Biophysical Insights into the Inhibitory Mechanism of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
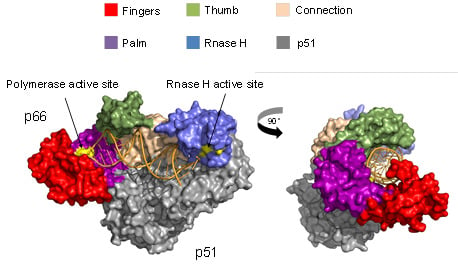
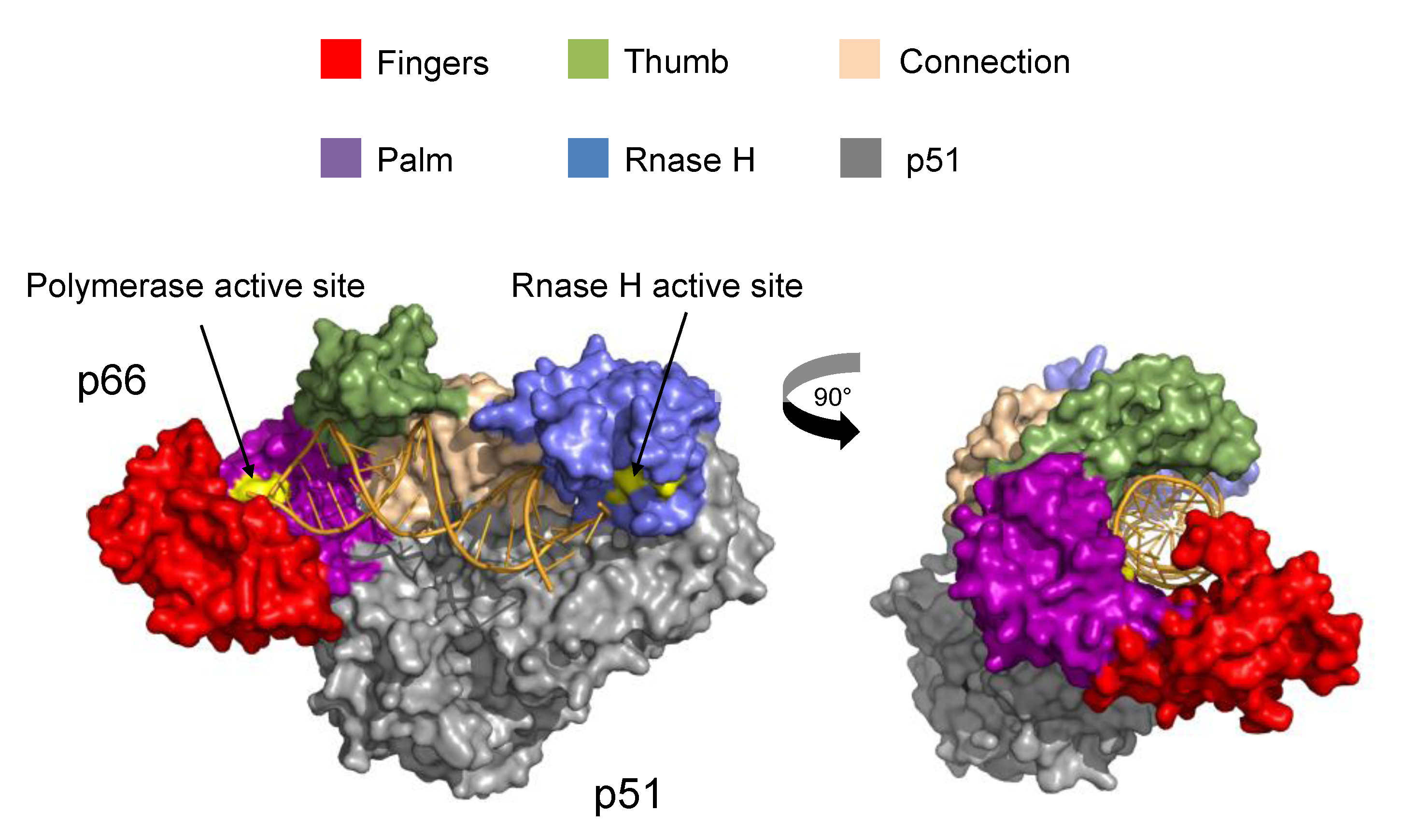
2. Structure and Function of HIV-1 RT
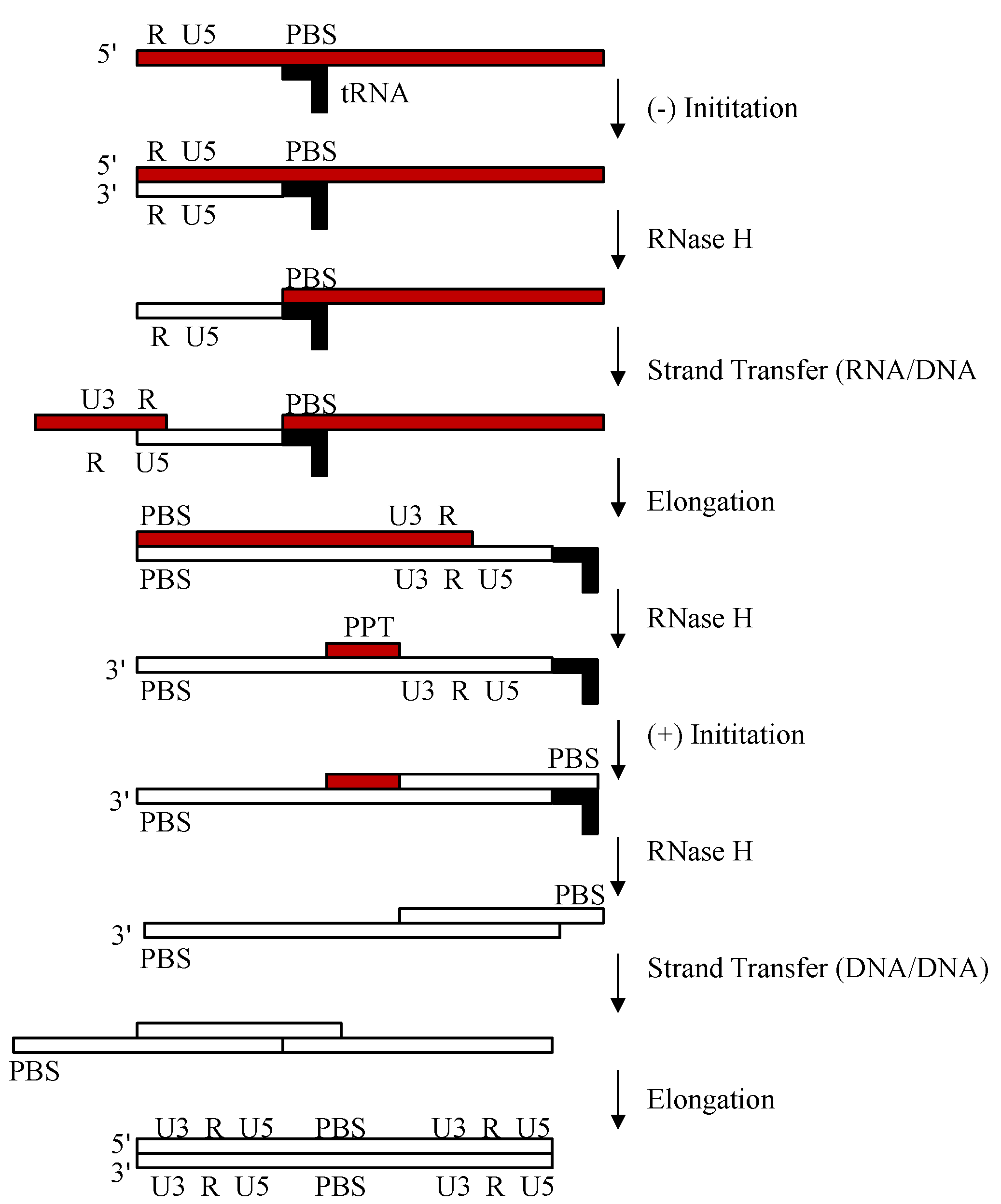
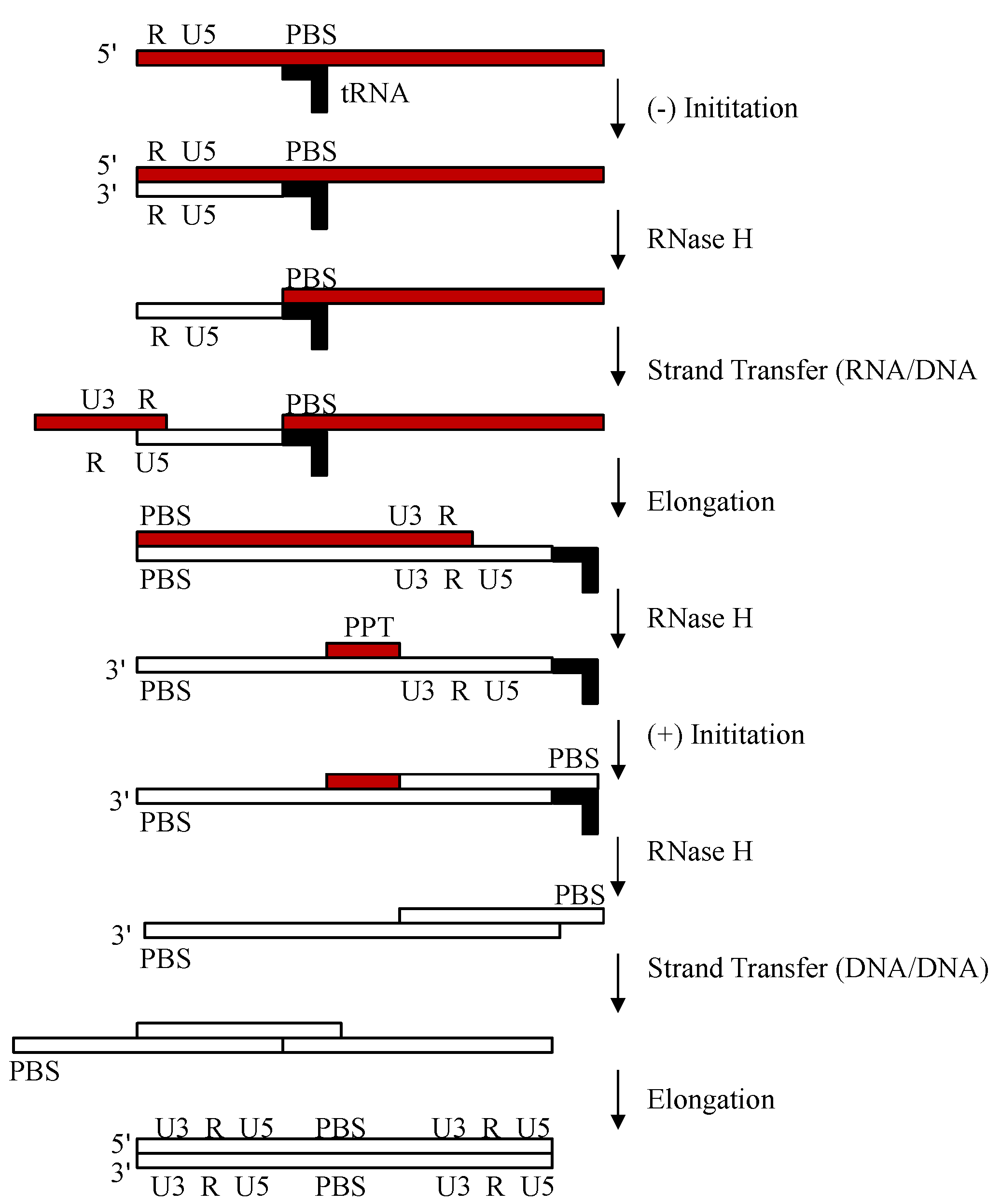
2.1. Reverse Transcription
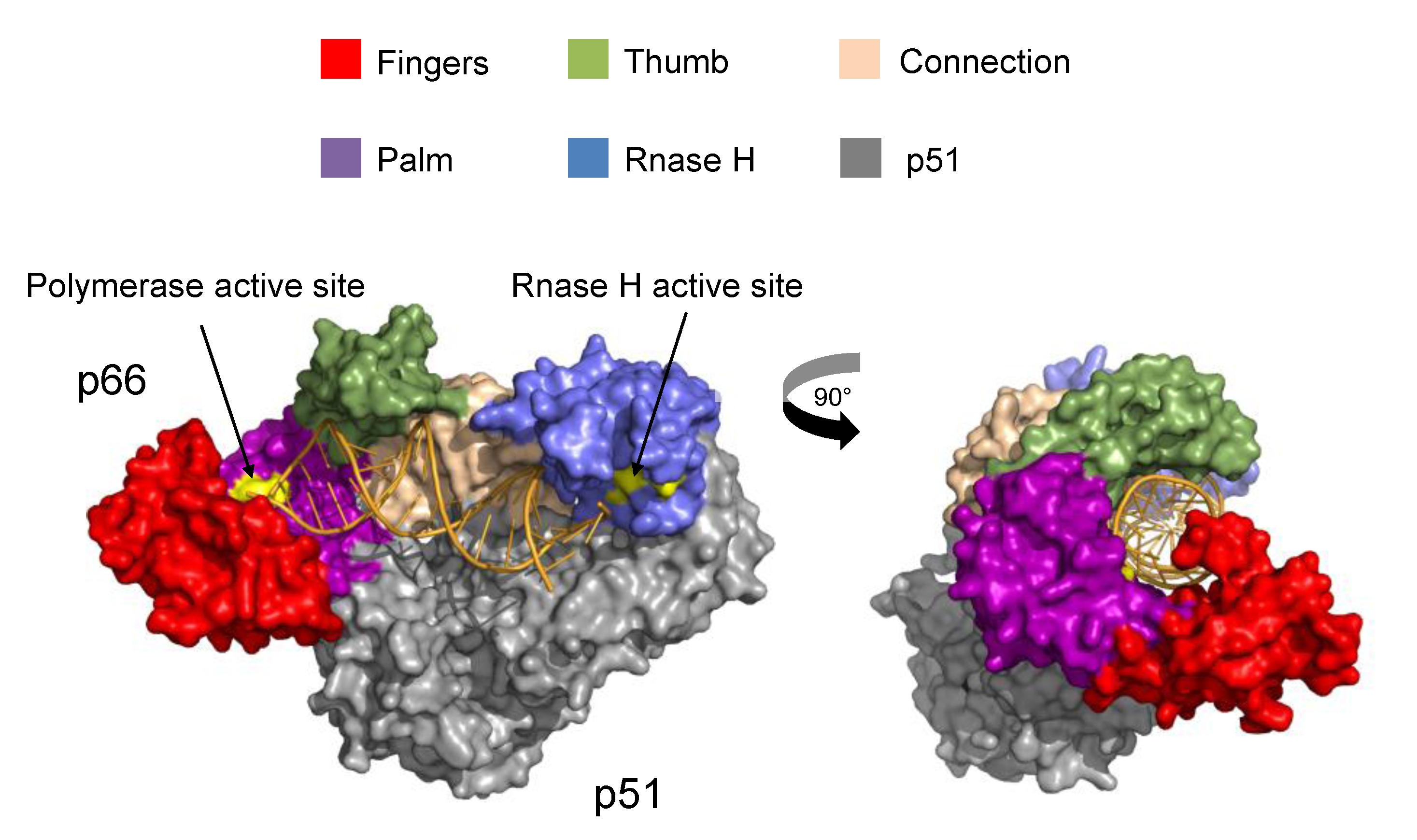
2.2. Structural and Biophysical Studies of RT
2.3. Conformational Dynamics of Reverse Transcription
3. NNRTIs and Their Mechanism of Action
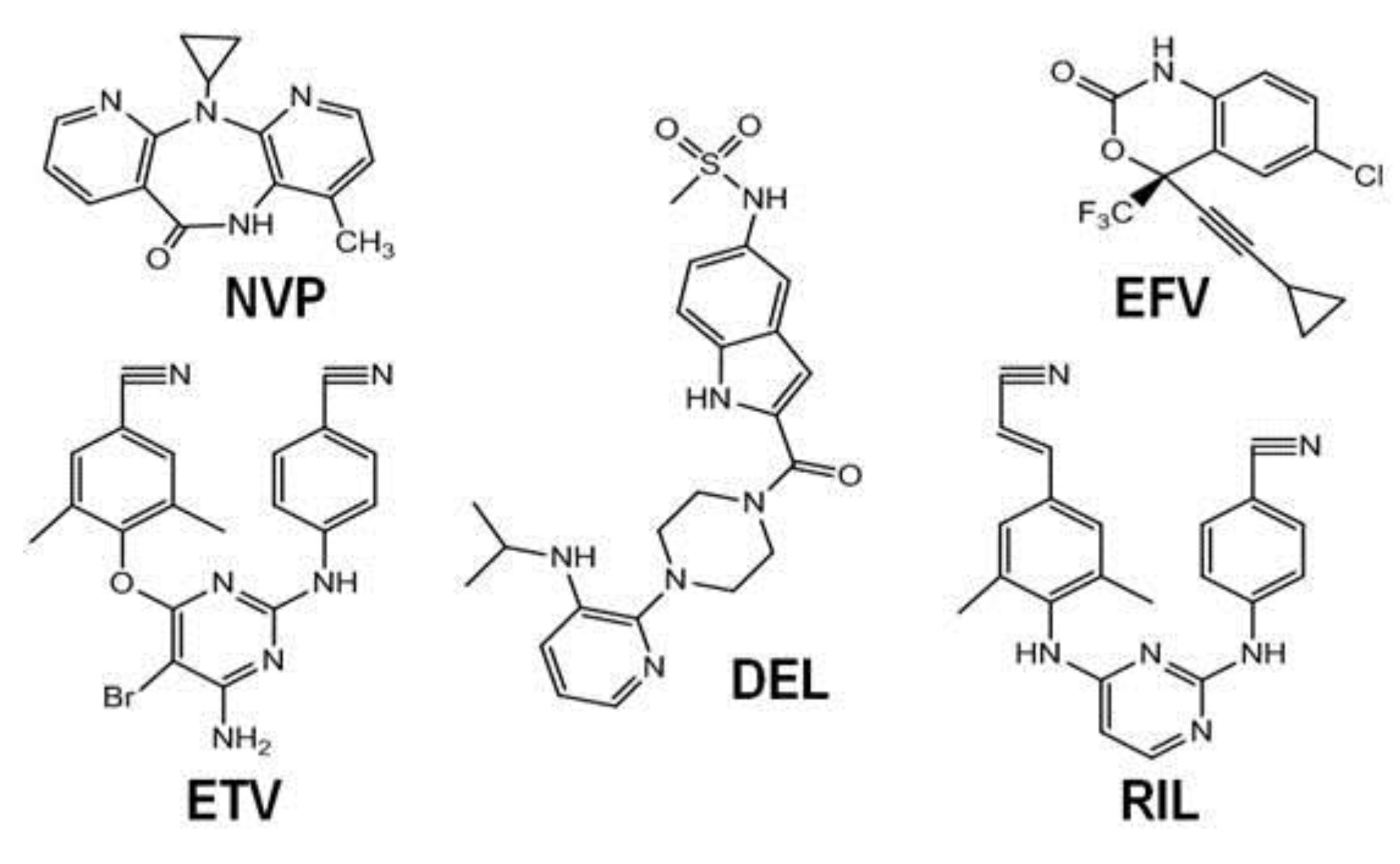
3.1. NNRTIs
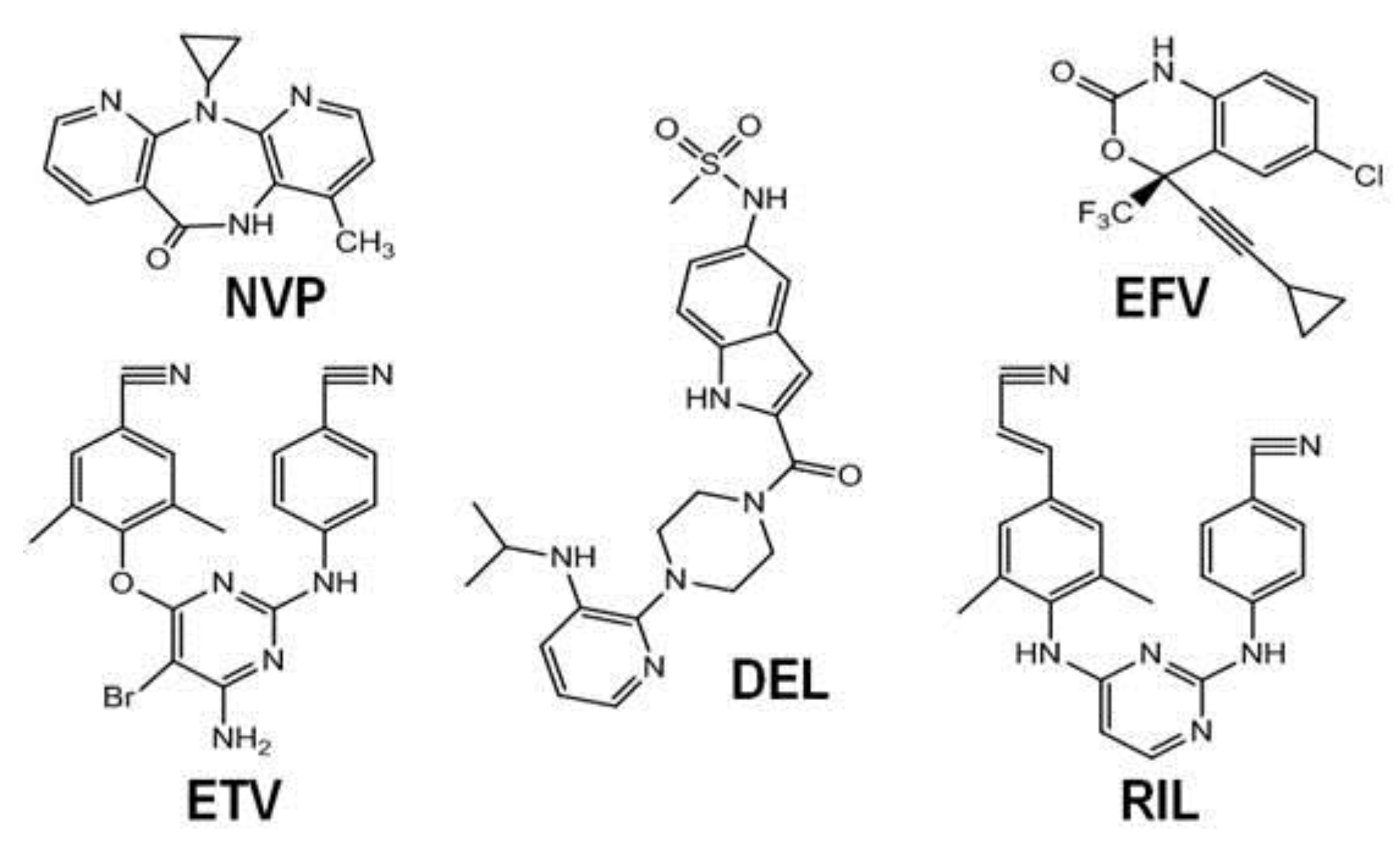
3.2. NNRTI Binding
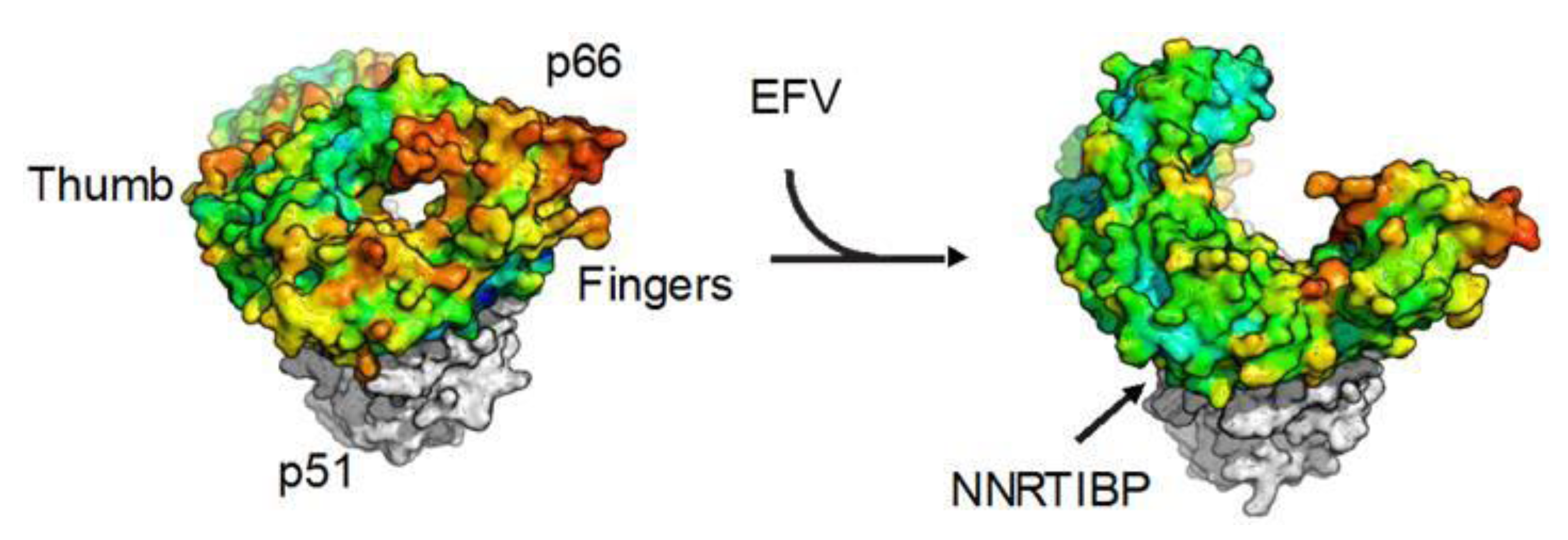
3.3. Putative Mechanisms of Inhibition by NNRTIs
3.4. Molecular Arthritis
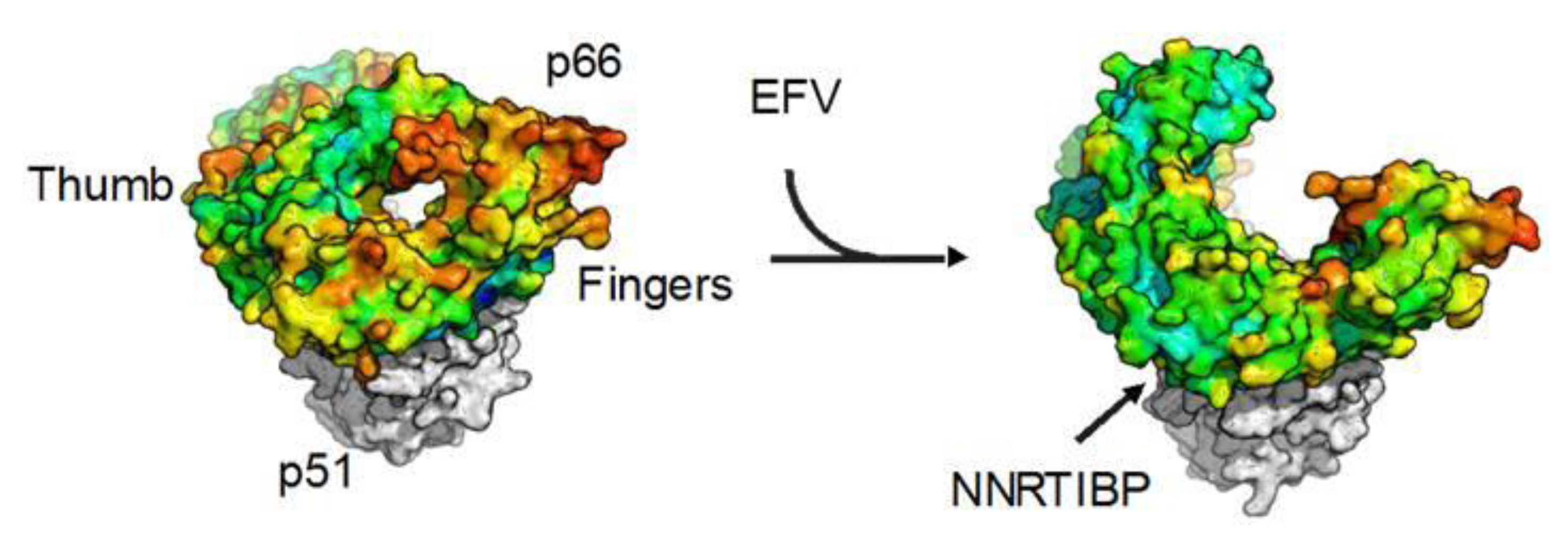
3.5. Effect of NNRTIs on RT Dimerization
3.6. NNRTI Resistance Mutations
3.6.1. K103N
3.7. Structures of NNRTI-Bound RT-T/P Complexes
3.8. Insights into RT Dynamics from Time-Resolved Single-Molecule Experiments
4. Conclusions and Future Directions
Acknowledgements
Conflicts of Interest
References
- Hu, W.-S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Tachedjian, G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008, 134, 147–156. [Google Scholar] [CrossRef]
- Divita, G.; Restle, T.; Goody, R.S. Characterization of the dimerization process of HIV-1 reverse transcriptase heterodimer using intrinsic protein fluorescence. FEBS Lett. 1993, 324, 153–158. [Google Scholar] [CrossRef]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar]
- Rodgers, D.W.; Gamblin, S.J.; Harris, B.A.; Ray, S.; Culp, J.S.; Hellmig, B.; Woolf, D.J.; Debouck, C.; Harrison, S.C. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1995, 92, 1222–1226. [Google Scholar] [CrossRef]
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Mol. Biol. 1995, 2, 303–308. [Google Scholar]
- KruhØfter, M.; Urbanke, C.; Grosse, F. Two step binding of HIV-1 reverse transcriptase to nucleic acid substrates. Nucleic Acids Res. 1993, 21, 3943–3949. [Google Scholar]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef]
- Grice, S.F.J.L. Human immunodeficiency virus reverse transcriptase: 25 Years of research, drug discovery, and promise. J. Biol. Chem. 2012, 287, 40850–40857. [Google Scholar] [CrossRef]
- Kati, W.M.; Johnson, K.A.; Jerva, L.F.; Anderson, K.S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992, 267, 25988–25997. [Google Scholar]
- Sarafianos, S.G.; Das, K.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Touching the heart of HIV-1 drug resistance: The fingers close down on the dNTP at the polymerase active site. Chem. Biol. 1999, 6, R137–R146. [Google Scholar] [CrossRef]
- Wöhrl, B.M.; Krebs, R.; Goody, R.S.; Restle, T. Refined model for primer/template binding by HIV-1 reverse transcriptase: Pre-steady-state kinetic analyses of primer/template binding and nucleotide incorporation events distinguish between different binding modes depending on the nature of the nucleic acid substrate. J. Mol. Biol. 1999, 292, 333–344. [Google Scholar] [CrossRef]
- Rothwell, P.J.; Berger, S.; Kensch, O.; Felekyan, S.; Antonik, M.; Wöhrl, B.M.; Restle, T.; Goody, R.S.; Seidel, C.A.M. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 1655–1660. [Google Scholar] [CrossRef]
- Sisamakis, E.; Valeri, A.; Kalinin, S.; Rothwell, P.J.; Seidel, C.A.M. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010, 475, 455–514. [Google Scholar] [CrossRef]
- Faradjian, A.K.; Elber, R. Computing time scales from reaction coordinates by milestoning. J. Chem. Phys. 2004, 120, 10880–10889. [Google Scholar] [CrossRef]
- Kirmizialtin, S.; Nguyen, V.; Johnson, K.A.; Elber, R. How conformational dynamics of DNA polymerase select correct substrates: Experiments and simulations. Structure 2012, 20, 618–627. [Google Scholar] [CrossRef]
- Götte, M.; Rausch, J.W.; Marchand, B.; Sarafianos, S.; Le Grice, S.F.J. Reverse transcriptase in motion: Conformational dynamics of enzyme-substrate interactions. Biochim. Biophys. Acta 2010, 1804, 1202–1212. [Google Scholar]
- Sarafianos, S.G.; Clark, A.D.; Das, K.; Tuske, S.; Birktoft, J.J.; Ilankumaran, P.; Ramesha, A.R.; Sayer, J.M.; Jerina, D.M.; Boyer, P.L.; et al. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 2002, 21, 6614–6624. [Google Scholar]
- Lu, H.; Macosko, J.; Habel-Rodriguez, D.; Keller, R.W.; Brozik, J.A.; Keller, D.J. Closing of the fingers domain generates motor forces in the HIV reverse transcriptase. J. Biol. Chem. 2004, 279, 54529–54532. [Google Scholar]
- Sluis-Cremer, N.; Temiz, N.A.; Bahar, I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr. HIV Res. 2004, 2, 323–332. [Google Scholar] [CrossRef]
- De Béthune, M.-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antiviral Res. 2010, 85, 75–90. [Google Scholar] [CrossRef]
- Zhou, H.-X. From induced fit to conformational selection: A continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J. 2010, 98, L15–L17. [Google Scholar] [CrossRef]
- Basavapathruni, A.; Anderson, K.S. Reverse transcription of the HIV-1 pandemic. FASEB J. 2007, 21, 3795–3808. [Google Scholar] [CrossRef]
- Elinder, M.; Selhorst, P.; Vanham, G.; Oberg, B.; Vrang, L.; Danielson, U.H. Inhibition of HIV-1 by non-nucleoside reverse transcriptase inhibitors via an induced fit mechanism-importance of slow dissociation and relaxation rates for antiviral efficacy. Biochem. Pharmacol. 2010, 80, 1133–1140. [Google Scholar]
- Geitmann, M.; Unge, T.; Danielson, U.H. Biosensor-based kinetic characterization of the interaction between HIV-1 reverse transcriptase and non-nucleoside inhibitors. J. Med. Chem. 2006, 49, 2367–2374. [Google Scholar] [CrossRef]
- Bakan, A.; Bahar, I. The intrinsic dynamics of enzymes plays a dominant role in determining the structural changes induced upon inhibitor binding. Proc. Natl. Acad. Sci. USA 2009, 106, 14349–14354. [Google Scholar] [CrossRef]
- Braz, V.A.; Barkley, M.D.; Jockusch, R.A.; Wintrode, P.L. Efavirenz binding site in HIV-1 reverse transcriptase monomers. Biochemistry 2010, 49, 10565–10573. [Google Scholar] [CrossRef]
- Braz, V.A.; Holladay, L.A.; Barkley, M.D. Efavirenz binding to HIV-1 reverse transcriptase monomers and dimers. Biochemistry 2010, 49, 601–610. [Google Scholar] [CrossRef]
- Zheng, X.; Mueller, G.A.; DeRose, E.F.; London, R.E. Solution characterization of [methyl-13C]methionine HIV-1 reverse transcriptase by NMR spectroscopy. Antiviral Res. 2009, 84, 205–214. [Google Scholar] [CrossRef]
- Chennubhotla, C.; Rader, A.J.; Yang, L.-W.; Bahar, I. Elastic network models for understanding biomolecular machinery: From enzymes to supramolecular assemblies. Phys. Biol. 2005, 2, S173–S180. [Google Scholar] [CrossRef]
- Radi, M.; Maga, G.; Alongi, M.; Angeli, L.; Samuele, A.; Zanoli, S.; Bellucci, L.; Tafi, A.; Casaluce, G.; Giorgi, G.; et al. Discovery of chiral cyclopropyl dihydro-alkylthio-benzyl-oxopyrimidine (S-DABO) derivatives as potent HIV-1 reverse transcriptase inhibitors with high activity against clinically relevant mutants. J. Med. Chem. 2009, 52, 840–851. [Google Scholar] [CrossRef]
- Geitmann, M.; Unge, T.; Danielson, U.H. Interaction kinetic characterization of HIV-1 reverse transcriptase non-nucleoside inhibitor resistance. J. Med. Chem. 2006, 49, 2375–2387. [Google Scholar] [CrossRef]
- Burnouf, D.; Ennifar, E.; Guedich, S.; Puffer, B.; Hoffmann, G.; Bec, G.; Disdier, F.; Baltzinger, M.; Dumas, P. kinITC: A new method for obtaining joint thermodynamic and kinetic data by isothermal titration calorimetry. J. Am. Chem. Soc. 2012, 134, 559–565. [Google Scholar]
- Bec, G.; Meyer, B.; Gerard, M.-A.; Steger, J.; Fauster, K.; Wolff, P.; Burnouf, D.Y.; Micura, R.; Dumas, P.; Ennifar, E. Thermodynamics of HIV-1 reverse transcriptase in action elucidates the mechanism of action of non-nucleoside inhibitors. J. Am. Chem. Soc. 2013, 135, 9743–9752. [Google Scholar] [CrossRef]
- Rittinger, K.; Divita, G.; Goody, R.S. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA 1995, 92, 8046–8049. [Google Scholar] [CrossRef]
- Spence, R.; Kati, W.; Anderson, K.; Johnson, K. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 1995, 267, 988–993. [Google Scholar] [CrossRef]
- Divita, G.; Mueller, B.; Immendoerfer, U.; Gautel, M.; Rittinger, K.; Restle, T.; Goody, R.S. Kinetics of interaction of HIV reverse transcriptase with primer/template. Biochemistry 1993, 32, 7966–7971. [Google Scholar]
- Seckler, J.M.; Barkley, M.D.; Wintrode, P.L. Allosteric suppression of HIV-1 reverse transcriptase structural dynamics upon inhibitor binding. Biophys. J. 2011, 100, 144–153. [Google Scholar] [CrossRef]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 reverse transcriptase still remains a new drug target: Structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012, 2012, 1–23. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef]
- Ren, J.; Nichols, C.E.; Stamp, A.; Chamberlain, P.P.; Ferris, R.; Weaver, K.L.; Short, S.A.; Stammers, D.K. Structural insights into mechanisms of non-nucleoside drug resistance for HIV-1 reverse transcriptases mutated at codons 101 or 138. FEBS J. 2006, 273, 3850–3860. [Google Scholar] [CrossRef]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef]
- Das, K.; Ding, J.; Hsiou, Y.; Clark, A.D., Jr.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; et al. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [Google Scholar] [CrossRef]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Pathak, V.K. The “connection” between HIV drug resistance and RNase H. Viruses 2010, 2, 1476–1503. [Google Scholar] [CrossRef]
- Lapkouski, M.; Tian, L.; Miller, J.T.; Le Grice, S.F.J.; Yang, W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat. Struct. Mol. Biol. 2013, 20, 230–236. [Google Scholar] [CrossRef]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Grice, S.F.J.L.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef]
- Bahar, I.; Erman, B.; Jernigan, R.L.; Atilgan, A.R.; Covell, D.G. Collective motions in HIV-1 reverse transcriptase: Examination of flexibility and enzyme function. J. Mol. Biol. 1999, 285, 1023–1037. [Google Scholar] [CrossRef]
- Ivetac, A.; McCammon, J.A. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J. Mol. Biol. 2009, 388, 644–658. [Google Scholar] [CrossRef]
- Temiz, N.A.; Bahar, I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins Struct. Funct. Genet. 2002, 49, 61–70. [Google Scholar]
- Wright, D.W.; Sadiq, S.K.; de Fabritiis, G.; Coveney, P.V. Thumbs down for HIV: Domain level rearrangements do occur in the NNRTI-bound HIV-1 reverse transcriptase. J. Am. Chem. Soc. 2012, 134, 12885–12888. [Google Scholar]
- Shaw-Reid, C.A.; Feuston, B.; Munshi, V.; Getty, K.; Krueger, J.; Hazuda, D.J.; Parniak, M.A.; Miller, M.D.; Lewis, D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry 2005, 44, 1595–1606. [Google Scholar] [CrossRef]
- Radzio, J.; Sluis-Cremer, N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008, 73, 601–606. [Google Scholar] [CrossRef]
- Tachedjian, G.; Goff, S.P. The effect of NNRTIs on HIV reverse transcriptase dimerization. Curr. Opin. Investig. Drugs 2003, 4, 966–973. [Google Scholar]
- Tachedjian, G.; Orlova, M.; Sarafianos, S.G.; Arnold, E.; Goff, S.P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2001, 98, 7188–7193. [Google Scholar]
- Venezia, C.F.; Howard, K.J.; Ignatov, M.E.; Holladay, L.A.; Barkley, M.D. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry 2006, 45, 2779–2789. [Google Scholar] [CrossRef]
- Tachedjian, G.; Moore, K.L.; Goff, S.P.; Sluis-Cremer, N. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 2005, 579, 379–384. [Google Scholar] [CrossRef]
- Figueiredo, A.; Moore, K.L.; Mak, J.; Sluis-Cremer, N.; de Bethune, M.-P.; Tachedjian, G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006, 2, e119. [Google Scholar]
- Xia, Q.; Radzio, J.; Anderson, K.S.; Sluis-Cremer, N. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007, 16, 1728–1737. [Google Scholar]
- Menéndez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral Res. 2013, 98, 93–120. [Google Scholar]
- Ren, J.; Nichols, C.; Bird, L.; Chamberlain, P.; Weaver, K.; Short, S.; Stuart, D.I.; Stammers, D.K. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001, 312, 795–805. [Google Scholar] [CrossRef]
- Huang, W.; Gamarnik, A.; Limoli, K.; Petropoulos, C.J.; Whitcomb, J.M. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J. Virol. 2003, 77, 1512–1523. [Google Scholar]
- Yap, S.-H.; Sheen, C.-W.; Fahey, J.; Zanin, M.; Tyssen, D.; Lima, V.D.; Wynhoven, B.; Kuiper, M.; Sluis-Cremer, N.; Harrigan, P.R.; et al. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 2007, 4, e335. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Stammers, D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008, 134, 157–170. [Google Scholar] [CrossRef]
- Clotet, B. Efavirenz: Resistance and cross-resistance. Int. J. Clin. Pract. Suppl. 1999, 103, 21–25. [Google Scholar]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A.D., Jr.; Boyer, P.L.; Lewi, P.; Janssen, P.A.; Kleim, J.-P.; Rösner, M.; Hughes, S.H.; et al. The Lys103Asn mutation of HIV-1 RT: A novel mechanism of drug resistance. J. Mol. Biol. 2001, 309, 437–445. [Google Scholar]
- Das, K.; Sarafianos, S.G.; Clark, A.D., Jr.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structures of clinically relevant Lys103Asn/Tyr181Cys double mutant HIV-1 reverse transcriptase in complexes with ATP and non-nucleoside inhibitor HBY 097. J. Mol. Biol. 2007, 365, 77–89. [Google Scholar]
- Andries, K.; Azijn, H.; Thielemans, T.; Ludovici, D.; Kukla, M.; Heeres, J.; Janssen, P.; de Corte, B.; Vingerhoets, J.; Pauwels, R.; et al. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48, 4680–4686. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Clark, A.D.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 1466–1471. [Google Scholar] [CrossRef]
- Azijn, H.; Tirry, I.; Vingerhoets, J.; de Béthune, M.-P.; Kraus, G.; Boven, K.; Jochmans, D.; van Craenenbroeck, E.; Picchio, G.; Rimsky, L.T. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob. Agents Chemother. 2010, 54, 718–727. [Google Scholar] [CrossRef]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): Implications for drug design. J. Med. Chem. 2010, 53, 4295–4299. [Google Scholar] [CrossRef]
- Rawal, R.K.; Murugesan, V.; Katti, S.B. Structure-activity relationship studies on clinically relevant HIV-1 NNRTIs. Curr. Med. Chem. 2012, 19, 5364–5380. [Google Scholar] [CrossRef]
- Kalinin, S.; Peulen, T.; Sindbert, S.; Rothwell, P.J.; Berger, S.; Restle, T.; Goody, R.S.; Gohlke, H.; Seidel, C.A.M. A toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat. Methods 2012, 9, 1218–1225. [Google Scholar] [CrossRef]
- Kim, S.; Blainey, P.C.; Schroeder, C.M.; Xie, X.S. Multiplexed single-molecule assay for enzymatic activity on flow-stretched DNA. Nat. Methods 2007, 4, 397–399. [Google Scholar]
- Kim, S.; Schroeder, C.M.; Xie, X.S. Single-molecule study of DNA polymerization activity of HIV-1 reverse transcriptase on DNA templates. J. Mol. Biol. 2010, 395, 995–1006. [Google Scholar] [CrossRef]
- Abbondanzieri, E.A.; Bokinsky, G.; Rausch, J.W.; Zhang, J.X.; Le Grice, S.F.J.; Zhuang, X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 2008, 453, 184–189. [Google Scholar] [CrossRef]
- Fagerburg, M.V.; Leuba, S.H. Optimal practices for surface-tethered single molecule total internal reflection fluorescence resonance energy transfer analysis. Methods Mol. Biol. 2011, 749, 273–289. [Google Scholar] [CrossRef]
- Hwang, H.; Kim, H.; Myong, S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc. Natl. Acad. Sci. USA 2011, 108, 7414–7418. [Google Scholar] [CrossRef]
- Marko, R.A.; Liu, H.-W.; Ablenas, C.J.; Ehteshami, M.; Götte, M.; Cosa, G. Binding kinetics and affinities of heterodimeric versus homodimeric HIV-1 reverse transcriptase on DNA—DNA substrates at the single-molecule level. J. Phys. Chem. B 2013, 117, 4560–4567. [Google Scholar]
- Seckler, J.M.; Howard, K.J.; Barkley, M.D.; Wintrode, P.L. Solution structural dynamics of HIV-1 reverse transcriptase heterodimer. Biochemistry 2009, 48, 7646–7655. [Google Scholar] [CrossRef]
- Moore, G.E. Cramming More Components onto Integrated Circuits; McGraw-Hill: New York, NY, USA, 1965. [Google Scholar]
- Van Oijen, A.M. Single-molecule approaches to characterizing kinetics of biomolecular interactions. Curr. Opin. Biotechnol. 2011, 22, 75–80. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schauer, G.; Leuba, S.; Sluis-Cremer, N. Biophysical Insights into the Inhibitory Mechanism of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. Biomolecules 2013, 3, 889-904. https://doi.org/10.3390/biom3040889
Schauer G, Leuba S, Sluis-Cremer N. Biophysical Insights into the Inhibitory Mechanism of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. Biomolecules. 2013; 3(4):889-904. https://doi.org/10.3390/biom3040889
Chicago/Turabian StyleSchauer, Grant, Sanford Leuba, and Nicolas Sluis-Cremer. 2013. "Biophysical Insights into the Inhibitory Mechanism of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors" Biomolecules 3, no. 4: 889-904. https://doi.org/10.3390/biom3040889