Research Applications of Proteolytic Enzymes in Molecular Biology


Abstract
:
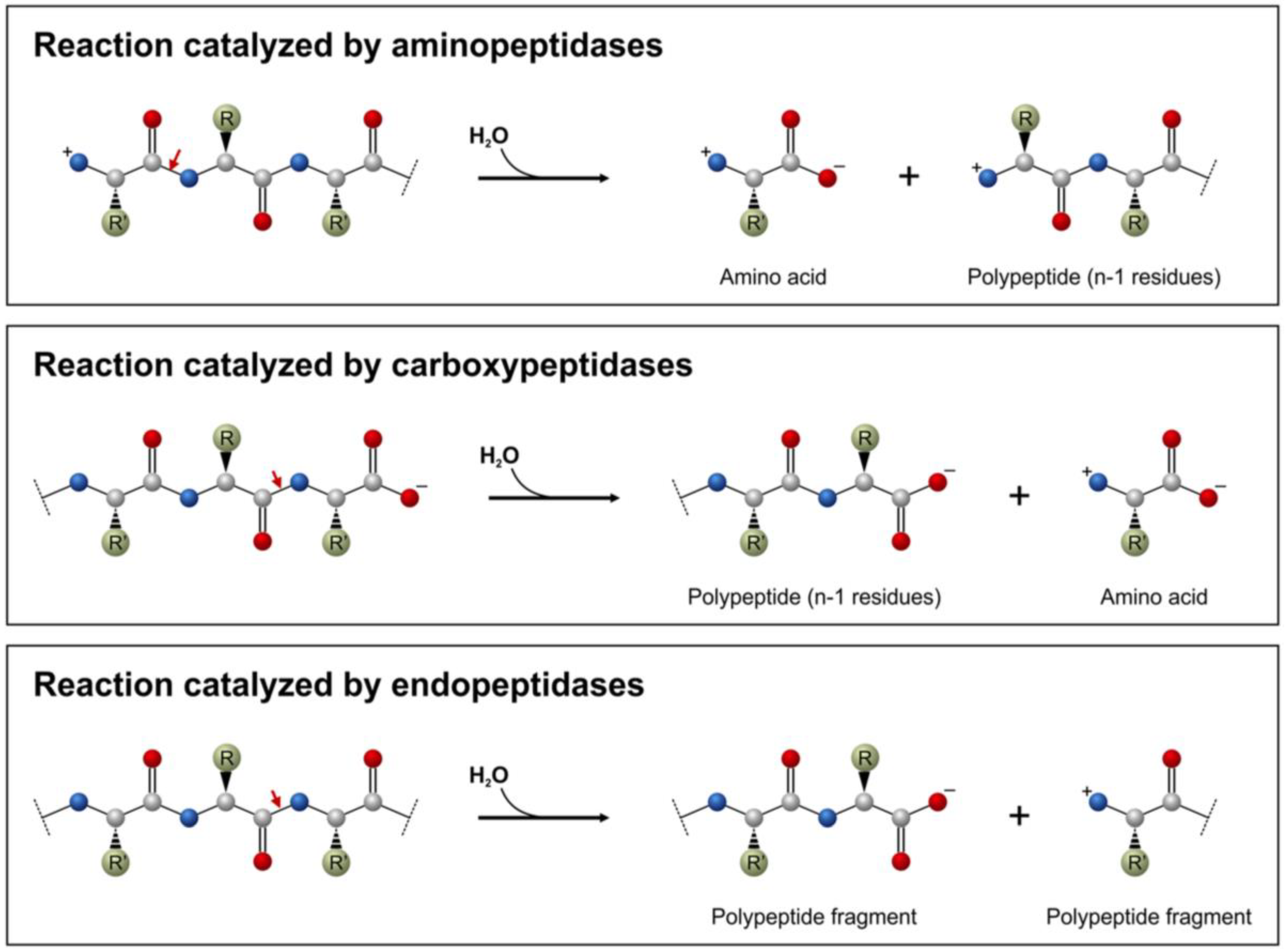
1. Scope of the Review

2. Molecular Biology Research Applications
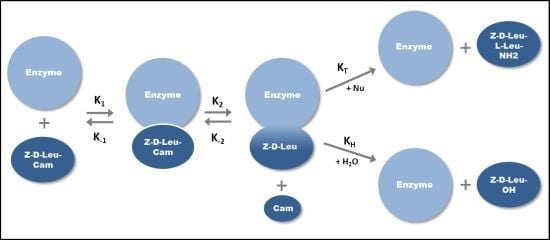
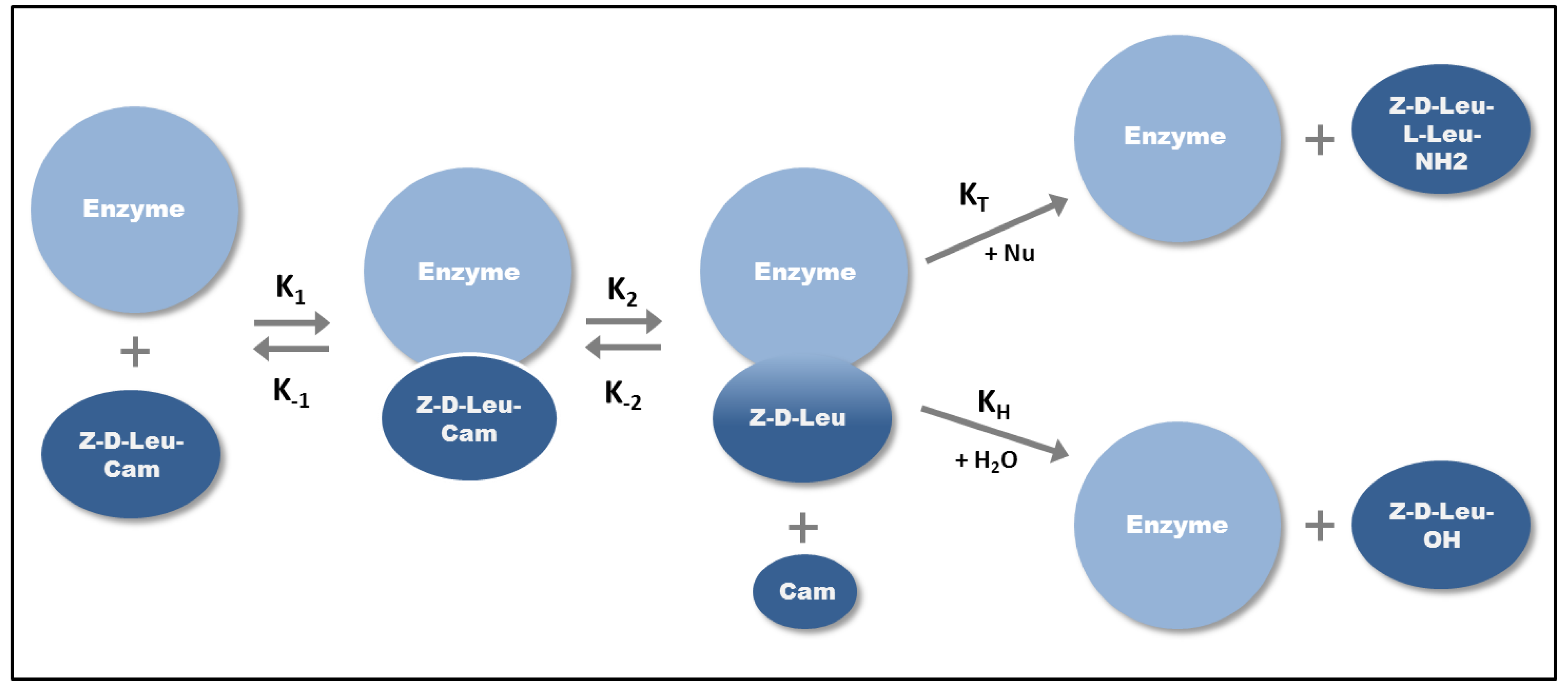
2.1. Klenow Fragment Production

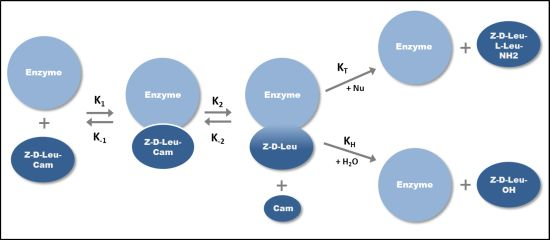{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Main source | Cleavage site |
|---|---|---|
| Endopeptidases | ||
| Serine proteases | ||
| Trypsin | bovine | -Arg or Lys↓nonspecific- |
| Chymotrypsin | bovine | -Trp (or Phe, Leu, Tyr)↓nonspecific- |
| Enterokinase | bovine | Asp-Asp-Asp-Lys↓nonspecific- |
| Endoproteinase Arg-C | microbial | -Arg↓nonspecific- |
| Endoproteinase Glu-C | microbial | -Glu (or Asp)↓nonspecific- |
| Endoproteinase Lys-C | microbial | -Lys↓nonspecific- |
| Elastase | porcine | -Ala (or Gly or Val)↓nonspecific- |
| Subtilisin | microbial | -Trp (or Tyr, Phe, Leu)↓nonspecific- |
| Proteinase K | fungal | -aromatic, aliphatic or hydrophobic ↓nonspecific- |
| Thrombin | bovine | -Arg (or Lys)↓nonspecific-specific for -Leu-Val-Pro-Arg-↓Gly-Ser- |
| Factor Xa | bovine | -Arg (or Lys)↓nonspecific-specific for -Leu-Val-Pro-Arg-↓Gly-Ser- |
| WNV protease | E. coli | -Lys (or Arg)-Arg↓Gly-Ser- |
| Cysteine proteases | ||
| Bromelain | plant | -nonspecific↓nonspecific- |
| Papain | plant | -Arg (or Lys)↓nonspecific- |
| Ficin (ficain) | plant | -nonspecific↓nonspecific- |
| Rhinovirus 3C | E. coli | Gly-Pro dipeptide after the scissile bondhighly specific for -Leu-Glu-Val-Leu-Phe-Gln↓Gly-Pro- |
| TEV protease | E. coli | specific for -Gln-Asn-Leu-Tyr-Phe-Gln↓Gly- |
| TVMV protease | E. coli | specific for -Glu-Thr-Val-Arg-Phe-Gln↓Ser- |
| Metalloproteases | ||
| Endoproteinase Asp-N | microbial | -nonspecific↓Asp- |
| Thermolysin | microbial | -Leu (or Phe)↓Leu (or Phe, Val, Met, Ala, Ile)- |
| Collagenase | microbial | -Pro-neutral↓Gly-Pro- |
| Dispase | microbial | -nonspecific↓non-polar- |
| Aspartic proteases | ||
| Pepsin | porcine | -Phe (or Tyr, Leu, Trp)↓Trp (or Phe, Tyr, Leu)- |
| Cathepsin D | bovine | -Phe (or Leu)↓nonspecific (not Val, Ala)- |
| Exopeptidases | ||
| Serine proteases | ||
| Carboxypeptidase Y | yeast | -nonspecific↓nonspecific |
| Cysteine proteases | ||
| Cathepsin C | bovine | removes N-terminal dipeptide |
| DAPase | porcine | removes N-terminal dipeptide |
| Metalloproteases | ||
| Carboxypeptidase A | bovine | -nonspecific↓aromatic or branched preferred |
| Carboxypeptidase B | porcine | specific for C-terminal Arg or Lys |
2.2. Enzymatic Peptide Synthesis
| Peptide | Sequence | Enzyme(s) | Reference |
|---|---|---|---|
| Aspartame | Asp-Phe | Thermolysin | [18] |
| Nutritional peptide | Tyr-Trp-Val | α-Chymotrypsin, papain | [19] |
| Somatostatin | Ala-Gly-Cys-Lys-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys | Thermolysin, chymotrypsin | [20] |
| Vasopressin | Tyr-Phe-Phe-Gln | Thermolysin, chymotrypsin | [21] |
| Oxytocin | Cys-Tyr Tyr-Ile Pro-Leu Leu-Gly | Papain, thermolysin, chymotrypsin | [21] |
| mouse EGF (21–31) | His-Ile-Glu-Ser-Leu-Asp-SerTyr-Thr-Cys | Papain, trypsin | [22] |
2.2.1. Kinetically Controlled Peptide Synthesis
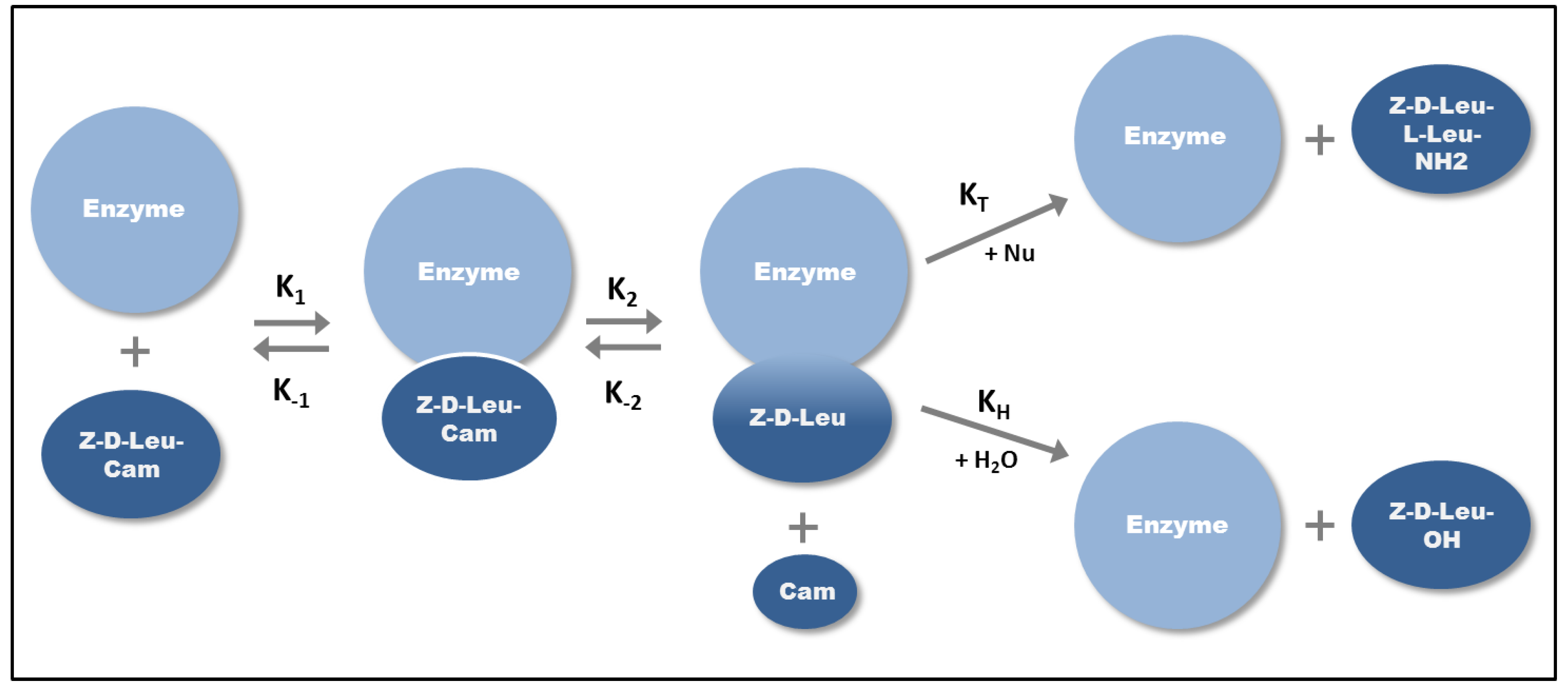
2.2.2. Equilibrium-Controlled Synthesis
2.2.3. Strategies Used in Enzymatic Synthesis
2.3. Nucleic Acid Isolation
2.4. Cell Isolation and Tissue Dissociation
2.5. Cell Culturing
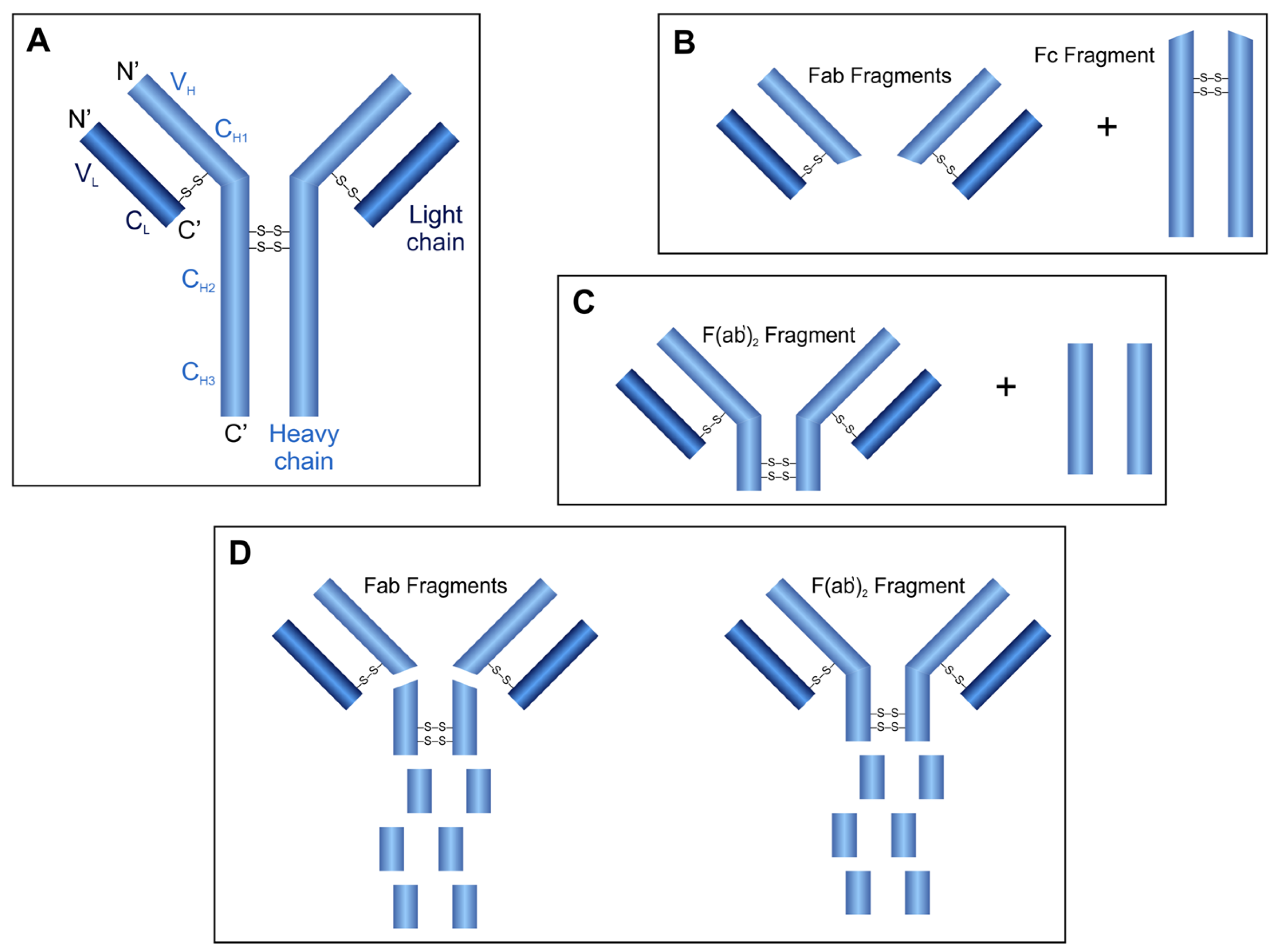
2.6. Antibody Fragment Production

2.7. Structural Studies
2.8. Fusion Tag Removal
2.8.1. Endoproteases
2.8.2. Exopeptidases
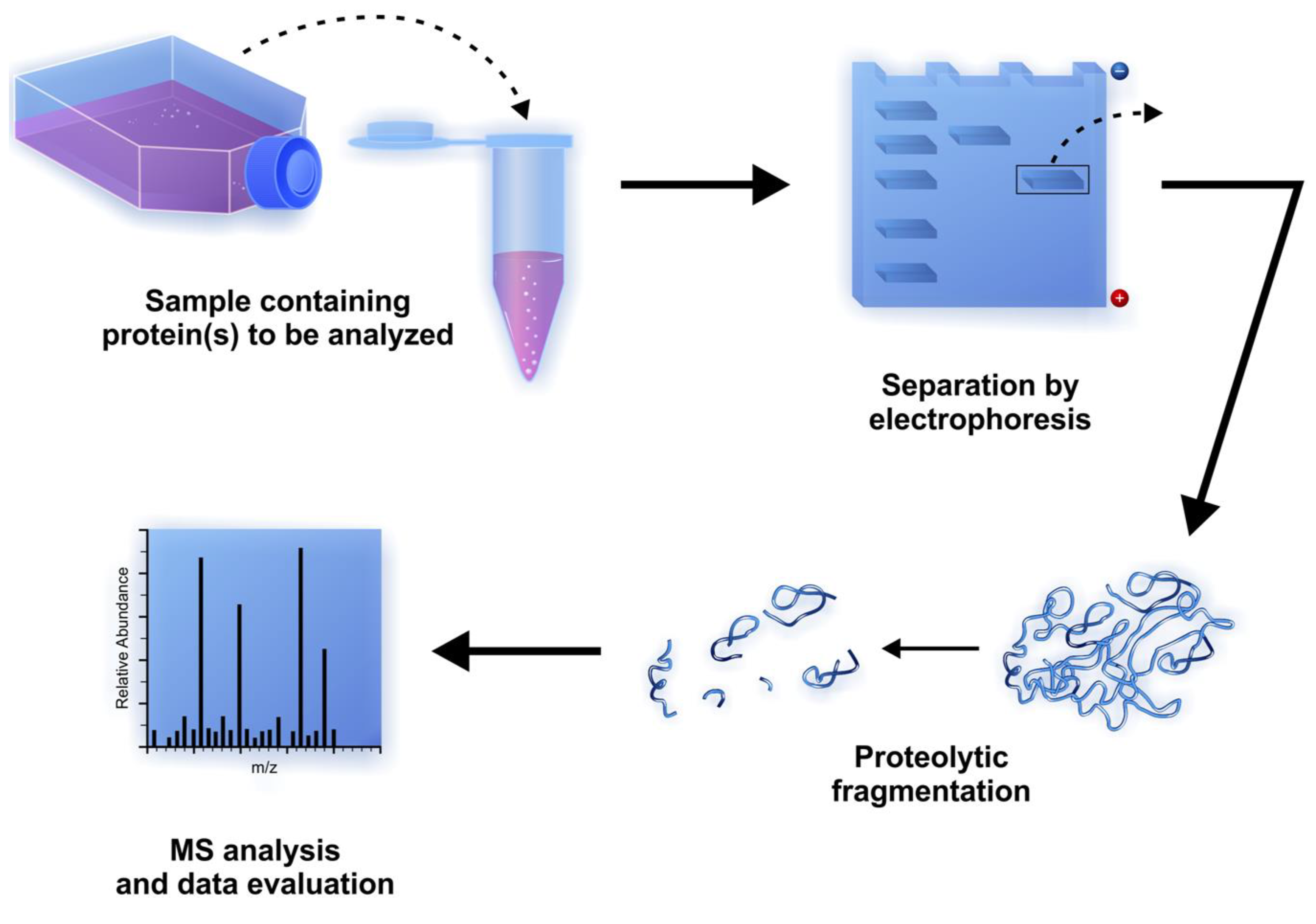
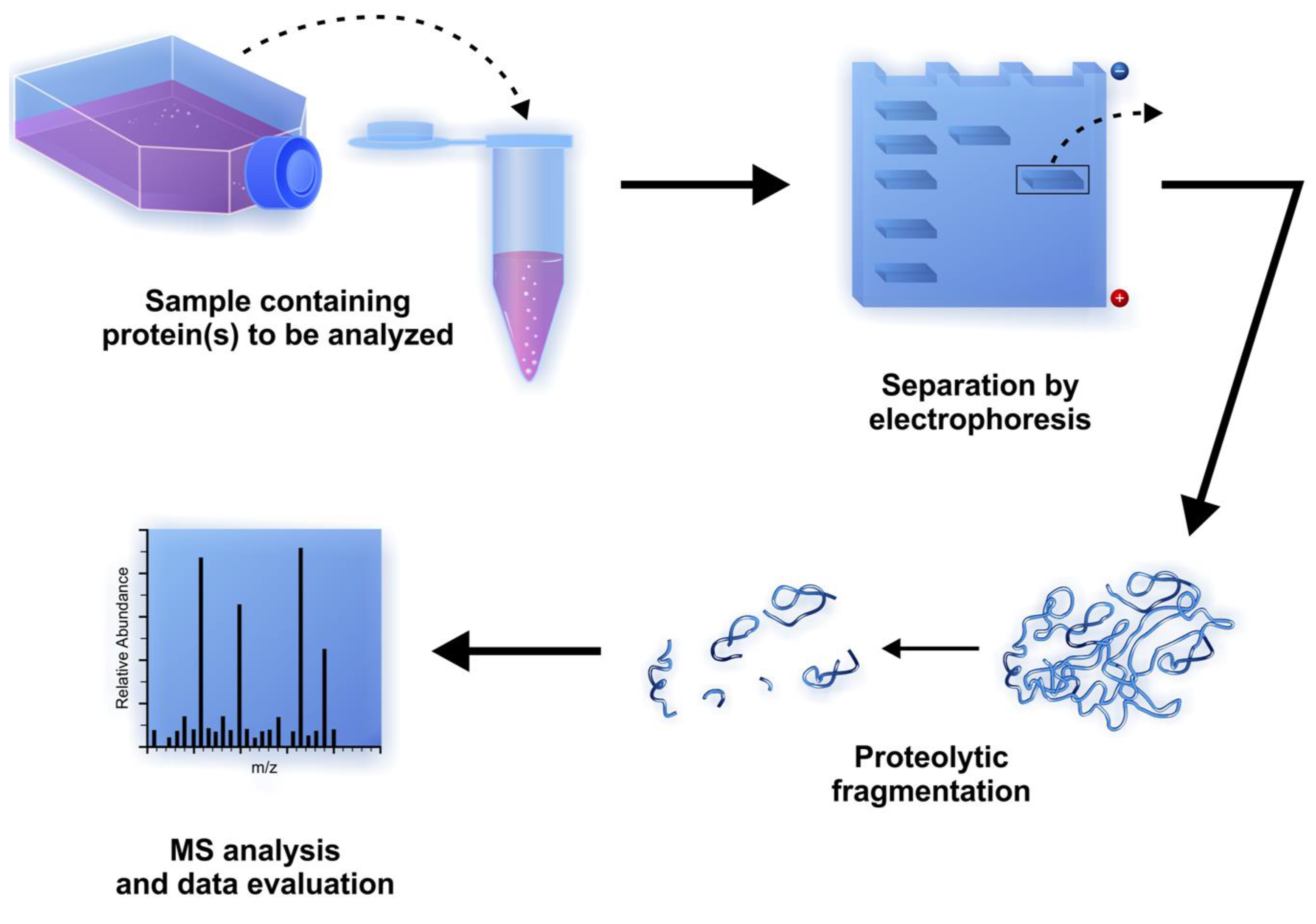
2.9. Proteomic Applications
3. Summary
Acknowledgments
Conflicts of Interest
References
- Barrett, A.J.; McDonald, J.K. Nomenclature: Protease, proteinase and peptidase. Biochem. J. 1986, 237, 935. [Google Scholar]
- Rao, M.B.; Tanksale, A.M.; Ghatge, M.S.; Deshpande, V.V. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 1998, 62, 597–635. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012, 40, D343–D350. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205–218. [Google Scholar]
- Neurath, H.; Walsh, K.A. Role of proteolytic enzymes in biological regulation (a review). Proc. Natl. Acad. Sci. USA 1976, 73, 3825–3832. [Google Scholar] [CrossRef]
- Devlin, T.M. Textbook of Biochemistry with Clinical Correlations, 5th ed.; Wiley & Sons: New York, NY, USA, 2002. [Google Scholar]
- Li, Q.; Yi, L.; Marek, P.; Iverson, B.L. Commercial proteases: Present and future. FEBS Lett. 2013, 587, 1155–1163. [Google Scholar] [CrossRef]
- Craik, C.S.; Page, M.J.; Madison, E.L. Proteases as therapeutics. Biochem. J. 2011, 435, 1–16. [Google Scholar] [CrossRef]
- Antonelli, G.; Turriziani, O. Antiviral therapy: Old and current issues. Int. J. Antimicrob. Agents 2012, 40, 95–102. [Google Scholar] [CrossRef]
- Kirk, O.; Borchert, T.V.; Fuglsang, C.C. Industrial enzyme applications. Curr. Opin. Biotechnol. 2002, 13, 345–351. [Google Scholar] [CrossRef]
- Rani, K.; Rana, R.; Datt, S. Review on latest overview of proteases. Int. J. Curr. Life Sci. 2012, 2, 12–18. [Google Scholar]
- Ray, A. Protease enzyme- potential industrial scope. Int. J. Technol. 2012, 2, 1–4. [Google Scholar]
- Sarrouh, B.; Santos, T.M.; Miyoshi, A.; Dias, R.; Azevedo, V. Up-to-date insight on industrial enzymes applications and global market. J. Bioprocess. Biotech. 2012, S4:002. [Google Scholar]
- Klenow, H.; Henningsen, I. Selective elimination of the exonuclease activity of the deoxyribonucleic acid polymerase from Escherichia coli B by limited proteolysis. Proc. Natl. Acad. Sci. USA 1970, 65, 168–175. [Google Scholar] [CrossRef]
- Morihara, K. Using proteases in peptide synthesis. Trends Biotechnol. 1987, 5, 164–170. [Google Scholar] [CrossRef]
- Bhalla, T.C.; Kumar, D.; Gajju, H.; Agrawal, H.O. Thermophilic bacterial proteases. J. Punjab Acad. Sci. 1999, 1, 77–91. [Google Scholar]
- Kumar, D.; Bhalla, T.C. Microbial proteases in peptide synthesis: Approaches and applications. Appl. Microbiol. Biotechnol. 2005, 68, 726–736. [Google Scholar] [CrossRef]
- Kühn, D.; Dürrschmidt, P.; Mansfeld, J.; Ulbrich-Hofmann, R. Biolysin and thermolysin in dipeptide synthesis: A comparative study. Biotechnol. Appl. Biochem. 2002, 36, 71–76. [Google Scholar] [CrossRef]
- Kimura, Y.; Muraya, K.; Araki, Y.; Matsuoka, H.; Nakanishi, K.; Matsuno, R. Synthesis peptides consisting of essential amino acids by a reactor system using three proteinases and an organic solvent. Agric. Biol. Chem. 1990, 54, 3331–3333. [Google Scholar] [CrossRef]
- Bille, V.; Ripak, C.; van Aasche, I.; Forni, I.; Degelaen, L.; Searso, A. A Semi-Enzymatic Synthesis of Somatostatin. In Proceedings of 21st European Peptide Symposium, ESCOM, Leiden, The Netherlands, 1991; Giralt, E., Andreu, D., Eds.; pp. 253–254.
- Rizo, J.; Gierarch, L.M. Constrained peptides: Models of bioactive peptides and protein structures. Ann. Rev. Biochem. 1992, 61, 387–418. [Google Scholar] [CrossRef]
- Widmer, F.; Bayne, S.; Houen, G.; Rigby, R.B.; Whittaker, R.G.; Johansen, J.T. Use of Proteolytic Enzymes for the Synthesis of Fragments of Mouse Epidermal Growth Factor. In Proceedings of Peptides 1984, Almquist, Stockholm, 1985; Ragnurrson, U., Ed.; pp. 193–196.
- Salam, S.M.; Kagawa, K.; Kawashiro, K. Alpha-chymotrypsin-catalyzed peptide synthesis using N-protected D-amino acid carbamoylmethyl esters as acyl donors. Biotechnol. Lett. 2005, 27, 1199–1203. [Google Scholar] [CrossRef]
- Guzmán, F.; Barberis, S.; Illanes, A. Peptide synthesis: Chemical or enzymatic. Electron. J. Biotechnol. 2007, 10, 279–314. [Google Scholar]
- Sergeeva, M.V.; Paradkar, V.M.; Dordick, J.S. Peptide synthesis using proteases dissolved in organic solvents. Enzyme Microb. Technol. 1997, 20, 623–628. [Google Scholar] [CrossRef]
- Dordick, J.S. Enzymatic catalysis in monophasic organic solvents. Enzyme Microb. Technol. 1989, 11, 194–211. [Google Scholar] [CrossRef]
- Khemlnitski, Y.L.; Levashov, A.V.; Klyachko, N.L.; Martinek, K. Engineering biocatalytic systems in organic media with low water content. Enzyme Microb. Technol. 1988, 10, 710–724. [Google Scholar] [CrossRef]
- Gill, I.; Fandino, R.L.; Jobra, X.; Vulfson, E.N. Biologically active peptides and enzymatic approaches to their production. Enzyme Microb. Technol. 1996, 18, 162–183. [Google Scholar] [CrossRef]
- Shen, H.Y.; Tian, G.L.; Ye, Y.H. Synthesis of demorphin (1–4) derivatives catalysed by proteases in organic solvents. J. Pept. Res. 2004, 65, 143–148. [Google Scholar] [CrossRef]
- Wells, J.A.; Estell, D.A. Subtilisin: An enzyme designed to be engineered. Trends Biochem. Sci. 1988, 13, 291–297. [Google Scholar] [CrossRef]
- Ogino, H.; Tsuchiyama, S.; Yasuda, M.; Doukyu, N. Enhancement of the aspartame precursor synthetic activity of an organic solvent-stable protease. Protein Eng. Des. Sel. 2010, 23, 147–152. [Google Scholar] [CrossRef]
- Xu, J.X.; Jiang, M.; Sun, H.L.; He, B.F. An organic solvent-stable protease from organic solvent-tolerant Bacillus cereus WQ9–2: Purification, biochemical properties, and potential application in peptide synthesis. Bioresour. Technol. 2010, 101, 7991–7994. [Google Scholar] [CrossRef]
- Abrahmsén, L.; Tom, J.; Burnier, J.; Butcher, K.A.; Kossiakoff, A.; Wells, J.A. Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry 1991, 30, 4151–4159. [Google Scholar] [CrossRef]
- Adamczak, M.; Krishna, S.H. Enzyme for efficient biocatalysis. Food Technol. Biotechnol. 2004, 42, 251–264. [Google Scholar]
- Ebeling, W.; Hennrich, N.; Klockow, M.; Metz, H.; Orth, H.D.; Lang, H. Proteinase K from Tritirachium album Limber. Eur. J. Biochem. 1974, 47, 91–97. [Google Scholar] [CrossRef]
- Carpi, F.M.; di Pietro, F.; Vincenzetti, S.; Mignini, F.; Napolioni, V. Human DNA extraction methods: Patents and applications. Recent Pat. DNA Gene Seq. 2011, 5, 1–7. [Google Scholar] [CrossRef]
- Sigma-Aldrich Corporation. Enzymes for cell dissociation and lysis. Biofiles Life Sci. Res. 2006. Available online: http://www.sigmaaldrich.com/ etc/medialib/docs/Sigma/General_Information/2/biofiles_issue2.Par.0001.File.tmp/biofiles_issue2.pdf (accessed on 20 July 2013).
- Santangelo, C. Worthington biochemical online tissue dissociation guide. Worthington Biochem. Corp. 2011. Available online: http://www.worthington-biochem.com/tissuedissociation/default.html (accessed on 20 July 2013).
- Mandl, I.; Maclennan, J.D.; Howes, E.L. Isolation and characterization of proteinase and collagenase from Cl. histolyticum. J. Clin. Investig. 1953, 32, 1323–1329. [Google Scholar] [CrossRef]
- Kin, T.; Johnson, P.R.; Shapiro, A.M.; Lakey, J.R. Factors influencing the collagenase digestion phase of human islet isolation. Transplantation 2007, 83, 7–12. [Google Scholar] [CrossRef]
- Canavan, H.E.; Cheng, X.; Graham, D.J.; Ratner, B.D.; Castner, D.G. Cell sheet detachment affects the extracellular matrix: A surface science study comparing thermal liftoff, enzymatic, and mechanical methods. J. Biomed. Mater. Res. A 2005, 75, 1–13. [Google Scholar]
- Huang, H.L.; Hsing, H.W.; Lai, T.C.; Chen, Y.W.; Lee, T.R.; Chan, H.T.; Lyu, P.C.; Wu, C.L.; Lu, Y.C.; Lin, S.T.; et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J. Biomed. Sci. 2012, 17. [Google Scholar] [CrossRef]
- Danhier, P.; Copetti, T.; de Preter, G.; Leveque, P.; Feron, O.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Influence of cell detachment on the respiration rate of tumor and endothelial cells. PLoS One 2013, 8, e53324. [Google Scholar] [CrossRef]
- Rader, C. Overview on concepts and applications of Fab antibody fragments. Curr. Protoc. Protein Sci. 2009, 55, 6.9.1–6.9.14. [Google Scholar]
- Flanagan, R.J.; Jones, A.L. Fab antibody fragments: Some applications in clinical toxicology. Drug Saf. 2004, 27, 1115–1133. [Google Scholar] [CrossRef]
- De Marco, A. Biotechnological applications of recombinant single-domain antibody fragments. Microb. Cell Fact. 2011, 10. [Google Scholar] [CrossRef]
- Zhao, Y.; Gutshall, L.; Jiang, H.; Baker, A.; Beil, E.; Obmolova, G.; Carton, J.; Taudte, S.; Amegadzie, B. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr. Purif. 2009, 67, 182–189. [Google Scholar] [CrossRef]
- Hunte, C.; Michel, H. Crystallisation of membrane proteins mediated by antibody fragments. Curr. Opin. Struct. Biol. 2002, 12, 503–508. [Google Scholar] [CrossRef]
- Kovari, L.C.; Momany, C.; Rossmann, M.G. The use of antibody fragments for crystallization and structure determinations. Structure 1995, 3, 1291–1293. [Google Scholar] [CrossRef]
- Caffrey, M. Membrane protein crystallization. J. Struct. Biol. 2003, 42, 108–132. [Google Scholar] [CrossRef]
- Bill, R.M.; Henderson, P.J.; Iwata, S.; Kunji, E.R.; Michel, H.; Neutze, R.; Newstead, S.; Poolman, B.; Tate, C.G.; Vogel, H. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 2011, 29, 335–340. [Google Scholar]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001, 414, 43–48. [Google Scholar] [CrossRef]
- Rasmussen, S.G.F.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Bagossi, P.; Sperka, T.; Fehér, A.; Kádas, J.; Zahuczky, G.; Miklóssy, G.; Boross, P.; Tőzsér, J. Amino acid preferences for a critical substrate binding subsite of retroviral proteases in type 1 cleavage sites. J. Virol. 2005, 79, 4213–4218. [Google Scholar] [CrossRef]
- Eizert, H.; Bander, P.; Bagossi, P.; Sperka, T.; Miklóssy, G.; Boross, P.; Weber, I.T.; Tőzsér, J. Amino acid preferences of retroviral proteases for amino-terminal positions in a type 1 cleavage site. J. Virol. 2008, 82, 10111–10117. [Google Scholar] [CrossRef]
- Tőzsér, J. Comparative studies on retroviral proteases: Substrate specificity. Viruses 2010, 2, 147–165. [Google Scholar] [CrossRef]
- Patick, A.K.; Potts, K.E. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 1998, 11, 614–627. [Google Scholar]
- Wlodawer, A.; Gustchina, A. Structural and biochemical studies of retroviral proteases. Biochim. Biophys. Acta 2000, 1477, 16–34. [Google Scholar]
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2000, 1, 13–25. [Google Scholar] [CrossRef]
- Wlodawer, A. Structure-based design of AIDS drugs and the development of resistance. Vox Sang. 2002, 83 (Suppl. 1), 23–26. [Google Scholar] [CrossRef]
- Pogson, M.; Georgiou, G.; Iverson, B.L. Engineering next generation proteases. Curr. Opin. Biotechnol. 2009, 20, 390–397. [Google Scholar] [CrossRef]
- Lim, E.J.; Sampath, S.; Coll-Rodriguez, J.; Schmidt, J.; Ray, K.; Rodgers, D.W. Swapping the substrate specificities of the neuropeptidases neurolysin and thimet oligopeptidase. J. Biol. Chem. 2007, 282, 9722–9732. [Google Scholar]
- Villa, J.P.; Bertenshaw, G.P.; Bond, J.S. Critical amino acids in the active site of meprin metalloproteinases for substrate and peptide bond specificity. J. Biol. Chem. 2003, 278, 42545–42550. [Google Scholar]
- Derewenda, Z.S. The use of recombinant methods and molecular engineering in protein crystallization. Methods 2004, 34, 354–363. [Google Scholar] [CrossRef]
- Waugh, D.S. Making the most of affinity tags. Trends Biotechnol. 2005, 23, 316–320. [Google Scholar] [CrossRef]
- Renzi, F.; Panetta, G.; Vallone, B.; Brunori, M.; Arceci, M.; Bozzoni, I.; Laneve, P.; Caffarelli, E. Large-scale purification and crystallization of the endoribonuclease XendoU: Troubleshooting with His-tagged proteins. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 298–301. [Google Scholar] [CrossRef]
- Horchani, H.; Ouertani, S.; Gargouri, Y.; Sayari, A. The N-terminal His-tag and the recombination process affect the biochemical properties of Staphylococcus aureus lipase produced in Escherichia coli. J. Mol. Catal. BEnzym. 2009, 61, 194–201. [Google Scholar] [CrossRef]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar]
- Jenny, R.J.; Mann, K.G.; Lundblad, R.L. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 2003, 31, 1–11. [Google Scholar] [CrossRef]
- Arnau, J.; Lauritzen, C.; Petersen, G.E.; Pedersen, J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr. Purif. 2005, 48, 1–13. [Google Scholar]
- Vergis, J.M.; Wiener, M.C. The variable detergent sensitivity of proteases that are utilized for recombinant protein affinity tag removal. Protein Expr. Purif. 2011, 78, 139–142. [Google Scholar] [CrossRef]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef]
- Nallamsetty, S.; Kapust, R.B.; Tőzsér, J.; Cherry, S.; Tropea, J.E.; Copeland, T.D.; Waugh, D.S. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr. Purif. 2004, 38, 108–115. [Google Scholar] [CrossRef]
- Huang, Q.; Li, Q.; Chen, A.S.; Kang, C. West Nile virus protease activity in detergent solutions and application for affinity tag removal. Anal. Biochem. 2013, 435, 44–46. [Google Scholar] [CrossRef]
- Zhang, D.; Tőzsér, J.; Waugh, D.S. Molecular cloning, overproduction, purification and biochemical characterization of the p39 nsp2 protease domains encoded by three alphaviruses. Protein Expr. Purif. 2009, 64, 89–97. [Google Scholar] [CrossRef]
- Austin, B.P.; Tőzsér, J.; Bagossi, P.; Tropea, J.E.; Waugh, D.S. The substrate specificity of Metarhizium anisopliae and Bos taurus carboxypeptidases A: Insights into their use as tools for the removal of affinity tags. Protein Expr. Purif. 2011, 77, 53–61. [Google Scholar] [CrossRef]
- Steen, H.; Mann, M. The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef]
- Granvogl, B.; Plöscher, M.; Eichacker, L.A. Sample preparation by in-gel digestion for mass spectrometry-based proteomics. Anal. Bioanal. Chem. 2007, 389, 991–1002. [Google Scholar] [CrossRef]
- Meyers, A.; Trauger, S.; Webb, W.; Reisdorph, N.; Wranik, C.; Peters, E.; Siuzdak, G. Protein identification and profiling with mass spectrometry. Spectroscopy 2003, 17, 1–15. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mótyán, J.A.; Tóth, F.; Tőzsér, J. Research Applications of Proteolytic Enzymes in Molecular Biology. Biomolecules 2013, 3, 923-942. https://doi.org/10.3390/biom3040923
Mótyán JA, Tóth F, Tőzsér J. Research Applications of Proteolytic Enzymes in Molecular Biology. Biomolecules. 2013; 3(4):923-942. https://doi.org/10.3390/biom3040923
Chicago/Turabian StyleMótyán, János András, Ferenc Tóth, and József Tőzsér. 2013. "Research Applications of Proteolytic Enzymes in Molecular Biology" Biomolecules 3, no. 4: 923-942. https://doi.org/10.3390/biom3040923
APA StyleMótyán, J. A., Tóth, F., & Tőzsér, J. (2013). Research Applications of Proteolytic Enzymes in Molecular Biology. Biomolecules, 3(4), 923-942. https://doi.org/10.3390/biom3040923