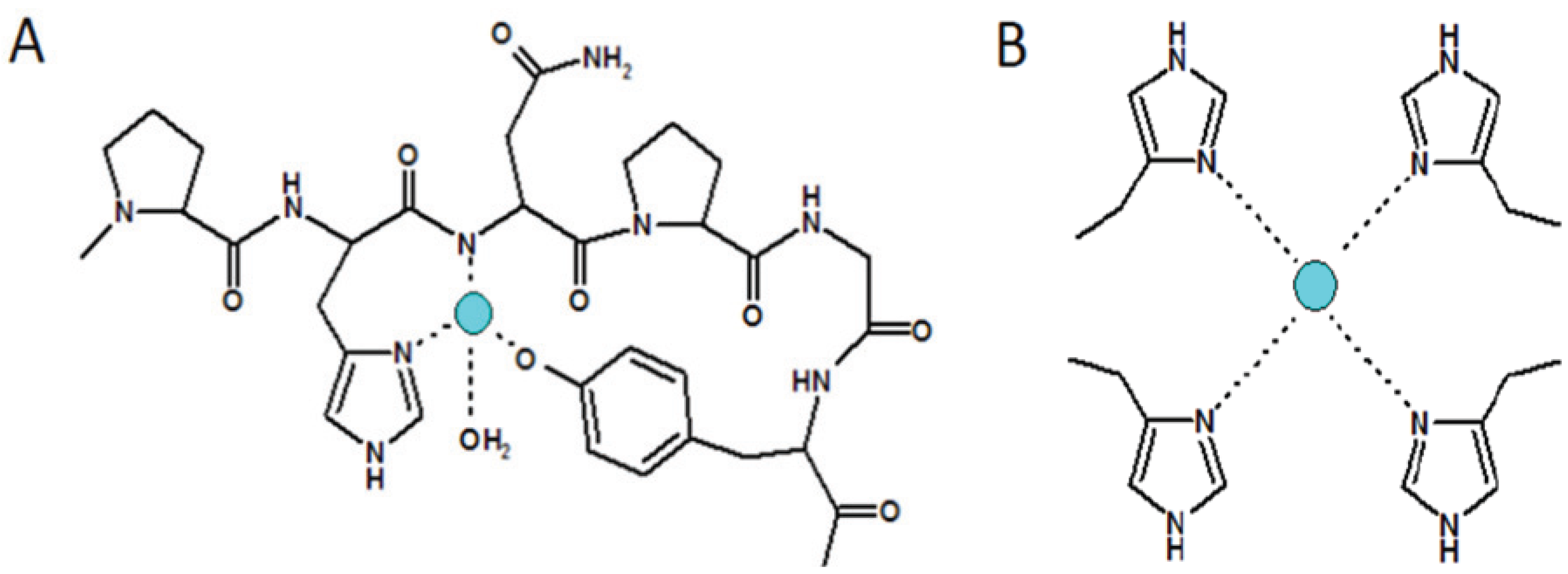
4.2. Other Species

5. Metal Binding Coordination Environment outside the Repeat Domain
6. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Williams, R.J.P.; Frausto da Silva, J.J.R. The Chemistry of Evolution; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Crichton, R.R.; Pierre, J.L. Old iron, young copper: From mars to venus. Biometals 2001, 14, 99–112. [Google Scholar] [CrossRef]
- Canfield, D.E.; Poulton, S.W.; Narbonne, G.M. Late neoproterozoic deep ocean oxygenation and the rise of animal life. Science 2007, 315, 92–95. [Google Scholar] [CrossRef]
- Williams, R.J.P. Chemical advances in evolution by and changes in use of space during time. J. Theor. Biol. 2011, 268, 146–159. [Google Scholar] [CrossRef]
- Williams, R.J.P. Life, the environment and our ecosystem. J. Inorg. Biochem. 2007, 101, 1550–1561. [Google Scholar] [CrossRef]
- Williams, R.J.P. The biological chemistry of the brain and its possible evolution. Inorg. Chim. Acta 2003, 356, 27–40. [Google Scholar] [CrossRef]
- Marx, G.; Gilon, C. The molecular basis of memory. ACS Chem. Neurosci. 2012, 3, 633–642. [Google Scholar]
- Ong, W.Y.; Farooqui, A.A. Iron, neuroinflammation, and Alzheimer’s disease. J. Alzheimers Dis. 2005, 8, 183–200. [Google Scholar]
- Zatta, P.; Frank, A. Copper deficiency and neurological disorders in man and animals. Brain Res. Rev. 2007, 54, 19–33. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion avalaibility. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 145–163. [Google Scholar]
- Travaglia, A.; Pietropaolo, A.; la Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The inorganic perspectives of neurotrophins and Alzheimer’s disease. J. Inorg. Biochem. 2012, 111, 130–137. [Google Scholar]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.W.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Van Rheede, T.; Smolenaars, M.M.W.; Madsen, O.; de Jong, W.W. Molecular evolution of the mammalian prion protein. Mol. Biol. Evol. 2003, 20, 111–121. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhofer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schatzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Gabriel, J.M.; Oesch, B.; Kretzschamer, H.; Scott, M.; Prusiner, S.B. Molecular cloning of a candidate chicken prion protein. Proc. Natl. Acad. Sci. USA 1992, 89, 9097–9101. [Google Scholar] [CrossRef]
- Simonic, T.; Duga, S.; Strumbo, B.; Asselta, R.; Ceciliani, F.; Ronchi, S. cDNA cloning of turtle prion protein. FEBS Lett. 2000, 469, 33–38. [Google Scholar] [CrossRef]
- Strumbo, B.; Ronchi, S.; Bolis, L.C.; Simonic, T. Molecular cloning of the cDNA coding for Xenopus laevis prion protein. FEBS Lett. 2001, 508, 170–174. [Google Scholar] [CrossRef]
- Suzuki, T.; Kurokawa, T.; Hashimoto, H.; Sugiyama, M. cDNA sequence and tissue expression of Fugu rubripes prion protein-like: A candidate for the teleost orthologue of tetrapod PrPs. Biochem. Biophys. Res. Commun. 2002, 294, 912–917. [Google Scholar] [CrossRef]
- Fournier, J.C.; Escaig-Haye, F.; Grigoriev, V. Ultrastructural localization of prion proteins: Physiological and pathological implications. Microsc. Res. Tech. 2000, 50, 76–88. [Google Scholar] [CrossRef]
- Piccardo, P.; Safar, J.; Ceroni, M.; Gajdusek, D.C.; Gibbs, C.J., Jr. Immunohistochemical localization of prion protein in spongiform encephalopathies and normal brain tissue. Neurology 1990, 40, 518–522. [Google Scholar] [CrossRef]
- Atoji, Y.; Ishiguro, N. Distribution of the cellular prion protein in the central nervous system of the chicken. J. Chem. Neuroanat. 2009, 38, 292–301. [Google Scholar] [CrossRef]
- Brown, D.R.; Clive, C.; Haswell, S.J. Antioxidant activity related to copper binding of native prion protein. J. Neurochem. 2001, 76, 69–76. [Google Scholar] [CrossRef]
- Alfaidy, N.; Chauvet, S.; Donadio-Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffman, P.; Andrieux, A.; Moulis, J.M.; et al. Prion protein expression and functional importance in developmental angiogenesis: Role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 2013, 18, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef]
- Herms, J.; Tings, T.; Gall, S.; Madlung, A.; Giese, A.; Siebert, H.; Schurmann, P.; Windl, O.; Brose, N.; Kretzschmar, H. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999, 19, 8866–8875. [Google Scholar]
- Guillot-Sestier, M.V.; Checler, F. Cellular prion and its catabolites in the brain: Production and function. Curr. Mol. Med. 2012, 12, 304–315. [Google Scholar] [CrossRef]
- Brown, D.R. Copper and prion disease. Brain Res. Bull. 2001, 55, 165–173. [Google Scholar] [CrossRef]
- Quaglio, E.; Chiesa, R.; Harris, D.A. Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J. Biol. Chem. 2001, 276, 11432–11438. [Google Scholar] [CrossRef]
- Choi, C.J.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Interaction of metals with prion protein: Possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology 2006, 27, 777–787. [Google Scholar] [CrossRef]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar]
- Brown, D.R.; Sasson, J. Copper-dependent functions for the prion protein. Mol. Biotechnol. 2002, 22, 165–178. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef]
- Allsop, D.; Mayes, J.; Moore, S.; Masad, A.; Tabner, B.J. Metal-dependent generation of reactive oxygen species from amyloid proteins implicated in neurodegenerative disease. Biochem. Soc. Trans. 2008, 36, 1293–1298. [Google Scholar] [CrossRef]
- Singh, N.; Das, D.; Singh, A.; Mohan, M.L. Prion protein and metal interaction: Physiological and pathological implications. Curr. Issues Mol. Biol. 2010, 12, 99–107. [Google Scholar]
- Milardi, D.; Rizzarelli, E. Neurodegeneration: Metallostasis and Proteostasis; RSC Publishing: Cambridge, UK, 2011. [Google Scholar]
- Rossi, L.; Lombardo, M.F.; Ciriolo, M.R.; Rotilio, G. Mitochondrial dysfunction in neurodegenerativediseases associated with copper imbalance. Neurochem. Res. 2004, 29, 493–504. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruc, T.; von Bohlen, A.; Schultz Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar]
- Perera, W.S.; Hooper, N.M. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr. Biol. 2001, 11, 519–523. [Google Scholar] [CrossRef]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.; Hooper, N.M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005, 118, 5141–5153. [Google Scholar] [CrossRef]
- Pauly, P.C.; Harris, D.A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998, 273, 33107–33110. [Google Scholar] [CrossRef]
- Brown, D.R. Prion and prejudice: Normal protein and the synapse. Trends Neurosci. 2001, 24, 85–90. [Google Scholar] [CrossRef]
- Brown, D.R.; Besinger, A. Prion protein expression and superoxide dismutase activity. Biochem. J. 1998, 334, 423–429. [Google Scholar]
- Brown, D.R.; Wong, B.S.; Hafiz, F.; Clive, C.; Haswell, S.J.; Jones, I.M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999, 344, 1–5. [Google Scholar] [CrossRef]
- Wong, B.S.; Pan, T.; Liu, T.; Li, R.; Gambetti, P.; Sy, M.S. Differential contribution of superoxidedismutase activity by prion protein in vivo. Biochem. Biophys. Res. Commun. 2000, 273, 136–139. [Google Scholar] [CrossRef]
- Li, A.; Barmada, S.J.; Roth, K.A.; Harris, D.A. N-terminal deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J. Neurosci. 2007, 27, 852–859. [Google Scholar] [CrossRef]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rulicke, T.; Burkle, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Noltig, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef]
- Malaisé, M.; Schatzl, H.M.; Burkle, A. The octarepeat region of prion protein, but not the TM1 domain, is important for the antioxidant effect of prion protein. Free Radic. Biol. Med. 2008, 45, 1622–1630. [Google Scholar] [CrossRef]
- Brazier, M.W.; Davies, P.; Player, E.; Marken, F.; Viles, J.H.; Brown, D.R. Manganese binding to the prion protein. J. Biol. Chem. 2008, 283, 12831–12839. [Google Scholar] [CrossRef]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef]
- Treiber, C.; Thompsett, A.R.; Pipkorn, R.; Brown, D.R.; Multhaup, G. Real-time kinetics of discontinuous and highly conformational metal-ion binding sites of prion protein. J. Biol. Inorg. Chem. 2007, 12, 711–720. [Google Scholar] [CrossRef]
- Viles, J.H.; Klewpatinond, M.; Nadal, R.C. Copper and structural biology of the prion protein. Biochem. Soc. Trans. 2008, 36, 1288–1292. [Google Scholar] [CrossRef]
- Rivera-Milla, E.; Oidtmann, B.; Panagiotidis, C.H.; Baier, M.; Sklaviadis, T.; Hoffman, R.; Zhou, Y.; Solis, G.P.; Stuermer, C.A.; Malaga-Trillo, E. Disparate evolution of prion protein domains and the distinct origin of doppel- and prion-related loci revealed by fish to mammal comparisons. FASEB J. 2006, 20, 317–319. [Google Scholar]
- Malaga-Trillo, E.; Salta, E.; Figueras, A.; Panagiotidis, C.; Sklaviadis, T. Fish models in prion biology: Underwater issues. Biochim. Biophys. Acta 2011, 1812, 402–414. [Google Scholar]
- Stahl, N.; Prusiner, S.B. Prions and prion proteins. FASEB J. 1991, 5, 2799–2807. [Google Scholar]
- Riek, R.; Wider, G.; Billeter, M.; Hornemann, S.; Glockshuber, R.; Wüthrich, K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc. Natl. Acad. Sci. USA 1998, 95, 11667–11672. [Google Scholar]
- Calzolai, L.; Lysek, D.A.; Perez, D.R.; Guntert, P.; Wüthrich, K. Prion protein NMR structures of chickens, turtles, and frogs. Proc. Natl. Acad. Sci. USA 2005, 102, 651–655. [Google Scholar] [CrossRef]
- Santo, K.P.; Berjanskii, M.; Wishart, D.S.; Stepanova, M. Comparative analysis of essential collective dynamics and NMR-derived flexibility profiles in evolutionarily diverse prion proteins. Prion 2011, 5, 188–200. [Google Scholar] [CrossRef]
- Marcotte, E.M.; Eisemberg, D. Chicken prion tandem repeats form a stable, protease resistant domain. Biochemistry 1999, 38, 667–676. [Google Scholar] [CrossRef]
- Russo, L.; Raiola, L.; Campitiello, M.A.; Magrì, A.; Fattorusso, R.; Malgieri, G.; Pappalardo, G.; la Mendola, D.; Isernia, C. Probing the residual structure in avian prion hexarepeats by CD, NMR and MD techniques. Molecules 2013, 18, 11467–11484. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Raiola, L.; Muccioli, L.; Tiberio, G.; Zannoni, C.; Fattorusso, R.; Isernia, C.; la Mendola, D.; Pappalardo, G.; Rizzarelli, E. An NMR and molecular dynamics investigation of the avian prion hexarepeat conformational features in solution. Chem. Phys. Lett. 2007, 442, 110–118. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Muccioli, L.; Zannoni, C.; la Mendola, D.; Maccarrone, G.; Pappalardo, G.; Rizzarelli, E. Unveiling the role of histidine and tyrosine residues on the conformation of the avian prion hexarepeat domain. J. Phys. Chem. B 2008, 112, 5182–5188. [Google Scholar] [CrossRef]
- Stanczak, P.; Valensin, D.; Juszczyk, P.; Grzonka, Z.; Migliorini, C.; Molteni, E.; Valensin, G.; Gaggelli, E.; Kozlowski, H. Structure and stability of the Cu(II) complexes with tandem repeats of the chicken prion. Biochemistry 2005, 44, 12940–12954. [Google Scholar] [CrossRef]
- Marcotte, E.M.; Pellegrini, M.; Yeates, T.O.; Eisenberg, D. A census of protein repeats. J. Mol. Biol. 1998, 293, 151–160. [Google Scholar]
- Millhauser, G.L. Copper and the prion protein: Methods, structures, function and disease. Annu. Rev. Phys. Chem. 2007, 58, 299–320. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Walter, E.D.; Newell, D.J.; Jackson, P.J.; Aronoff-Spencer, E.; Peisach, J.; Gerfen, G.J.; Bennett, B.; Antholine, W.E.; Millhauser, G.L. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 2005, 127, 12647–12656. [Google Scholar] [CrossRef]
- Burns, C.S.; Aronoff-Spencer, E.; Dunham, C.M.; Lario, P.; Avdievich, N.I.; Olmstead, M.M.; Vrielink, A.; Gerfen, G.J.; Peisach, J.; Scott, W.G.; et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 2002, 413, 991–4001. [Google Scholar]
- Luczkowski, M.; Kozlowski, H.; Stawikowski, M.; Rolka, K.; Gaggelli, E.; Valensin, D.; Valensin, G. Is the monomeric prion octapeptide repeat PHGGGWGQ a specific ligand for Cu2+ ions? Dalton Trans. 2002. [Google Scholar] [CrossRef]
- Bonomo, R.P.; Cucinotta, V.; Giuffrida, A.; Impellizzeri, G.; Magrì, A.; Pappalardo, G.; Rizzarelli, E.; Santoro, A.M.; Tabbì, G.; Vagliasindi, L.I. A re-investigation of copper coordination in the octa-repeats region of the prion protein. Dalton Trans. 2005. [Google Scholar] [CrossRef]
- Valensin, D.; Luczkowski, M.; Mancini, F.M.; Legowska, A.; Gaggelli, E.; Valensin, G.; Rolka, K.; Kozlowski, H. The dimeric and tetrameric octarepeat fragments of prion protein behave differently to its monomeric unit. Dalton Trans. 2004. [Google Scholar] [CrossRef]
- Walter, E.D.; Chattopadhyay, M.; Millhauser, G.L. The affinity of copper binding to the prion protein octarepeat domain: Evidence for negative cooperativity. Biochemistry 2006, 45, 13083–13092. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, D.; McDonald, A.; Hao, Y.; Millhauser, G.L.; Zhou, F. Copper redox cycling in the prion protein depends critically on binding mode. J. Am. Chem. Soc. 2011, 133, 12229–12237. [Google Scholar] [CrossRef]
- Zhou, F.; Millhauser, G.L. The rich electrochemistry and redox reactions of the copper sites in the cellular prion protein. Coord. Chem. Rev. 2012, 256, 2285–2296. [Google Scholar] [CrossRef]
- Stanczak, P.; Valensin, D.; Porciatti, E.; Jankowska, E.; Grzonka, Z.; Molteni, E.; Gaggelli, E.; Valensin, G.; Kozlowski, H. Tandem repeat-like domain of “similar to prion protein” (StPrP) of japanese pufferfish binds Cu(II) as effectively as the mammalian protein. Biochemistry 2006, 45, 12227–12239. [Google Scholar] [CrossRef]
- Gaggelli, E.; Jankowska, E.; Kozlowski, H.; Marcinkowska, A.; Migliorini, C.; Stanczak, P.; Valensin, D.; Valensin, G. Structural characterization of the intra-and inter-repeat copper binding modes within the N-terminal region of “prion related protein” (PrP-rel-2) of zebrafish. J. Phys. Chem. B 2008, 112, 15140–15150. [Google Scholar] [CrossRef]
- Valensin, D.; Szyrwiel, L.; Camponeschi, F.; Rowinska-Zyrek, M.; Molteni, E.; Jankowska, E.; Szymanska, A.; Gaggelli, E.; Valensin, G.; Kozlowski, H. Heteronuclear and homonuclear Cu2+ and Zn2+ complexes with multihistidine peptides based on zebrafish prion-like protein. Inorg. Chem. 2009, 48, 7330–7340. [Google Scholar] [CrossRef]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M. Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem. Biophys. Res. Commun. 1995, 207, 621–629. [Google Scholar] [CrossRef]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M.; Lakey, J.H. Copper binding to the N-terminal tandem repeat region of mammalian and avian prion protein: Structural studies using synthetic peptides. Biochem. Biophys. Res. Commun. 1995, 214, 993–999. [Google Scholar] [CrossRef]
- Garnett, A.P.; Viles, J.H. Copper binding to the octarepeats of the prion protein. Affinity, specificity, folding, and cooperativity: Insights from circular dichroism. J. Biol. Chem. 2003, 278, 6795–6802. [Google Scholar] [CrossRef]
- Stanczak, P.; Luczkowski, M.; Juszczyk, P.; Grzonka, Z.; Kozlowski, H. Interactions of Cu2+ ions with chicken prion tandem repeats. Dalton Trans. 2004. [Google Scholar] [CrossRef]
- La Mendola, D.; Bonomo, R.P.; Impellizzeri, G.; Maccarrone, G.; Pappalardo, G.; Rizzarelli, E.; Pietropaolo, A.; Zito, V. Copper(II) complexes with chicken prion repeats: Influence of proline and tyrosine residues on the coordination features. J. Biol. Inorg. Chem. 2005, 10, 463–475. [Google Scholar] [CrossRef]
- Redecke, L.; Meyer-Klaucke, W.; Koker, M.; Clos, J.; Georgieva, D.; Genov, N.; Echner, H.; Kalbacher, H.; Perbandt, M.; Bredehorst, R.; et al. Comparative analysis of the human and chicken prion protein copper binding regions at pH 6.5. J. Biol. Chem. 2005, 280, 13987–13992. [Google Scholar] [CrossRef]
- La Mendola, D.; Bonomo, R.P.; Carminati, S.; di Natale, G.; Emmi, S.S.; Hansson, O.; Maccarrone, G.; Pappalardo, G.; Pietropaolo, A.; Rizzarelli, E. Copper(II) complexes with avian prion N-terminal region and their potential SOD-like activity. J. Inorg. Biochem. 2009, 103, 195–204. [Google Scholar] [CrossRef]
- Di Natale, G.; Pappalardo, G.; Milardi, D.; Sciacca, M.F.; Attanasio, F.; la Mendola, D.; Rizzarelli, E. Membrane interactions and conformational preferences of human and avian prion N-terminal tandem repeats: The role of copper(II) ions, pH and membrane mimicking environments. J. Phys. Chem. B 2010, 114, 13830–13838. [Google Scholar] [CrossRef]
- Kaim, W.; Schwederski, B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life; John Wiley & Sons: Chichester, UK, 1994; p. 209. [Google Scholar]
- Stanczak, P.; Kozlowski, H. Can chicken and human PrPs possess SOD-like activity after beta-cleavage? Biochem. Biophys. Res. Commun. 2007, 352, 198–202. [Google Scholar] [CrossRef]
- Stanczak, P.; Juszczyk, P.; Grzonka, Z.; Kozlowski, H. The whole hexapeptide repeats domain from avian PrP displays untypical hallmarks in aspect of the Cu2+ complexes formation. FEBS Lett. 2007, 581, 4544–4548. [Google Scholar]
- Hutter, G.; Heppner, F.L.; Aguzzi, A. No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 2003, 384, 1279–1285. [Google Scholar]
- Jones, S.; Batchelor, M.; Bhelt, D.; Clarke, A.R.; Collinge, J.; Jackson, G.S. Recombinant prion protein does not possess SOD-1 activity. Biochem. J. 2005, 392, 309–312. [Google Scholar] [CrossRef]
- Thompsett, A.R.; Abdelraheim, S.R.; Daniels, M.; Brown, D.R. High affinity binding between copper and full-length prion protein identified by two different techniques. J. Biol. Chem. 2005, 280, 42750–42758. [Google Scholar] [CrossRef]
- Klewpatinond, M.; Viles, J.H. Fragment length influences affinity for Cu2+ and Ni2+ binding to His96 or His111 of the prion protein and spectroscopic evidence for a multiple histidine binding only at low pH. Biochem. J. 2007, 404, 393–402. [Google Scholar] [CrossRef]
- Osz, K.; Nagy, Z.; Pappalardo, G.; di Natale, G.; Sanna, D.; Micera, G.; Rizzarelli, E.; Sovago, I. Copper(II) interaction with prion peptide fragments encompassing histidine residues within and outside the octarepeat domain: Speciation, stability constants and binding details. Chem. Eur. J. 2007, 13, 7129–7143. [Google Scholar] [CrossRef]
- Gralka, E.; Valensin, D.; Porciatti, E.; Gajda, C.; Gaggelli, E.; Valensin, G.; Kamysz, W.; Nadolny, R.; Guerrini, R.; Bacco, D.; et al. Cu(II) binding sites located at His-96 and His-111 of the human prion protein: Thermodynamic and spectroscopic studies on model peptides. Dalton Trans. 2008. [Google Scholar] [CrossRef]
- Di Natale, G.; Osz, K.; Nagy, Z.; Sanna, D.; Micera, G.; Pappalardo, G.; Sovago, I.; Rizzarelli, E. Interaction of copper(II) with the prion peptide fragment HuPrP(76-114) encompassing four histidyl residues within and outside the octarepeat domain. Inorg. Chem. 2009, 48, 4239–4250. [Google Scholar] [CrossRef]
- Fioriti, L.; Quaglio, E.; Massignan, T.; Colombo, L.; Stewart, R.S.; Salmona, M.; Harris, D.A.; Forloni, G.; Chiesa, R. The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol. Cell Neurosci. 2005, 28, 165–176. [Google Scholar] [CrossRef]
- Jobling, M.F.; Huang, X.; Stewart, L.R.; Barnham, K.J.; Curtain, C.; Volitakis, I.; Perugini, M.; White, A.R.; Cherny, R.A.; Masters, C.L.; et al. Copper and zinc binding modulates the aggregation and neurotoxic properties of the prion peptide PrP106–126. Biochemistry 2001, 40, 8073–8084. [Google Scholar] [CrossRef]
- Jones, C.E.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Preferential Cu2+ coordination by His96 and His111 induces β-sheet formation in the unstructured amyloidogenic region of the prion protein. J. Biol. Chem. 2004, 279, 32018–32027. [Google Scholar]
- Di Natale, G.; Grasso, G.; Impellizzeri, G.; la Mendola, D.; Micera, G.; Mihala, N.; Nagy, Z.; Osz, K.; Pappalardo, G.; Rigo, V.; et al. Copper(II) interaction with unstructured prion domain outside the octarepeat region: Speciation, stability, and binding details of copper(II) complexes with PrP106–126 peptides. Inorg. Chem. 2005, 44, 7214–7225. [Google Scholar] [CrossRef]
- Gralka, E.; Valensin, D.; Gajda, K.; Bacco, D.; Szyrwiel, L.; Remelli, M.; Valensin, G.; Kamasz, W.; Baranska-Rybak, W.; Kozlowski, H. Copper(II) coordination outside the tandem repeat region of an unstructerd domain of chicken prion protein. Mol. Biosyst. 2009, 5, 497–510. [Google Scholar] [CrossRef]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbe, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Haakon, B.N.; Strittmatter, S.M. Cellular prion protein mediates the toxicity of β-amyloid oligomers. Arch. Neurol. 2009, 66, 1325–1328. [Google Scholar]
- Kenward, A.G.; Bortolotti, L.J.; Burns, C.S. Copper and zinc promote interactions between membrane-anchored peptides of the metal binding domain of the prion protein. Biochemistry 2007, 46, 4261–4271. [Google Scholar] [CrossRef]
- Vagliasindi, L.I.; Arena, G.; Bonomo, R.P.; Pappalardo, G.; Tabbì, G. Copper complex species within a fragment of the N-terminal repeat region in opossum PrP protein. Dalton Trans. 2011. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
La Mendola, D.; Rizzarelli, E. Evolutionary Implications of Metal Binding Features in Different Species’ Prion Protein: An Inorganic Point of View. Biomolecules 2014, 4, 546-565. https://doi.org/10.3390/biom4020546
La Mendola D, Rizzarelli E. Evolutionary Implications of Metal Binding Features in Different Species’ Prion Protein: An Inorganic Point of View. Biomolecules. 2014; 4(2):546-565. https://doi.org/10.3390/biom4020546
Chicago/Turabian StyleLa Mendola, Diego, and Enrico Rizzarelli. 2014. "Evolutionary Implications of Metal Binding Features in Different Species’ Prion Protein: An Inorganic Point of View" Biomolecules 4, no. 2: 546-565. https://doi.org/10.3390/biom4020546