Biocatalysis for Biobased Chemicals


Abstract
:1. Introduction
2. Enzymes in Food Industry
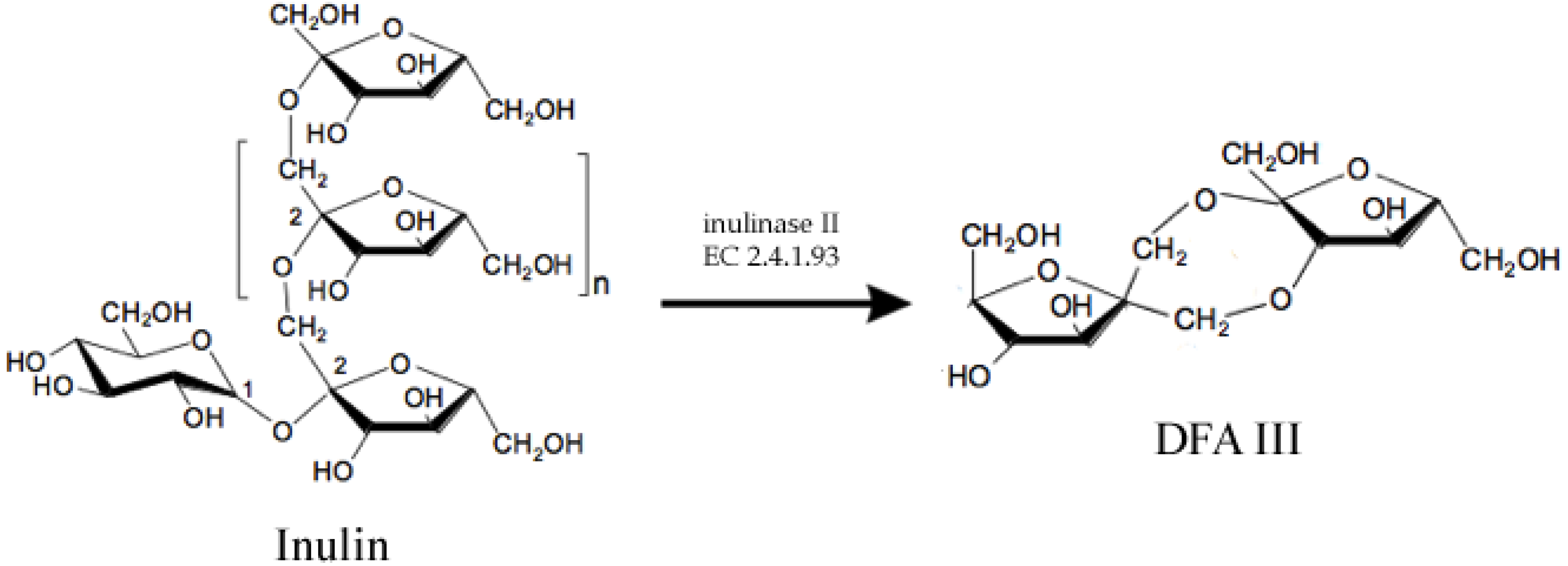
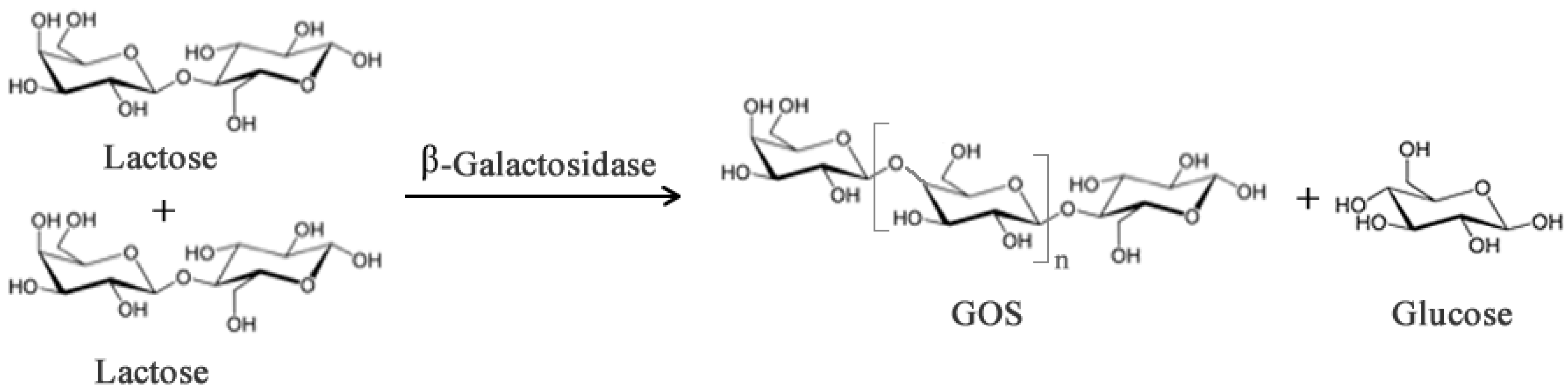
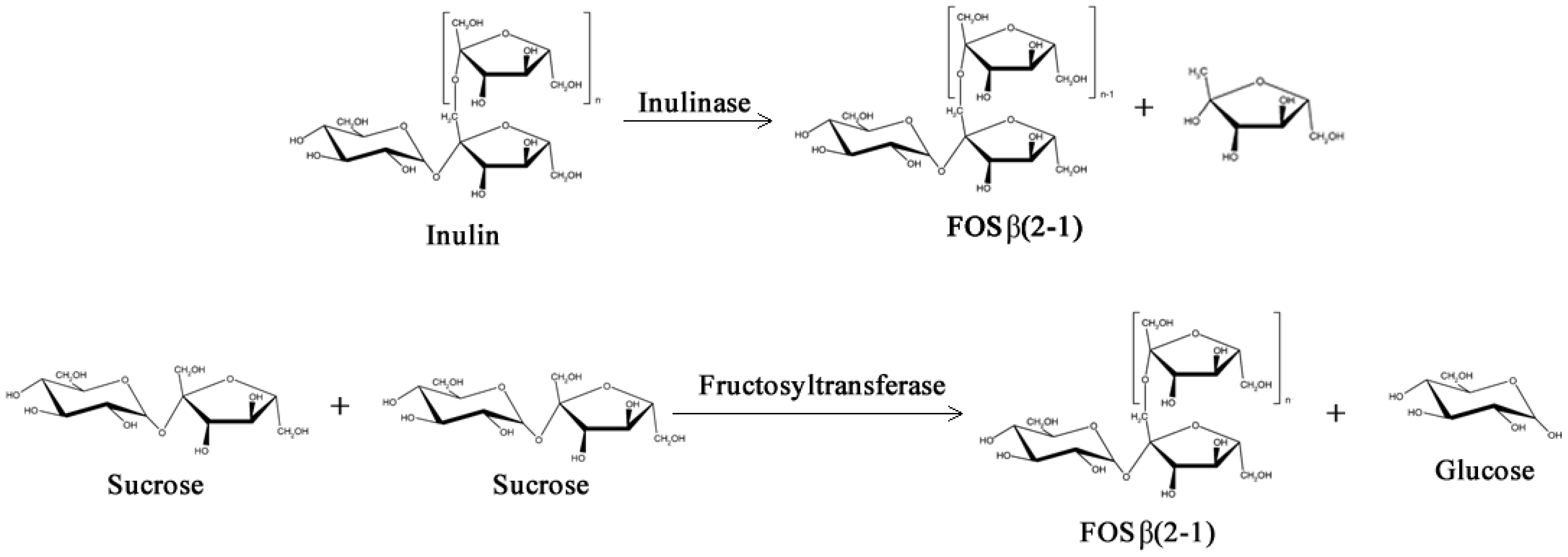
2.1. Prebiotics and Sweeteners
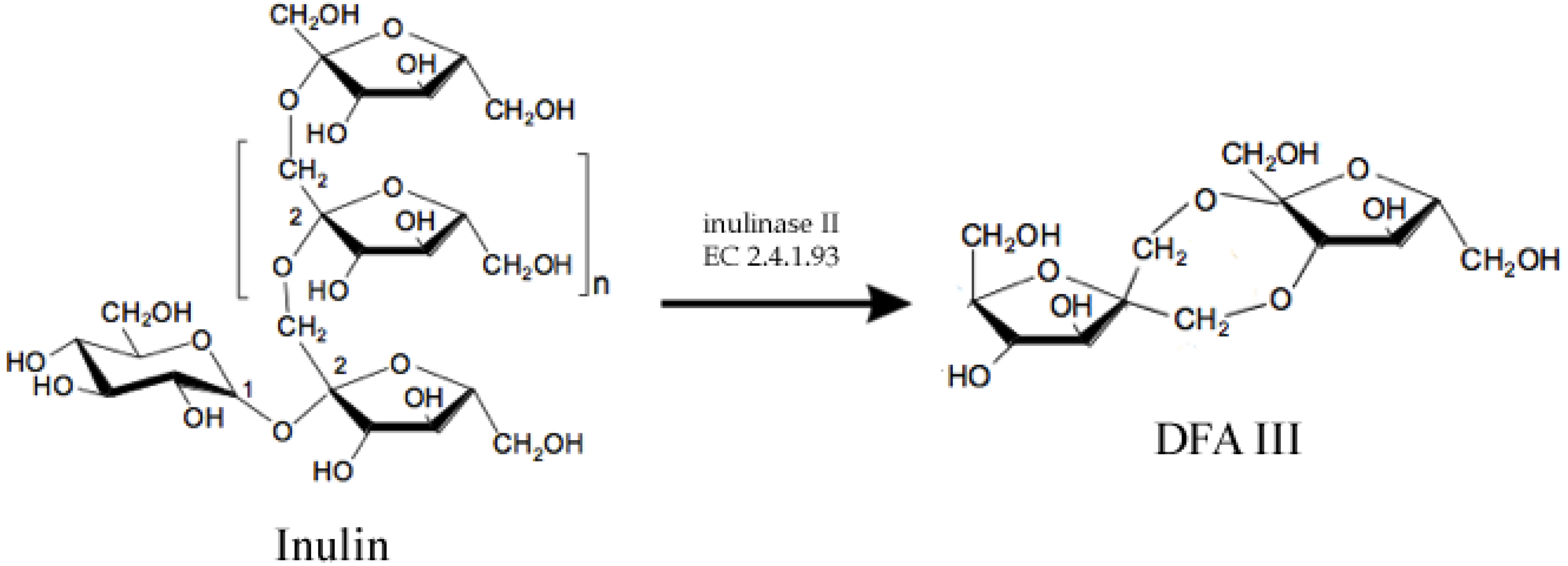
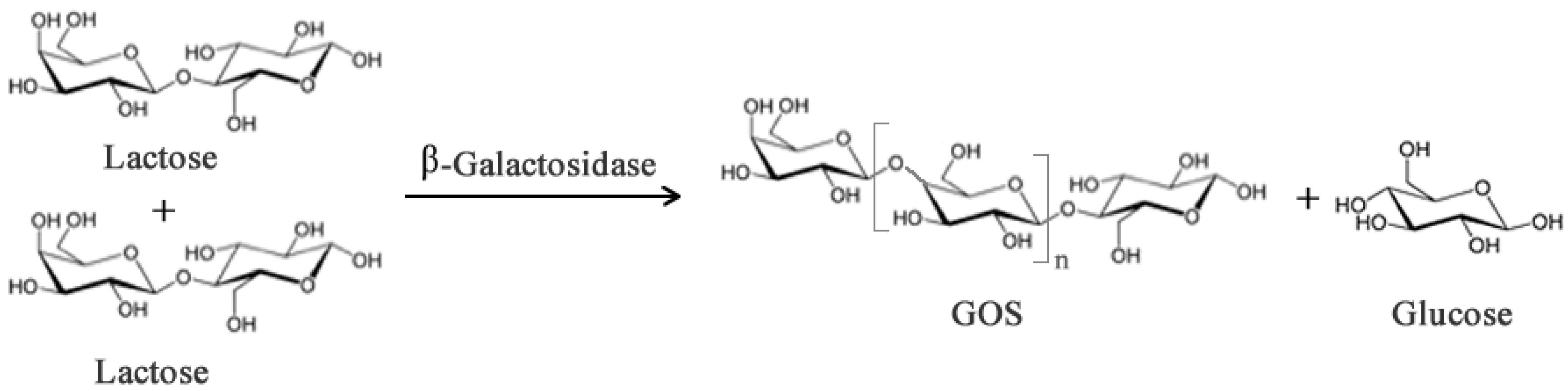
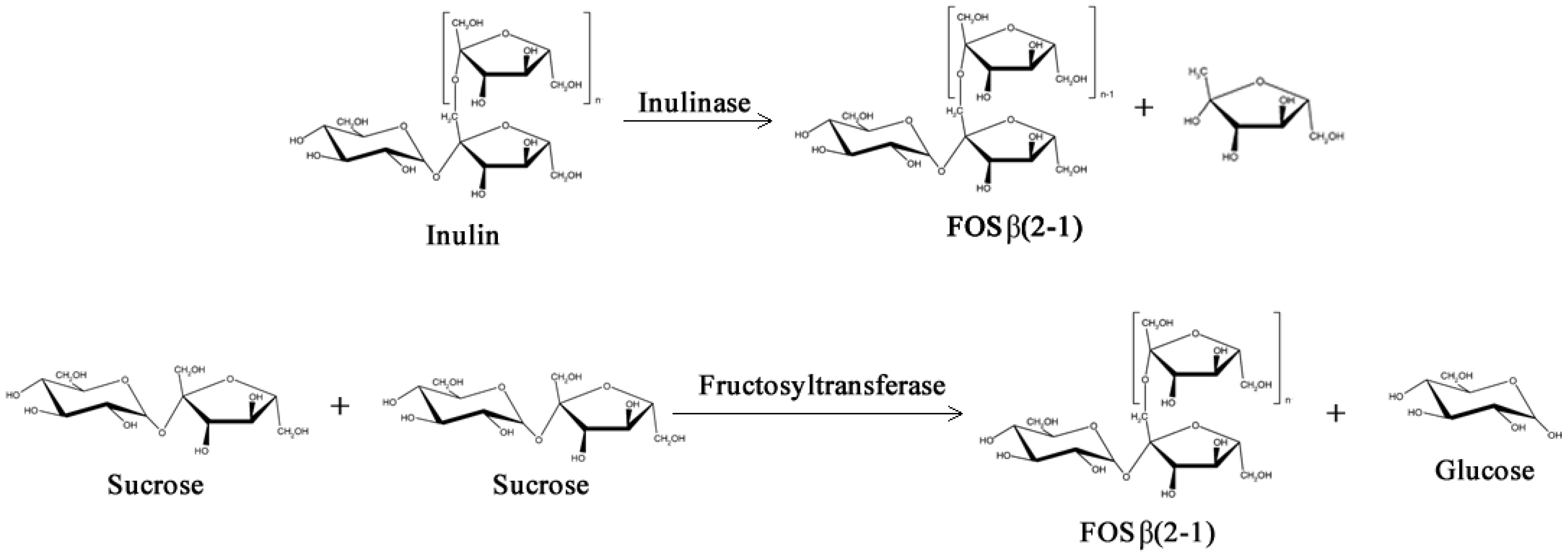
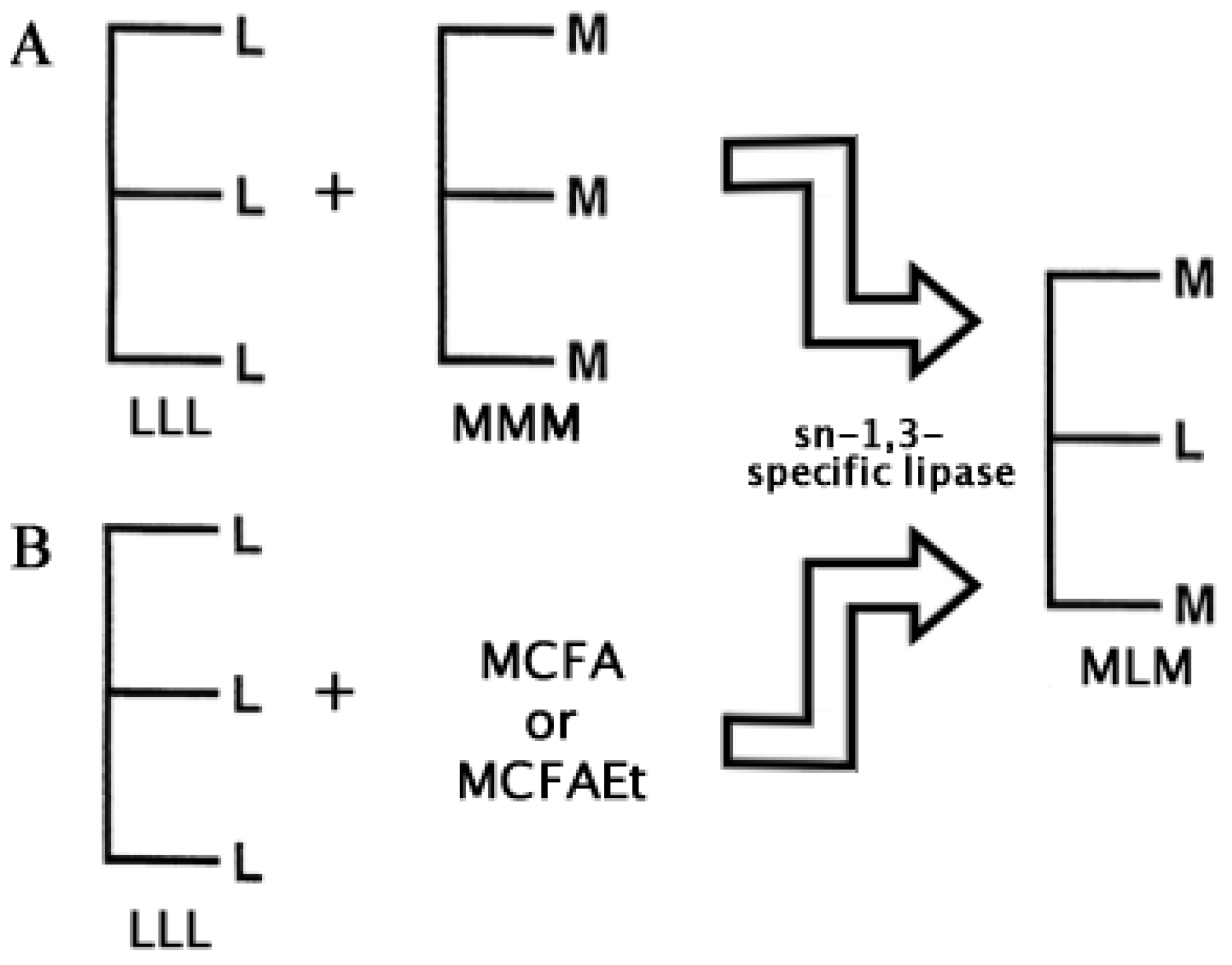
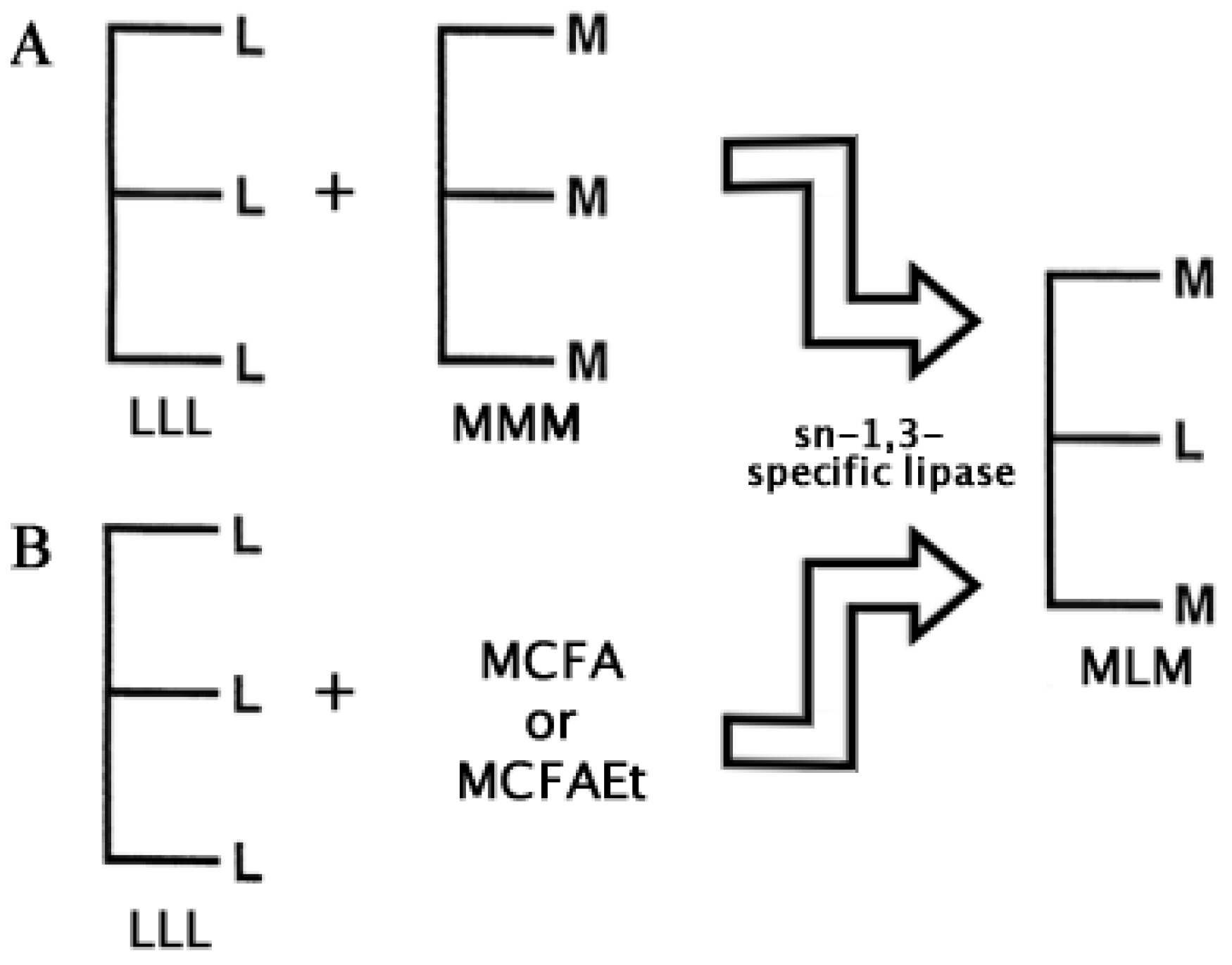
2.2. Structured Lipids
3. Enzymes in the Bulk Chemistry Industry
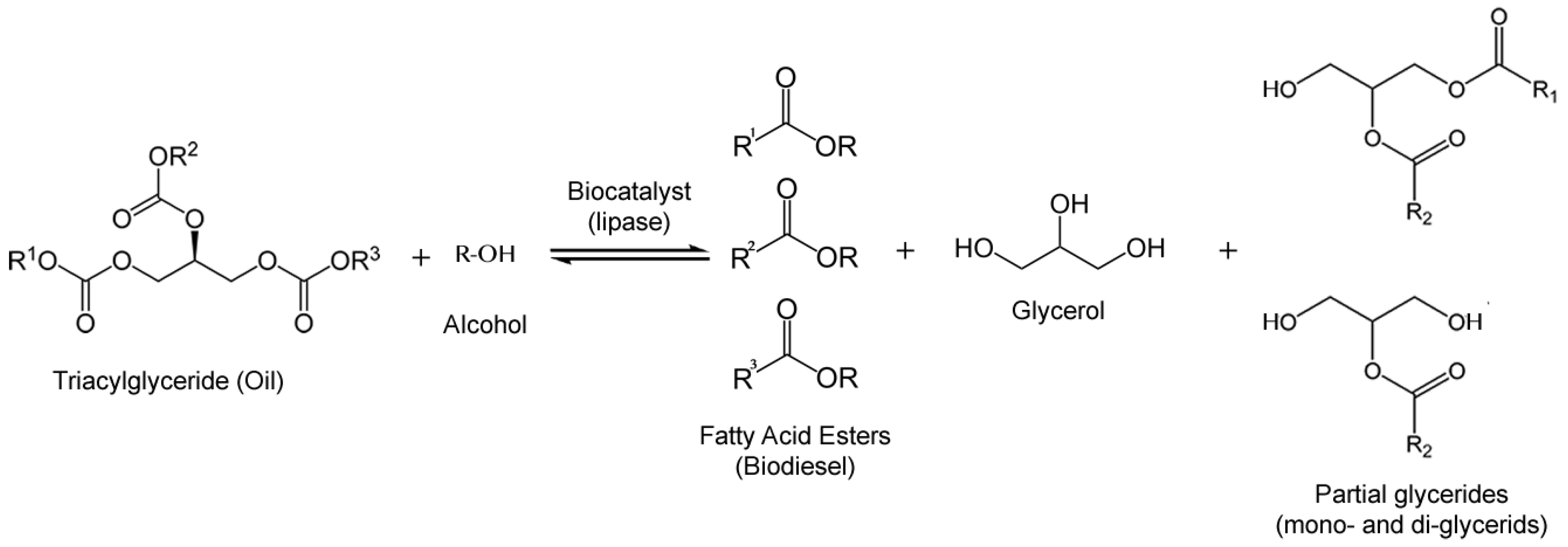
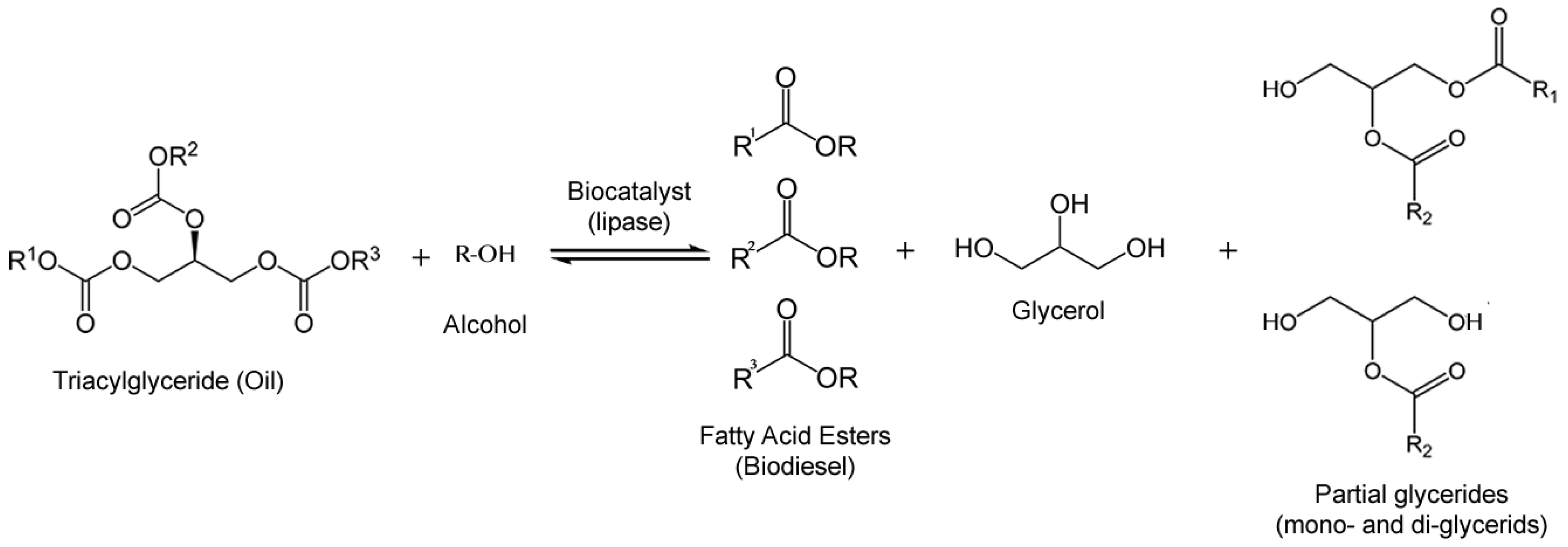
3.1. Biodiesel
3.2. Industrial Polymers
3.2.1. Transferases
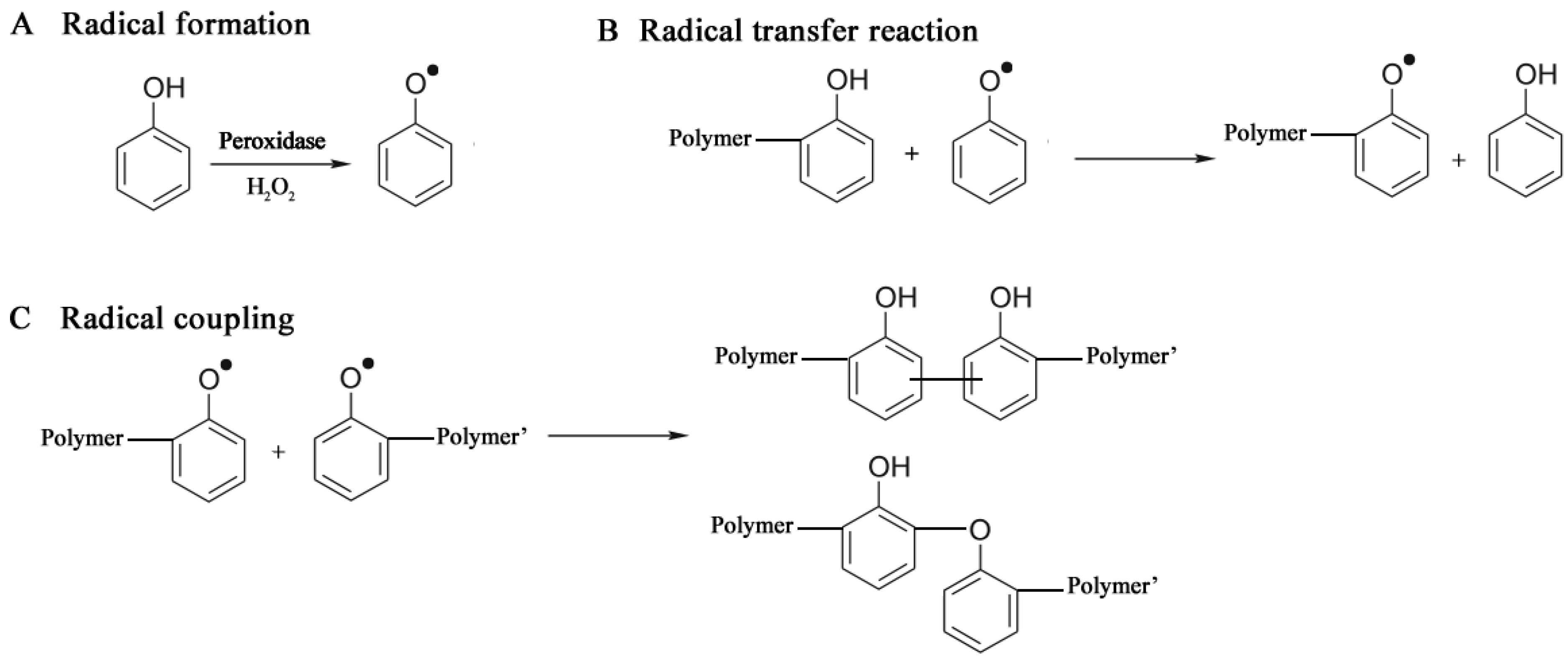
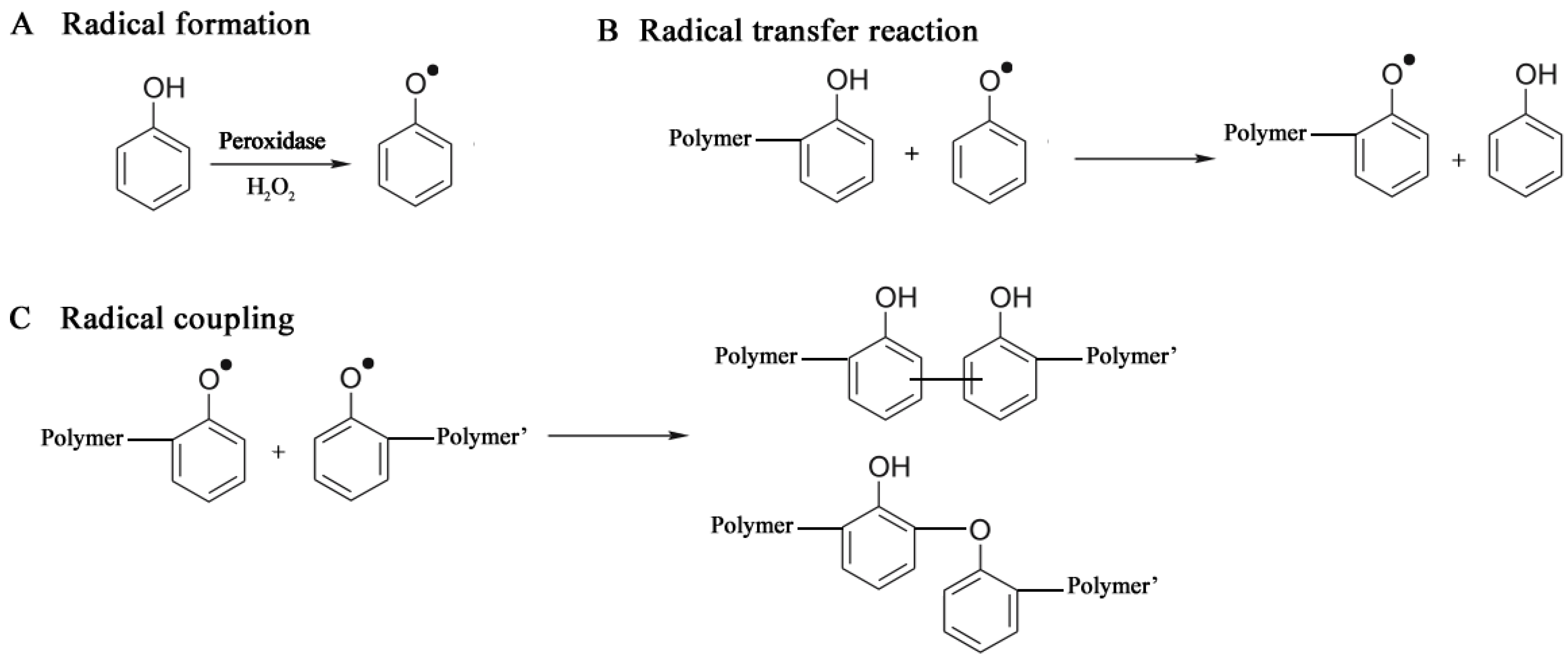
3.2.2. Oxidoreductases

| Enzymes | Polymers synthesized |
|---|---|
| Oxidoreductases | |
| Peroxidases Laccases Tyrosinases Glucose oxidases | Polyphenols, polyanilines, vinyl polymers |
| Transferases | |
| Glycosyl transferases Acyl transferases | Polysaccharides, cyclic oligosaccharides, polyesters |
| Hydrolases | |
| Glycosidases Lipases Peptidases Proteases | Polysaccharides, polyesters, polycarbonates, polyamides, polythioesters, polyphosphates |
3.2.2.1. Polyaromatics
3.2.3. Hydrolases
3.2.4. Polyesters
3.2.5. Polycondensations
3.2.5.1. Lipase-Catalyzed Polycondensations
3.2.5.2. Protease-Catalyzed Polycondensations
3.2.6. Polyamides
3.2.6. Vinyl Polymers
3.2.7. Nylons
3.2.8. Acrylate and Styrene Polymerization
3.2.9. Ring-Opening Polymerizations (ROP)
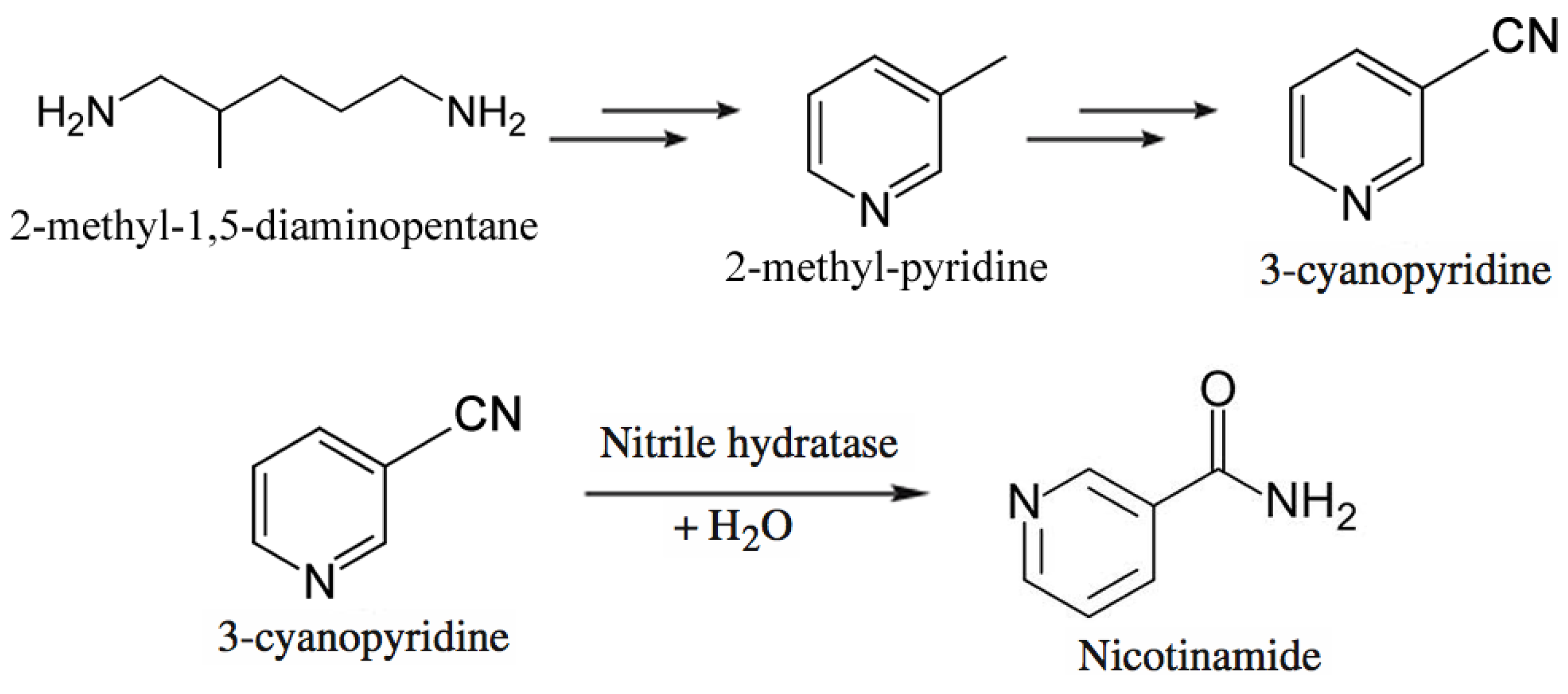
3.3. Commodity Chemicals
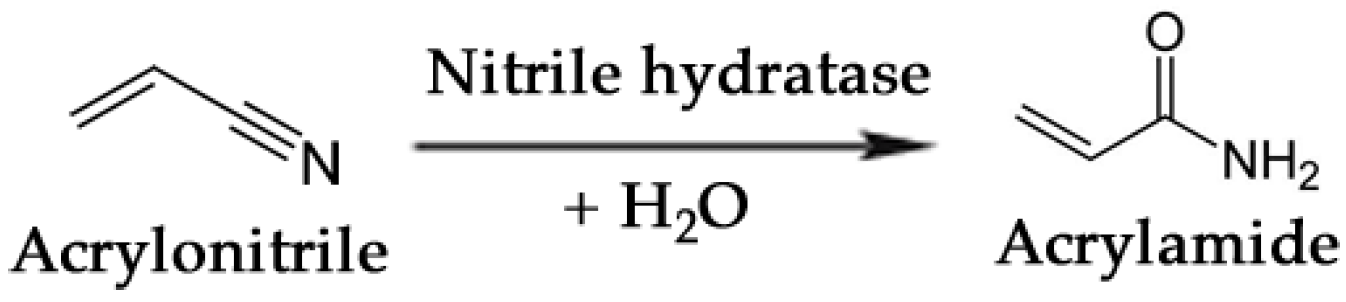
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
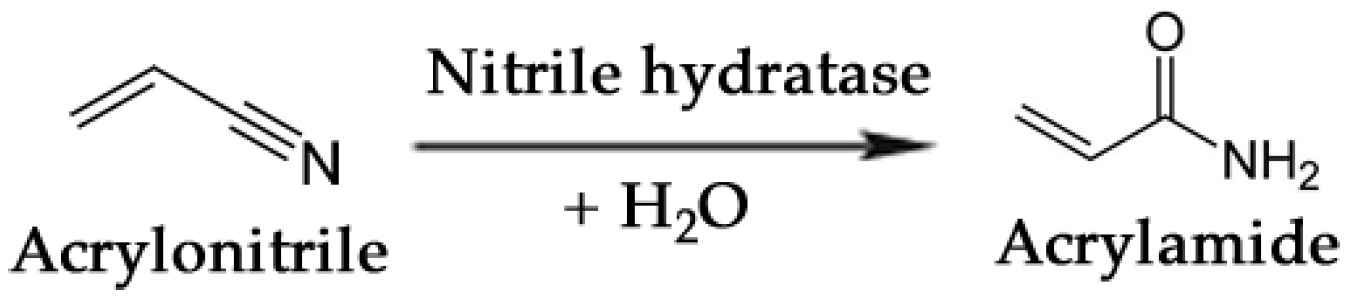{kind=link}
{kind=link}
{kind=link}
4. Enzymes in the Fine Chemicals Industry
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Council of European Union. En Route to the Knowledge-Based Bio-Economy; German Presidency of the Council of European Union: Cologne, Germany, 2007. [Google Scholar]
- Sandoval, G.; Plou, F.G. Obtención enzimática de compuestos bioactivos a partir de recursos naturales iberoamericanos; Consejo Superior de Instigaciones Cientificas (CSIC): Madrid, Spain, 2012; pp. 1–336. [Google Scholar]
- Jahnz, U.; Schubert, M.; Baars, H.; Klaus, V. Process for producing the potential food ingredient DFA III from inulin-screening, genetic engineering, fermentation and immobilisation of inulase II. Int. J. Pharm. 2003, 256, 199–206. [Google Scholar] [CrossRef]
- Uchiyama, T.; Niwa, S.; Tanaka, K. Purification and properties of Arthrobacter ureafaciens inulase II. Biochim. Biophys. Acta 1973, 315, 412–420. [Google Scholar] [CrossRef]
- Kim, G.E.; Lee, S. Efficient production of DFA III (di-d-fructofuranose-dianhydride) from chicory root. In Abstracts of the World Congress on Biotechnology; DECHEMA: Berlin, Germany, 2000; Volume 272. [Google Scholar]
- Yokota, A.; Hirayama, S.; Enomoto, K.; Miura, Y.; Takao, S.; Tomita, F. Production of inulin fructotransferase (depolymerizing) by Arthrobacter sp. H65–7 and preparation of DFA III from inulin by the enzyme. J. Ferm. Bioeng 1991, 72, 258–261. [Google Scholar] [CrossRef]
- Kawamura, M.; Takahashi, S.; Uchiyama, T. Purification and some properties of inulin fructotransferase (depolymerizing) from Arthrobacter ilicis. Agric. Biol. Chem. 1988, 52, 3209–3210. [Google Scholar]
- Gosling, A.; Stevens, G.W.; Barber, A.R.; Kentish, S.E.; Gras, S.L. Recent advances refining galactooligosaccharide production from lactose. Food Chem. 2010, 121, 307–318. [Google Scholar] [CrossRef]
- Rastall, R.A.; Gibson, G.R.; Gill, H.S.; Guarner, F.; Klaenhammer, T.R.; Pot, B.; Reid, G.; Rowland, I.R.; Sanders, M.E. Modulation of the microbial ecology of the human colon by probiotics, prebiotics and synbiotics to enhance human health: An overview of enabling science and potential applications. FEMS Microbiol. Ecol. 2005, 52, 145–152. [Google Scholar] [CrossRef]
- Sanz, J.I. Production of Glactooligo-Sacchariddes from Lactose by Immobilized β-Galactosidase and Posterioir Chromatographic Spearation. Ph.D. Thesis, Ohio State University, Columbus, OH, USA, 2009. [Google Scholar]
- Gullón, B.; Gómez, B.; Martínez-Sabajanesb, M.; Yáñez, R.; Parajó, J.C.; Alonso, J.L. Pectic oligosaccharides: Manufacture and functional properties. Trends Food Sci. Technol. 2013, 30, 153–161. [Google Scholar] [CrossRef]
- Neri, D.F.M.; Balcão, V.M.; Costa, R.S.; Rocha, I.C.A.P.; Ferreira, E.M.F.C.; Torres, D.P.M.; Rodrigues, L.R.M.; Carvalho, L.B., Jr.; Teixeira, J.A. Galacto-oligosaccharides production during lactose hydrolysis by free Aspergillus oryzae β-galactosidase and immobilized on magnetic polysiloxane-polyvinyl alcohol. Food Chem. 2009, 115, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Delattre, C.; Michaud, P. Production of oligosaccharides as promising new food additive generation. Food Technol. Biotechnol. 2006, 44, 323–333. [Google Scholar]
- Gaur, R.; Pant, H.; Jain, R.; Khare, S.K. Galacto-oligosaccharide synthesis by immobilized Aspergillus oryzae β-Galactosidase. Food Chem. 2006, 97, 426–430. [Google Scholar] [CrossRef]
- Whisner, C.M.; Weaver, C.M. Galacto-oligosaccharides: Prebiotic effects on calcium absorption and bone health. In Nutritional Influences on Bone Health; Burckhardt, P., Dawson-Hughes, B., Weaver, C.M., Eds.; Springer: London, UK, 2013; pp. 315–323. [Google Scholar]
- Perugino, G.; Trincone, A.; Rossi, M.; Moracci, M. Oligosaccharide synthesis by glycosynthases. Trends Biotech. 2004, 22, 31–37. [Google Scholar] [CrossRef]
- Dias, L.G.; Veloso, A.C.A.; Correia, D.M.; Rocha, O.; Torres, D.; Rocha, I.; Rodrigues, L.R.; Peres, A.M. UV spectrophotometry method for the monitoring of galacto-oligosaccharides production. Food Chem. 2009, 113, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, B.; Poveda, A.; Jiménez, J.; Ballesteros, A.O.; Plou, F.J. Galacto-oligosaccharide synthesis from lactose solution or skim milk using the β-galactosidase from Bacillus circulans. J. Agric. Food Chem. 2012, 60, 6391–6398. [Google Scholar]
- Torres, D.; Gonçalves, M.D.P.F.; Teixeira, J.A.; Rodrigues, L. Galacto-oligosaccharides: Production, properties, applications, and significance as prebiotics. Compr. Rev. Food Sci. F. 2010, 9, 438–454. [Google Scholar] [CrossRef] [Green Version]
- Tzortzis, G.; Vulevic, J. Galacto-oligosaccharide prebiotics. In Prebiotics and Probiotics Science and Technology; Charalampopoulos, D., Rastall, R.A., Eds.; Springer: New York, NY, USA, 2009; pp. 207–244. [Google Scholar]
- Park, H.Y.; Kim, H.J.; Lee, J.K.; Kim, D.; Oh, D.K. Galactooligosaccharide production by a thermostable β-galactosidase from Sulfolobus solfataricus. World J. Microb. Biot. 2008, 24, 1553–1558. [Google Scholar] [CrossRef]
- Rodriguez, B.; de Abreu, M.A.; Fernandez, L.; de Beer, R.; Poveda, A.; Jimenez, J.; Haltrich, D.; Ballesteros, A.O.; Fernandez, M.; Plou, F.J. Production of galacto-oligosaccharides by the β-galactosidase from Kluyveromyces lactis: Comparative analysis of permeabilized cells versus soluble enzyme. J. Agric. Food Chem. 2011, 59, 10477–10484. [Google Scholar] [CrossRef]
- Hsu, C.A.; Lee, S.L.; Chou, C.C. Enzymatic production of galactooligosaccharides by β-galactosidase from Bifidobacterium longum BCRC 15708. J. Agric. Food Chem. 2007, 55, 2225–2230. [Google Scholar] [CrossRef]
- Hansson, T.; Adlercreutz, P. Optimization of galactooligosaccharide production from lactose using β-glycosidases from hyperthermophiles. Food Biotechnol. 2001, 15, 79–97. [Google Scholar]
- UBIC Europe Marketing Consulting. In The World GOS Market; UBIC Europe Press: Sierre, Switzerland, 2010.
- Padalino, M.; Perez-Conesa, D.; López-Nicolás, R.; Frontela-Saseta, C.; Berruezo, G. Effect of fructooligosaccharides and galactooligosaccharides on the folate production of some folate-producing bacteria in media cultures or milk. Int. Dairy J. 2012, 27, 27–33. [Google Scholar] [CrossRef]
- Fanaro, S.; Boehm, G.; Garssen, J.; Knol, J.; Mosca, F.; Stahl, B.; Vigi, V. Galacto-oligosaccharides and long-chain fructo-oligosaccharides as prebiotics in infant formulas: A review. Acta Paediatr. Suppl. 2005, 94, 22–26. [Google Scholar] [CrossRef]
- Watzl, B.; Girrbach, S.; Roller, M. Inulin, oligofructose and immunomodulation. Br. J. Nutr. 2005, 93, S49–S55. [Google Scholar] [CrossRef]
- Bornet, F.R.; Brouns, F.; Tashiro, Y.; Duvillier, V. Nutritional aspects of short-chain fructooligosaccharides: Natural occurrence, chemistry, physiology and health implications. Dig. Liver Dis. 2002, 34, S111–S120. [Google Scholar] [CrossRef]
- Yun, J.W. Fructooligosaccharides—Occurrence, preparation, and application. Enzyme Microb. Tech. 1996, 19, 107–117. [Google Scholar] [CrossRef]
- Arrizon, J.; Urias-Silvas, J.E.; Sandoval, G.; Mancilla-Margalli, N.A.; Gschaedler, A.C.; Morel, S.; Monsan, P. Production and bioactivity of fructan-type oligosaccharides. In Food Oligosaccharides: Production, Analysis and Bioactivity; Moreno, F.J., Sanz, M.L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; in press. [Google Scholar]
- Beine, R.; Morarua, R.; Nimtz, M.; Na’amniehc, S.; Pawlowskic, A.; Buchholz, K.; Seibel, J. Synthesis of novel fructooligosaccharides by substrate and enzyme engineering. J. Biotechnol. 2008, 138, 33–41. [Google Scholar] [CrossRef]
- Castillo, E.; López-Munguía, A. Synthesis of levan in water-miscible organic solvents. J. Biotechnol. 2004, 114, 209–217. [Google Scholar] [CrossRef]
- Vega, R.J.; Zúniga, M.E. Potential application of commercial enzyme preparations for industrial production of short-chain fructooligosaccharides. J. Mol. Catal. B Enzym. 2012, 76, 44–51. [Google Scholar] [CrossRef]
- Singh, R.S.; Singh, R.P. Production of fructooligosaccharides from inulin by endoinulinases and their prebiotic potential. Food Technol. Biotech. 2010, 48, 435–450. [Google Scholar]
- Tanriseven, A.; Aslan, Y. Immobilization of Pectinex Ultra SP-L to produce fructooligosaccharides. Enzyme Microb. Technol. 2005, 36, 550–554. [Google Scholar] [CrossRef]
- Guío, F.; Rodríguez, M.A.; Alméciga, C.J.; Sánchez, O.F. Recent trends in fructooligosaccharides production. Recent Pat. Food Nutr. Agric. 2009, 1, 221–230. [Google Scholar] [CrossRef]
- Jala, R.C.R.; Hu, P.; Yang, T.; Jiang, Y.; Zheng, Y; Xu, X. Lipases as biocatalysts for the synthesis of structured lipids. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, USA, 2012; Volume 861, Chapter 23. [Google Scholar]
- Xu, X.; Akoh, C.C. Enzymatic production of Betapol and other specialty fats. In Lipid Biotechnology; Marcel Dekker: New York, NY, USA, 2002; pp. 461–478. [Google Scholar]
- Odle, J. New insights into the utilization of medium-chain triglycerides by the neonate: Observations from a piglet model. J. Nutr. 1997, 127, 1061–1067. [Google Scholar]
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1987, 86, 439–472. [Google Scholar]
- Osborn, H.T.; Akoh, C. Structured lipids: Novel fats with medical, nutraceutical, and food applications. Compr. Rev. Food Sci. F. 2002, 1, 110–120. [Google Scholar] [CrossRef]
- Xu, X.; Hoy, C.E.; Balchen, S.; Adler-Nissen, J. Specific-Structured Lipid: Nutritional Perspectives and Production Potentials. In Proceedings of International Symposium on the Approach to Functional Cereals and Oils, CCOA, Beijing, China, 9–14 November 1997.
- Trivedi, R.; Singh, R.P. Modification of oils and fats to produce structured lipids. J. Oleo Sci. 2005, 54, 423–430. [Google Scholar] [CrossRef]
- Hellner, G.; Tőke, E.R.; Nagy, V.; Szakács, G.; Poppe, L. Integrated enzymatic production of specific structured lipid and phytosterol ester compositions. Process Biochem. 2010, 45, 1245–1250. [Google Scholar] [CrossRef]
- Lee, K.T.; Akoh, C.C. Characterization of enzymatically synthesized structured lipids containing eicosapentaenoic, docosahexaenoic, and caprylic acids. J. Am. Oil Chem. Soc. 1998, 75, 495–499. [Google Scholar] [CrossRef]
- Akoh, C.C. Structured lipids. In Food Lipids Chemistry, Nutrition, and Biotechnology; Akoh, C.C., Min, D.B., Eds.; Marcel Dekker: New York, NY, USA, 1998; pp. 699–727. [Google Scholar]
- Iwasaki, Y.; Yamane, T. Enzymatic synthesis of structured lipids. J. Mol. Catal. B Enzym. 2000, 10, 129–140. [Google Scholar] [CrossRef]
- Khodadadi, M.; Aziz, S.; St-Louis, R.; Kermash, S. Lipase-catalyzed synthesis and characterization of flaxseed oil-based structured lipids. J. Funct. Foods 2013, 5, 424–433. [Google Scholar] [CrossRef]
- Casas-Godoy, L.; Marty, A.; Sandoval, G.; Ferreira-Dias, S. Optimization of medium chain length fatty acid incorporation into olive oil catalyzed by immobilized Lip2 from Yarrowia lipolytica. Biochem. Eng. J. 2013, 77, 20–27. [Google Scholar] [CrossRef]
- Villeneuve, P.; Barouh, N.; Baréa, B.; Piombo, G.; Figueroa-Espinoza, M.C.; Turon, M.C.; Pina, M.; Lago, R. Chemoenzymatic synthesis of structured triacylglycerols with conjugated linoleic acids (CLA) in central position. Food Chem. 2007, 100, 1443–1452. [Google Scholar] [CrossRef]
- Nagachinta, S.; Akoh, C.C. Enrichment of palm olein with long chain polyunsaturated fatty acids by enzymatic acidolysis. LWT Food Sci. Technol. 2012, 46, 29–35. [Google Scholar] [CrossRef]
- Tecelão, C.; Rivera, I.; Sandoval, G.; Ferreira-Dias, S. Carica papaya latex: A low-cost biocatalyst for human milk fat substitutes production. Eur. J. Lipid Sci. Technol. 2012, 114, 266–276. [Google Scholar] [CrossRef]
- Nagao, T.; Watanabe, Y.; Maruyama, M.; Momokawa, Y.; Kishimoto, N.; Shimada, Y. One-pot enzymatic synthesis of docosahexaenoic acid-rich triacylglycerols at the sn-1(3) position using by-product from selective hydrolysis of tuna oil. New Biotechnol. 2011, 28, 7–13. [Google Scholar]
- Buchholtz, K.; Volker, K.; Borscheuer, U.T. Biocatalysts and Enzyme Technology; Wiley-VCH: Berlin, Germany, 2005; pp. 1–465. [Google Scholar]
- Bornscheuer, U.T.; Kazlauskas, R.J. Hydrolases in Organic Synthesis—Regio- and Stereoselective Biotransformations; Wiley-VCH: Berlin, Germany, 1999; pp. 1–355. [Google Scholar]
- Cao, L. Carrier-Bound Immobilized Enzymes; Wiley-VCH: Berlin, Germany, 2005; pp. 1–578. [Google Scholar]
- Gog, A.; Roman, M.; Toşa, M.; Paizs, C.; Irimie, F.D. Biodiesel production using enzymatic transesterification—Current state and perspectives. Renew. Energ. 2012, 39, 10–16. [Google Scholar] [CrossRef]
- Véras, I.C.; Silva, F.A.; Ferrão-Gonzales, A.D.; Moreau, V.H. One-step enzymatic production of fatty acid ethyl ester from high-acidity waste feedstocks in solvent-free media. Bioresour. Technol. 2011, 102, 9653–9658. [Google Scholar] [CrossRef]
- Fan, X.; Niehus, X.; Sandoval, G. Lipases as biocatalyst for biodiesel production. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, NY, USA, 2012; Volume 861, Chapter 27. [Google Scholar]
- Fukuda, H.; Kondo, A.; Noda, H. Biodiesel fuel production by transesterification of oils. J. Biosci. Bioeng. 2001, 92, 405–416. [Google Scholar]
- Maleki, E.; Aroua, M.K.; Sulaiman, N.M.N. Improved yield of solvent free enzymatic methanolysis of palm and jatropha oils blended with castor oil. Appl. Energ. 2013, 104, 905–909. [Google Scholar] [CrossRef]
- Hama, S.; Kondo, A. Enzymatic biodiesel production: An overview of potential feedstocks and process development. Bioresour. Technol. 2013, 135, 386–395. [Google Scholar] [CrossRef]
- Al-Zuhair, S.; Hasan, M.; Ramachandran, K.B. Kinetics of the enzymatic hydrolysis of palm oil by lipase. Process Biochem. 2003, 38, 1155–1163. [Google Scholar]
- Salis, A.; Pinna, M.; Monduzzi, M.; Solinas, V. Biodiesel production from triolein and short chain alcohols through biocatalysis. J. Biotechnol. 2005, 119, 291–299. [Google Scholar]
- Shimada, Y.; Watanabe, Y.; Samukawa, T.; Sugihara, A.; Noda, H.; Fukuda, H.; Tominaga, Y. Conversion of vegetable oil to biodiesel using immobilized Candida antarctica lipase. J. Am. Oil Chem. Soc. 1999, 76, 789–793. [Google Scholar]
- Li, Q.; Xu, J.; Du, W.; Li, Y.; Liu, D. Ethanol as the acyl acceptor for biodiesel production. Renew. Sust. Energ. Rev. 2013, 25, 742–748. [Google Scholar]
- Modi, M.K.; Reddy, J.R.; Rao, B.V.; Prasad, R.B. Lipase mediated conversion of vegetable oils into biodiesel using ethyl acetate as acyl acceptor. Bioresour. Technol. 2007, 98, 1260–1264. [Google Scholar]
- Rivera, I.; Villanueva, G.; Sandoval, G. Biodiesel production from animal grease wastes by enzymatic catalysis. Grasas Aceites 2009, 60, 468–474. [Google Scholar]
- Royon, D.; Daz, M.; Ellenrieder, G.; Locatelli, S. Enzymatic production of biodiesel from cotton seed oil using t-butanol as a solvent. Bioresour. Technol. 2007, 98, 648–653. [Google Scholar]
- Abigor, R.D.; Uadia, P.O.; Foglia, T.A.; Haas, M.J.; Jones, K.C.; Okpefa, E.; Obibuzor, J.U.; Bafor, M.E. Lipase-catalysed production of biodiesel fuel from some Nigerian lauric oils. Biochem. Soc. Trans. 2000, 28, 979–981. [Google Scholar]
- Nelson, L.; Foglia, T.; Marmer, W.N. Lipase-catalyzed production of biodiesel. J. Am. Oil Chem. Soc. 1996, 73, 1191–1195. [Google Scholar] [CrossRef]
- Linko, Y.Y.; Lamsä, M.; Huhtala, A.; Rantanen, O. Lipase biocatalysis in the production of esters. J. Am. Oil Chem. Soc. 1995, 72, 1293–1299. [Google Scholar]
- Du, W.; Xu, Y.; Liu, D.; Zeng, J. Comparative study on lipase-catalyzed transformation of soybean oil for biodiesel production with different acyl acceptors. J. Mol. Catal. B Enzym. 2004, 30, 125–129. [Google Scholar] [CrossRef]
- Watanabe, Y.; Shimada, Y.; Sugihara, A.; Noda, H.; Fukuda, H.; Tominaga, Y. Continuous production of biodiesel fuel from vegetable oil using immobilized Candida antarctica lipase. J.Am. Oil Chem. Soc. 2000, 77, 355–360. [Google Scholar]
- Zhang, B.; Weng, Y.; Xu, H.; Mao, Z. Enzyme immobilization for biodiesel production. Appl. Microbiol. Biotechnol. 2012, 93, 61–70. [Google Scholar] [CrossRef]
- Soumanou, M.M.; Bornscheuer, U.T. Lipase-catalyzed alcoholysis of vegetable oils. Eur. J. Lipid Sci. Technol. 2003, 105, 656–660. [Google Scholar]
- Zhu, X.; Zhou, T.; Wu, X.; Cai, Y.; Yao, D.; Xie, C.; Liu, D. Covalent immobilization of enzymes within micro-aqueous organic media. J. Mol. Catal. B Enzym 2011, 3–4, 145–149. [Google Scholar]
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 1985, 82, 3192–3196. [Google Scholar] [CrossRef]
- Iso, M.; Chen, B.; Eguchi, M.; Kudo, T.; Shrestha, S. Production of biodiesel fuel from triglycerides and alcohol using immobilized lipase. J. Mol. Catal. B Enzym. 2001, 16, 53–58. [Google Scholar] [CrossRef]
- Chen, J.W.; Wu, W.T. Regeneration of immobilized Candida antarctica lipase for transesterification. J. Biosci. Bioeng. 2003, 95, 466–469. [Google Scholar]
- Nie, K.; Xie, F.; Wang, F.; Tan, T. Lipase catalyzed methanolysis to produce biodiesel: optimization of the biodiesel production. J. Mol. Catal. B Enzym. 2006, 43, 142–147. [Google Scholar] [CrossRef]
- Ciftci, O.N.; Temelli, F. Enzymatic conversion of corn oil into biodiesel in a batch supercritical carbon dioxide reactor and kinetic modeling. J. Supercrit. Fluids. 2013, 75, 172–180. [Google Scholar] [CrossRef]
- Lin, Y.C.; Yang, P.M.; Chen, S.C.; Lin, J.F. Improving biodiesel yields from waste cooking oil using ionic liquids as catalysts with a microwave heating system. Fuel Process. Technol. 2013, 115, 57–62. [Google Scholar] [CrossRef]
- Earle, M.J.; Plechkova, N.V.; Seddon, K.R. Green synthesis of biodiesel using ionic liquids. Pure Appl. Chem. 2009, 81, 2045–2057. [Google Scholar]
- Ha, S.H.; Lan, M.N.; Lee, S.H.; Hwang, S.M.; Koo, Y.M. Lipase-catalyzed biodiesel production from soybean oil in ionic liquids. Enzyme Microb. Technol. 2007, 41, 480–483. [Google Scholar]
- Koda, R.; Numata, T.; Hama, S.; Tamalampudi, S.; Nakashima, K.; Tanaka, T; Ogino, C.; Fukuda, H.; Kondo, A. Ethanolysis of rapeseed oil to produce biodiesel fuel catalyzed by Fusarium heterosporum lipase-expressing fungus immobilized whole-cell biocatalysts. J. Mol. Catal. B Enzym. 2010, 68, 101–104. [Google Scholar]
- Adachi, D.; Koha, F.; Hamab, S.; Ogino, C.; Kondo, A. A robust whole-cell biocatalyst that introduces a thermo- and solvent-tolerant lipase into Aspergillus oryzae cells: Characterization and application to enzymatic biodiesel production. Enzyme Microb. Technol. 2013, 52, 331–335. [Google Scholar] [CrossRef]
- Fukuda, H.; Hama, S.; Tamalampudi, S.; Noda, H. Whole-cell biocatalysts for biodiesel fuel production. Trends Biotechnol. 2008, 26, 668–673. [Google Scholar] [CrossRef]
- Atkinson, B.; Black, G.M.; Lewis, P.J.S.; Pinches, A. Biological particles of given size, shape, and density for use in biological reactors. Biotechnol. Bioeng. 1979, 21, 193–200. [Google Scholar]
- Meng, X.; Yang, J.; Xu, X.; Zhang, L.; Nie, Q.; Xian, M. Biodiesel production from oleaginous microorganisms. Renew. Energ. 2009, 34, 1–5. [Google Scholar] [CrossRef]
- Turner, N.J.; Truppo, M.D. Biocatalysis enters a new era. Curr. Opin. Chem. Biol. 2013, 17, 212–214. [Google Scholar] [CrossRef]
- Woodley, J. New opportunities for biocatalysis: Making pharmaceutical processes greener. Trends Biotechnol. 2008, 26, 321–327. [Google Scholar] [CrossRef]
- Kittl, R.; Withers, S.G. New approaches to enzymatic glycoside synthesis through directed evolution. Carbohyd. Res. 2010, 345, 1272–1279. [Google Scholar] [CrossRef]
- Hay, A.S. Polymerization by oxidative coupling: Discovery and commercialization of PPO®. J. Polym. Sci. Pol. Chem. 1998, 36, 505–517. [Google Scholar] [CrossRef]
- He, F.; Li, S.; Garreau, H.; Vert, M.; Zhuo, R. Enzyme-catalyzed polymerization and degradation of copolyesters of ε-caprolactone and γ-butyrolactone. Polymer 2005, 46, 12682–12688. [Google Scholar] [CrossRef]
- Salvachúa, D.; Prieto, A.; Mattinen, M.L.; Tamminen, T.; Liitiä, T.; Lille, M.; Willför, S.; Martínez, A.T.; Martínez, M.J.; Faulds, C.B. Versatile peroxidase as a valuable tool for generating new biomolecules by homogeneous and heterogeneous cross-linking. Enzyme Microb. Technol. 2013, 52, 303–311. [Google Scholar] [CrossRef]
- Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Enzymatic synthesis of soluble polyphenol. Bull. Chem. Soc. Jpn. 2000, 73, 1389–1396. [Google Scholar] [CrossRef]
- Dordick, J.S.; Marletta, M.A.; Klibanov, A.M. Polymerization of phenols catalyzed by peroxidase in nonaqueous media. Biotech. Bioeng. 1987, 30, 31–36. [Google Scholar]
- Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Soluble polyphenol. Macromol. Rapid Commun. 1999, 20, 401–403. [Google Scholar] [CrossRef]
- Mita, N.; Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Control of structure and molecular weight of polyphenols in enzymatic oxidative polymerization. Polymer Prepr. 2000, 41, 223–224. [Google Scholar]
- Kurioka, H.; Komatsu, I.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of alkylphenols. Macromol. Rapid Commun. 1994, 15, 507–510. [Google Scholar] [CrossRef]
- Uyama, H.; Kurioka, H.; Kobayashi, S. Preparation of polyphenol particles by dispersion polymerization using enzyme as catalyst. Chem. Lett. 1995, 24, 795–796. [Google Scholar]
- Uyama, H. Enzymatic polymerization. In Future Directions in Biocatalysis; Matsuda, T., Ed.; Elsevier Science: Cambridge, MA, USA, 2007; pp. 1–2. [Google Scholar]
- Reihmann, M.; Ritter, H. Synthesis of phenol polymers using peroxidases. Adv. Polym. Sci. 2006, 194, 1–49. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Ikeda, R. Artificial Urushi. Chem. Eur. J. 2001, 7, 4755–4760. [Google Scholar]
- Kim, Y.H.; An, E.S.; Song, B.K.; Kim, D.S.; Chelikani, R. Polymerization of cardanol using soybean peroxidase and its potential application as anti-biofilm coating material. Biotechnol. Lett. 2003, 25, 1521–1524. [Google Scholar] [CrossRef]
- Tonami, H.; Uyama, H.; Kobayashi, S.; Kubota, M. Peroxidase-catalyzed oxidative polymerization of m-substituted phenol derivatives. Macromol. Chem. Phys. 1999, 200, 2365–2371. [Google Scholar] [CrossRef]
- Kadota, J.; Fukuoka, T.; Uyama, H.; Hasegawa, K.; Kobayashi, S. New Positive-type photoresists based on enzymatically synthesized polyphenols. Macromol. Rapid Commun. 2004, 25, 441–444. [Google Scholar] [CrossRef]
- Antoniotti, S.; Santhanam, L.; Ahuja, D.; Hogg, M.G.; Dordick, J.S. Structural diversity of peroxidase-catalyzed oxidation products of o-methoxyphenols. Org. Lett. 2004, 6, 1975–1978. [Google Scholar] [CrossRef]
- Mita, N.; Tawaki, S.I.; Uyama, H.; Kobayashi, S. Laccase-catalyzed oxidative polymerization of phenols. Macromol. Biosci. 2003, 3, 253–257. [Google Scholar] [CrossRef]
- Ikeda, R.; Sugihara, J.; Uyama, H.; Kobayashi, S. Poly(2,6-dihydroxy-l,4-oxyphenylene synthesis of a new poly(phenylene oxide) derivative. Polym. Bull. 1997, 38, 273–277. [Google Scholar] [CrossRef]
- Ikeda, R.; Sugihara, J.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of 2,6-dimethylphenol. Macromolecules 1996, 29, 8702–8705. [Google Scholar] [CrossRef]
- Uyama, H.; Kurioka, H.; Sugihara, J.; Komatsu, I.; Kobayashi, S. Oxidative polymerization of p-alkylphenols catalyzed by horseradish peroxidase. J. Polym. Sci. Pol. Chem. 1997, 35, 1453–1459. [Google Scholar] [CrossRef]
- Wang, P.; Martin, B.D.; Parida, S.; Rethwisch, D.G.; Dordick, J.S. Multienzymic synthesis of poly(hydroquinone) for use as a redox polymer. J. Am. Chem. Soc. 1995, 117, 12885–12886. [Google Scholar] [CrossRef]
- Akkara, J.A.; Senecal, K.J.; Kaplan, D.L. Synthesis and characterization of polymers produced by horseradish peroxidase in dioxane. J. Polym. Sci. Pol. Chem. 1991, 29, 1561–1574. [Google Scholar] [CrossRef]
- Akita, M.; Tsutsumi, D.; Kobayashi, M.; Kise, H. Structural change and catalytic activity of horseradish peroxidase in oxidative polymerization of phenol. Biosci. Biotech. Bioch. 2001, 65, 1581–1588. [Google Scholar] [CrossRef]
- Angerer, P.S.; Studer, A.; Witholt, B.; Li, Z. Oxidative polymerization of a substituted phenol with ion-paired horseradish peroxidase in an organic solvent. Macromolecules 2005, 38, 6248–6250. [Google Scholar] [CrossRef]
- Eker, B.; Zagorevski, D.; Zhu, G.; Linhardt, R.J.; Dordick, J.S. Enzymatic polymerization of phenols in room-temperature ionic liquids. J. Mol. Catal. B Enzym. 2009, 59, 177–184. [Google Scholar] [CrossRef]
- Mita, N.; Tawaki, S.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of phenol in an aqueous solution in the presence of a catalytic amount of cyclodextrin. Macromol. Biosci. 2002, 3, 127–130. [Google Scholar]
- Kim, Y.J.; Uyama, H.; Kobayashi, S. Regioselective synthesis of poly(phenylene) as a complex with poly(ethylene glycol) by template polymerization of phenol in water. Macromolecules 2003, 36, 5058–5060. [Google Scholar] [CrossRef]
- Kommareddi, N.S.; Tata, M.; Karayigitoglu, C.; John, V.T.; McPherson, G.L.; Herman, M.F.; Oconnor, C.J.; Lee, Y.S.; Akkara, J.A.; Kaplan, D.L. Enzymatic polymerizations using surfactant microstructures and the preparation of polymer-ferrite composites. Appl. Biochem. Biotechnol. 1995, 51–52, 241–252. [Google Scholar] [CrossRef]
- Ghan, R.; Shutava, T.; Patel, A.; John, V.T.; Lvov, Y. Enzyme-catalyzed polymerization of phenols within polyelectrolyte microcapsules. Macromolecules 2004, 37, 4519–4524. [Google Scholar]
- Marín, F.R.; Frutos, M.J.; Pérez-Alvarez, J.A.; Martinez-Sánchez, F.; del Río, J.A. Flavonoids as nutraceuticals: Structural related antioxidant properties and their role on ascorbic acid preservation. Stud. Nat. Prod. Chem. 2002, 26, 741–778. [Google Scholar] [CrossRef]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Flavonoids in Biology and Medicine III—Current Issues in Flavonoids Research; Das, N.P.; Cheeseman, K.H. (Eds.) Informa Healthcare: London, UK, 1991; Volume 14, pp. 77–78.
- Mejias, L.; Reihmann, M.H.; Sepulveda-Boza, S.; Ritter, H. New polymers from natural phenols using horseradish or soybean peroxidase. Macromol. Biosci. 2002, 2, 24–32. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Laccase-catalyzed synthesis and antioxidant property of poly(catechin). Macromol. Biosci. 2003, 3, 758–764. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Oxidative coupling of epigallocatechin gallate amplifies antioxidant activity and inhibits xanthine oxidase activity. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Kim, Y.J.; Uyama, H.; Kobayashi, S. Amplification of antioxidant activity and xanthine oxidase inhibition of catechin by enzymatic polymerization. Biomacromolecules 2003, 4, 469–471. [Google Scholar] [CrossRef]
- Gonçalves, I.; Matamá, T.; Cavaco-Paulo, A.; Silva, C. Laccase coating of catheters with poly(catechin) for biofilm reduction. Biocatal. Biotransform 2013, in press. [Google Scholar]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and antioxidant properties of poly(rutin). Biomacromolecules 2003, 4, 1394–1399. [Google Scholar] [CrossRef]
- Božič, M.; Gorgieva, S.; Kokol, V. Laccase-mediated functionalization of chitosan by caffeic and gallic acids for modulating antioxidant and antimicrobial properties. Carbohyd. Polym. 2012, 87, 2388–2398. [Google Scholar] [CrossRef]
- Brzonova, I.; Steiner, W.; Zankel, A.; Nyanhongo, G.S. Enzymatic synthesis of catechol and hydroxyl-carboxic acid functionalized chitosan microspheres for iron overload therapy. Eur. J. Pharm. Biopharm. 2011, 79, 294–303. [Google Scholar] [CrossRef]
- Fras-Zemljič, L.; Kokol, V.; Čakara, D. Antimicrobial and antioxidant properties of chitosan-based viscose fibres enzymatically functionalized with flavonoids. Text. Res. J. 2011, 81, 1532–1540. [Google Scholar] [CrossRef]
- Sousa, F.; Guebitz, G.M.; Kokol, V. Antimicrobial and antioxidant properties of chitosan enzymatically functionalized with flavonoids. Process Biochem. 2009, 44, 749–756. [Google Scholar] [CrossRef]
- Pina-Luis, G.; Rosquete-Pina, G.; Valdés, A.C.; Ochoa, A.; Rivero, I.; Díaz-García, M.E. Morin functionalized Merrifield’s resin: A new material for enrichment and sensing heavy metals. React. Funct. Polym. 2012, 72, 61–68. [Google Scholar] [CrossRef]
- Donato, L.; Chiappetta, G.; Drioli, E. Naringin-imprinted polymer layer using photo polymerization method. Sep. Sci. Technol. 2011, 46, 1555–1562. [Google Scholar] [CrossRef]
- Spizzirri, U.G.; Parisi, O.I.; Iemma, F.; Cirillo, G.; Puoci, F.; Curcio, M.; Picci, N. Antioxidant-polysaccharide conjugates for food application by eco-friendly grafting procedure. Carbohyd. Polym. 2010, 79, 333–334. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar]
- Sharma, R.; Chisti, Y.; Banerjee, U.C. Production, purification, characterization and applications of lipases. Biotechnol. Adv. 2001, 19, 627–662. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.; Liu, C.; Zhao, Z.; Yang, Y.; Li, Q. Lipase/esterase-catalyzed synthesis of aliphatic polyesters via polycondensation: A review. Process Biochem. 2012, 47, 1027–1036. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, Y.; Zhang, Y.; Liu, C.; Shi, W.; Li, Q. Lipase/esterase-catalyzed ring-opening polymerization: A green polyester synthesis technique. Process Biochem. 2011, 46, 1900–1908. [Google Scholar] [CrossRef]
- He, F.; Wang, Y.P.; Liu, G.; Jia, H.L.; Feng, J.; Zhuo, R.X. Synthesis, characterization and ring-opening polymerization of a novel six-membered cyclic carbonate bearing pendent allyl ether group. Polymer 2008, 49, 1185–1190. [Google Scholar] [CrossRef]
- Runge, M.; O’Hagan, D.; Haufe, G. Lipase-catalyzed polymerization of fluorinated lactones and fluorinated hydroxycarboxylic acids. J. Polym. Sci. Pol. Chem. 2000, 38, 2004–2012. [Google Scholar] [CrossRef]
- Zinck, P. One-step synthesis of polyesters specialties for biomedical applications. Rev. Environ. Sci. Biotechnol. 2009, 8, 231–234. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Uyama, H.; Takamoto, T.; Kobayashi, S. Enzymatic synthesis of polyesters in ionic liquids. Polym. J. 2002, 34, 94–96. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Beckman, E.J.; Russell, A.J. Rational control of polymer molecular weight and dispersity during enzyme-catalyzed polyester synthesis in supercritical fluids. J. Am. Chem. Soc. 1995, 117, 3728–3733. [Google Scholar] [CrossRef]
- Okumura, S.; Iwai, M.; Tominaga, T. Synthesis of ester oligomer by Aspergillus niger lipase. Agr. Biol. Chem. (Tokyo) 1984, 48, 2805–2813. [Google Scholar] [CrossRef]
- Binns, F.; Roberts, S.M.; Taylor, A.; Williams, C.F. Enzymic polymerization of unactivated diol/diacid system. J. Chem. Soc. Perkin Trans. 1 1993, 1, 899–904. [Google Scholar]
- Linko, Y.Y.; Seppala, J. Producing high molecular weight biodegradable polyesters. Chem. Tech. 1996, 26, 25–31. [Google Scholar]
- Gómez-Patiño, M.B.; Cassani, J.; Jaramillo-Flores, M.E.; Zepeda-Vallejo, L.G.; Sandoval, G.; Jimenez-Estrada, M.; Arrieta-Baez, D. Oligomerization of 10,16-dihydroxyhexadecanoic acid and methyl 10,16-dihydroxyhexadecanoate catalyzed by lipases. Molecules 2013, 18, 9317–9333. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Suda, S.; Namekawa, S. Dehydration polymerization in aqueous medium catalyzed by lipase. Chem. Lett. 1997, 26, 105–107. [Google Scholar] [CrossRef]
- Poojari, Y.; Palsule, A.S.; Cai, M.; Clarson, S.J.; Gross, R.A. Synthesis of organosiloxane copolymers using enzymatic polyesterification. Eur. Polym. J. 2008, 44, 4139–4145. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, W.; Cai, J.; Hou, Y.; Ouyang, S.; Xie, W.; Gross, R.A. Poly(oleicdiacid-co-glycerol): Comparison of polymer structure resulting from chemical and lipase catalysis. Macromolecules 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Korupp, C.; Weberskirch, R.; Muller, J.J.; Liese, A.; Hilterhaus, L. Scaleup of lipase-catalyzed polyester synthesis. Org. Process. Res. Dev. 2010, 14, 1118–1124. [Google Scholar] [CrossRef]
- Fehling, E.; Bergander, K.; Klein, E.; Weber, N.; Vosmann, K. Thiol-functionalized copolymeric polyesters by lipase-catalyzed esterification and transesterification of 1,12-dodecanedioic acid and its diethyl ester, respectively, with 1-thioglycerol. Biotechnol. Lett. 2010, 32, 1463–1471. [Google Scholar] [CrossRef]
- Hu, J.; Gao, W.; Kulshrestha, A.S.; Gross, R.A. Sweet polyesters: Lipase-catalyzed condensation polymerization of alditols. Macromolecules 2006, 39, 6789–6792. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. Enzymatic synthesis of polyesters via polycondensation. Adv. Polym. Sci. 2006, 194, 133–158. [Google Scholar] [CrossRef]
- Brazwell, E.M.; Filos, D.Y.; Morrow, C.J. Biocatalytic synthesis of polymers. III. Formation of a high molecular weight polyester through limitation of hydrolysis by enzyme-bound water and through equilibrium control. J. Polym. Sci. Pol. Chem. 1995, 33, 89–95. [Google Scholar] [CrossRef]
- Kobayashi, S. Recent developments in lipase-catalyzed synthesis of polyesters. Macromol. Rapid Commun. 2009, 30, 237–266. [Google Scholar] [CrossRef]
- Azim, H.; Dekhterman, A.; Jiang, Z.; Gross, R.A. Candida antarctica lipase B-catalyzed synthesis of poly(butylene succinate): Shorter chain building blocks also work. Biomacromolecules 2006, 7, 3093–3097. [Google Scholar] [CrossRef]
- Mesiano, A.J.; Beckman, E.J.; Russell, A.J. Biocatalytic synthesis of fluorinated polyesters. Biotechnol. Prog. 2000, 16, 64–68. [Google Scholar] [CrossRef]
- Uyama, H.; Yaguchi, S.; Kobayashi, S. Lipase-catalyzed polycondensation of dicarboxylic acid-divinyl esters and glycols to aliphatic polyesters. J. Polym. Sci. Pol. Chem. 1999, 37, 2737–2745. [Google Scholar]
- Yao, D.; Li, G.; Kuila, T.; Li, P.; Kim, N.H.; Kim, S.I.; Lee, J.H. Lipase-catalyzed synthesis and characterization of biodegradable polyester containing l-malic acid unit in solvent system. J. Appl. Polym. Sci. 2011, 120, 1114–1120. [Google Scholar] [CrossRef]
- Steunenberg, P.; Uiterweerd, M.; Sijm, M.; Scott, E.L.; Zuilhof, H.; Sanders, J.P.M.; Franssen, M.C.R. Enzyme-catalyzed polymerization of β-alanine esters, a sustainable route towards the formation of poly-β-alanine. Curr. Org. Chem. 2013, 17, 682–690. [Google Scholar]
- Kumar, D.; Bhalla, T.C. Microbial proteases in peptide synthesis: Approaches and applications. Appl. Microbiol. Biotechnol. 2005, 68, 726–736. [Google Scholar] [CrossRef]
- Kuhn, D.; Durrschmidt, P.; Mansfeld, J.; Ulbrich-Hofmann, R. Boilysin and thermolysin in dipeptide synthesis: A comparative study. Biotechnol. Appl. Biochem. 2002, 36, 71–76. [Google Scholar] [CrossRef]
- Bille, V.; Ripak, C.; Assche, I.; Forni, L.; Degelaen, J.; Searso, J. Semi-enzymic synthesis of somatostatin. In Proceedings of 21st European Peptide Symposium, Platja d’Aro, Spain, 2–8 September 1990.
- Gu, Q.M.; Maslanka, W.W.; Cheng, H.N. Enzyme-catalyzed polyamides and their derivatives. In Polymer Biocatalysis and Biomaterials II; ACS Symposium Series 999; Cheng, H.N., Gross, R.A., Eds.; Oxford University Press: Washington, DC, USA, 2008; pp. 309–319. [Google Scholar]
- Poulhès, F.; Mouysset, D.; Gil, G.; Bertrand, M.P.; Gastaldi, S. Speeding-up enzyme-catalyzed synthesis of polyamides using ω-amino-a-alkoxy-acetate as monomer. Polymer 2013, 54, 3467–3471. [Google Scholar] [CrossRef]
- Ikeda, R.; Tanaka, H.; Uyama, H.; Kobayashi, S. Laccase-catalyzed polymerization of acrylamide. Macromol. Rapid Commun. 1998, 19, 423–425. [Google Scholar] [CrossRef]
- Hollmann, F.; Gumulya, Y.; Tölle, C.; Liese, A.; Thum, O. Evaluation of the laccase from Myceliophthora thermophila as industrial biocatalyst for polymerization reactions. Macromolecules 2008, 41, 8520–8524. [Google Scholar] [CrossRef]
- Emery, O.; Lalot, T.; Brigodiot, M.; Maréchal, E. Free-radical polymerization of acrylamide by horseradish peroxidase-mediated initiation. J. Polym. Sci. Pol. Chem. 1997, 35, 3331–3333. [Google Scholar] [CrossRef]
- Mucientes, A.E.; Santiago, F.; Carrero, A.M.; Talavera, B. Superabsorbent hydrogels of poly(sodium acrylate) with crude and exfoliated vermiculites. J. Polym. Eng. 2013, 33, 61–69. [Google Scholar]
- Jones, N.A.; Atkins, E.D.T.; Hill, M.J.; Cooper, S.J.; Franco, L. Chain-folded lamellar crystals of aliphatic polyamides. comparisons between nylons 44, 64, 84, 104, and 124. Macromolecules 1996, 29, 6011–6018. [Google Scholar] [CrossRef]
- Ragupathy, L.; Ziener, U.; Dyllick-Brenzinger, R.; von Vacano, B.; Landfester, K. Enzyme-catalyzed polymerizations at higher temperatures: Synthetic methods to produce polyamides and new poly(amide-co-ester)s. J. Mol. Catal. B Enzym. 2012, 76, 94–105. [Google Scholar] [CrossRef]
- Kalra, B.; Gross, R.A. Horseradish peroxidase mediated free radical polymerization of methyl methacrylate. Biomacromolecules 2000, 1, 501–505. [Google Scholar] [CrossRef]
- Singh, A.; Ma, D.C.; Kaplan, D.L. Enzyme-mediated free radical polymerization of styrene. Biomacromolecules 2000, 1, 592–596. [Google Scholar] [CrossRef]
- Sandoval, G.; Rivera, I.; Barrera-Rivera, K.A.; Martínez-Richa, A. Biopolymer synthesis catalyzed by tailored lipases. Macromol. Symp. 2010, 289, 135–139. [Google Scholar] [CrossRef]
- Varma, I.K.; Albertsson, A.C.; Rajkhowa, R.; Srivastava, R.K. Enzyme catalyzed synthesis of polyesters. Prog. Polym. Sci. 2005, 30, 949–981. [Google Scholar] [CrossRef]
- Kobayashi, S. Enzymatic polymerization: A new method of polymer synthesis. J. Polym. Sci. Pol. Chem. 1999, 37, 3041–3056. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Ohmae, M. Enzymatic polymerization for precision polymer synthesis. Bull. Chem. Soc. Jpn. 2001, 74, 613–635. [Google Scholar] [CrossRef]
- Uyama, H.; Kikuchi, H.; Takeya, K.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization and copolymerization of 15-pentadecanolide. Acta Polym. 1996, 47, 357–360. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, K.; Kobayashi, S. Enzymatic ring-opening polymerization of lactones to polyesters by lipase catalyst: Unusually high reactivity of macrolides. Bull. Chem. Soc. Jpn. 1995, 68, 56–61. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, K.; Hoshi, N.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of 12-dodecanolide. Macromolecules 1995, 28, 7046–7050. [Google Scholar] [CrossRef]
- Bisht, K.S.; Henderson, L.A.; Gross, R.A. Enzyme-catalyzed ring-opening polymerization of ω-pentadecalactone. Macromolecules 1997, 30, 2705–2711. [Google Scholar] [CrossRef]
- Hunsen, M.; Azim, A.; Mang, H.; Wallner, S.R.; Ronkvist, A.; Xie, W.C.; Gross, R.A. A cutinase with polyester synthesis activity. Macromolecules 2007, 40, 148–150. [Google Scholar] [CrossRef]
- Nishida, H.; Yamashita, M.; Nagashima, M.; Endo, T.; Tokiwa, Y. Synthesis of metal-free poly(1,4-dioxan-2-one) by enzyme-catalyzed ring-opening polymerization. J. Polym. Sci. Pol. Chem. 2000, 38, 1560–1567. [Google Scholar] [CrossRef]
- Jiang, Z.; Azim, H. Lipase-catalyzed copolymerization of ω-pentadecalactone with p-dioxanone and characterization of copolymer thermal and crystalline properties. Biomacromolecules 2007, 8, 2262–2269. [Google Scholar] [CrossRef]
- Fickers, P.; Marty, A.; Nicaud, J.M. The lipases from Yarrowia lipolytica: Genetics, production, regulation, biochemical characterization and biotechnological applications. Biotechnol. Adv. 2011, 29, 632–644. [Google Scholar] [CrossRef]
- Uyama, H.; Kikuchi, H.; Takeya, K.; Hoshi, N.; Kobayashi, S. Immobilized lipase showing high catalytic activity toward enzymatic ring-opening polymerization of macrolides. Chem. Lett. 1996, 25, 107–108. [Google Scholar]
- Kobayashi, S.; Uyama, H. Precision enzymatic polymerization to polyesters with lipase catalysts. Macromol. Symp. 1999, 144, 237–246. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Namekawa, S. In vitro biosynthesis of polyesters with isolated enzymes in aqueous systems and organic solvents. Polym. Degrad. Stabil. 1998, 59, 195–201. [Google Scholar] [CrossRef]
- Kumar, A.; Kalra, B.; Dekhterman, A.; Gross, R.A. Efficient ring-opening polymerization and copolymerization of ε-caprolactone and ω-pentadecalactone catalyzed by Candida antartica lipase B. Macromolecules 2000, 33, 6303–6309. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Namekawa, S.; Hayakawa, H. Enzymatic ring-opening polymerization and copolymerization of 8-octanolide by lipase catalyst. Macromolecules 1998, 31, 5655–5659. [Google Scholar]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of 16-hexadecanolide. P. Jpn. Acad. B 1998, 74, 65–68. [Google Scholar] [CrossRef]
- Ebata, H.; Toshima, K.; Matsumura, S. Lipase-catalyzed transformation of poly(ε-caprolactone) into cyclic dicaprolactone. Biomacromolecules 2000, 1, 511–514. [Google Scholar] [CrossRef]
- Sugihara, S.; Toshima, K.; Matsumura, S. New strategy for enzymatic synthesis of high-molecular-weight poly(butylene succinate) via cyclic oligomers. Macromol. Rapid Commun. 2006, 27, 203–207. [Google Scholar] [CrossRef]
- Kikuchi, H.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of substituted lactones. Polym. J. 2002, 34, 835–893. [Google Scholar] [CrossRef]
- Küllmer, K.; Kikuchi, H.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of α-methyl-δ-valerolactone and α-methyl-ε-caprolacton. Macromol. Rapid. Commun. 1998, 19, 127–130. [Google Scholar] [CrossRef]
- Cordova, A.; Iversen, T.; Martinelle, M. Lipase-catalysed formation of macrocycles by ring-opening polymerisation of ϵ-caprolactone. Polymer 1998, 39, 6519–6524. [Google Scholar] [CrossRef]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening and copolymerization of β-propiolactione. Polym. J. 1996, 28, 730–731. [Google Scholar] [CrossRef]
- Suzuki, Y.; Taguchi, S.; Hisano, T.; Toshima, K.; Matsumura, S.; Doi, Y. Correlation between structure of the lactones and substrate specificity in enzyme-catalyzed polymerization for the synthesis of polyesters. Biomacromolecules 2003, 4, 537–543. [Google Scholar] [CrossRef]
- Nagasawa, T.; Shimizu, H.; Yamada, H. The superiority of the third-generation catalyst, Rhodococcus rhodochrous J1 nitrile hydratase, for industrial production of acrylamide. Appl. Microbiol. Biotechnol. 1993, 40, 189–195. [Google Scholar]
- Thomas, S.M.; DiCosimo, R.; Nagarajan, V. Biocatalysis: Applications and potentials for the chemical industry. Trends Biotechnol. 2002, 20, 238–242. [Google Scholar] [CrossRef]
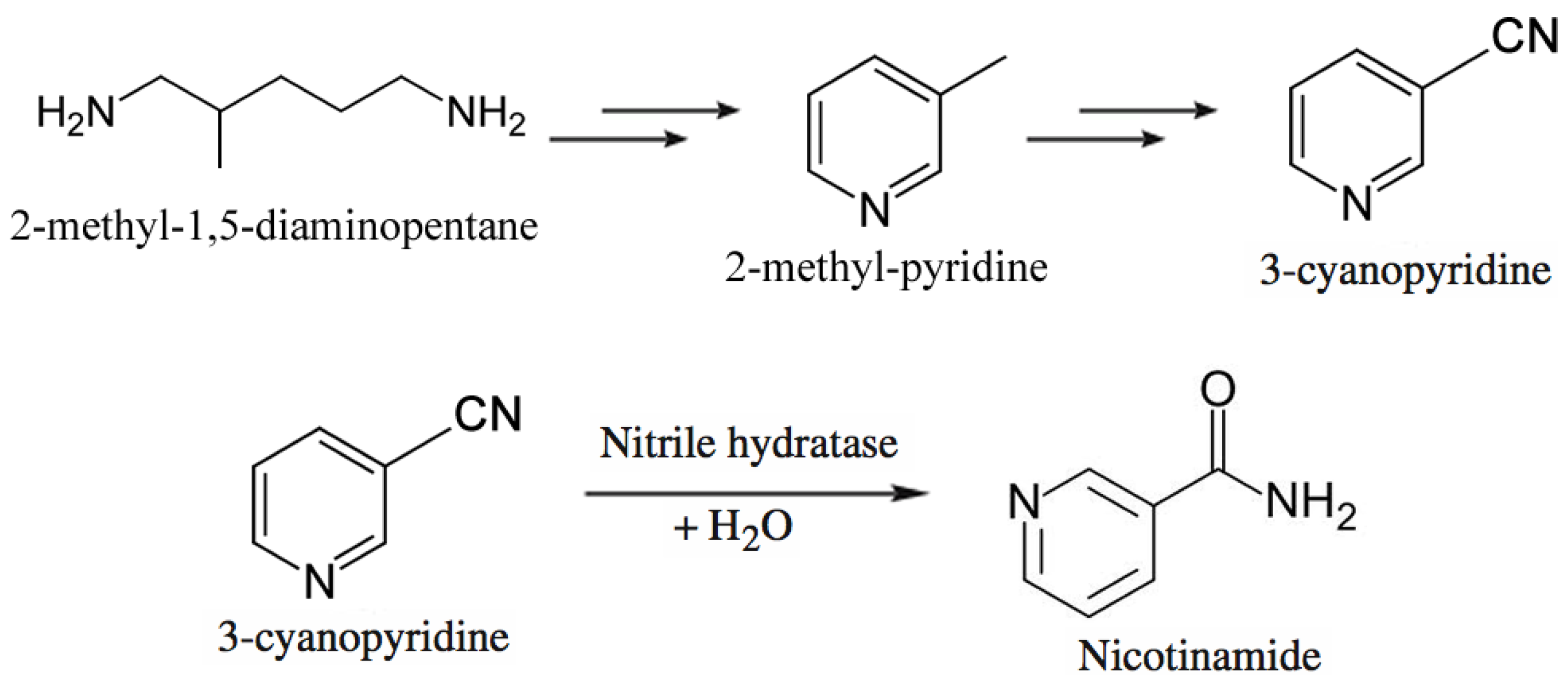
- Chassin, C. A biotechnological process for the production of nicotinamide. Chim. Oggi. 1996, 14, 9–12. [Google Scholar]
- Nagasawa, T.; Matthew, C.D.; Mauger, J.; Yamada, H. Nitrile hydratase-catalyzed production of nicotinamide from 3-cyanopyridine in Rhodococcus rhodochrous J1. Appl. Environ. Microbiol. 1988, 54, 1766–1769. [Google Scholar]
- Robins, K.T.; Nagasawa, T. Process for Preparing Amides. PCT Int. Appl. US 7,666,635 B2, 23 February 2010. [Google Scholar]
- Heveling, J.; Armbruster, E.; Utiger, L.; Rhoner, M.; Dettwiler, H.R.; Chuck, R.J. Process for Preparing Nicotinamide. US Patent 5,719,045, 17 February 1998. [Google Scholar]
- Straathof, A.J.J. Transformation of biomass into commodity chemicals using enzymes or cells. Chem. Rev. 2013. [Google Scholar] [CrossRef]
- Vuyyuru, K.R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: Aerobic oxidation of 5-hydroxymethyl-2-furfural into 2,5-furandicarboxylic acid with gold nanoparticle catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef]
- Koopman, F.; Wierckx, N.; de Winde, J.H.; Ruijssenaars, H.J. Efficient whole-cell biotransformation of 5-(hydroxymethyl)furfural into FDCA, 2,5-furandicarboxylic acid. Bioresour. Technol. 2010, 101, 6291–6296. [Google Scholar] [CrossRef]
- Krystof, M.; Pérez-Sánchez, M.; Domínguez de María, P. Lipase-mediated selective oxidation of furfural and 5-hydroxymethylfurfural. ChemSusChem 2013, 6, 826–830. [Google Scholar] [CrossRef]
- Van Deurzen, M.P.J.; van Rantwijk, F.; Sheldon, R.A. Chloroperoxidase-catalyzed oxidation of 5-hydroxymethylfurfural. J. Carbohydr. Chem. 1997, 16, 299–309. [Google Scholar]
- Panke, S.; Wubbolts, M. Advances in biocatalytic synthesis of pharmaceutical intermediates. Curr. Opin. Chem. Biol. 2005, 9, 188–194. [Google Scholar] [CrossRef]
- Ooshima, H.; Mori, H.; Harano, Y. Synthesis of aspartame precursor by solid thermolysin in organic solvent. Biotechnol. Lett. 1985, 7, 789–792. [Google Scholar] [CrossRef]
- Griengl, H.; Klempier, N.; Pochlauer, P.; Schmidt, M.; Shi, N.Y.; Zabelinskaja-Mackova, A.A. Enzyme catalysed formation of (S)-cyanohydrins derived from aldehydes and ketones in a biphasic solvent system. Tetrahedron 1998, 54, 14477–14486. [Google Scholar]
- Bieber, L.L. Carnitine. Annu. Rev. Biochem. 1988, 57, 261–283. [Google Scholar] [CrossRef]
- Fritz, I.B. Action of carnitine on long chain fatty acid oxidation by liver. Am. J. Physiol. 1959, 197, 297–304. [Google Scholar]
- Meyer, H.P.; Kiener, A.; Imwinkelried, R.; Shaw, N. Biotransformations for fine chemicals production. Chimia 1997, 51, 287–289. [Google Scholar]
- Bommarius, A.S.; Schwarm, M.; Stingl, K.; Kottenhahn, M.; Huthmacher, K.; Drauz, K. Synthesis and use of enantiomerically pure tert-leucine. Tetrahedron Asymmetry 1995, 6, 2851–2888. [Google Scholar]
- Menzel, A.; Werner, H.; Altenbuchner, J.; Gröger, H. From enzymes to “designer bugs” in reductive amination: A new process for the synthesis of l-tert-leucine using a whole cell-catalyst. Eng. Life Sci. 2004, 4, 573–576. [Google Scholar] [CrossRef]
- Krix, G.; Bommarius, A.S.; Drauz, K.; Kottenhahn, M.; Schwarm, M.; Kula, M.R. Enzymatic reduction of α-keto acids leading to l-amino acids, d- or l-hydroxy acids. J. Biotechnol. 1997, 53, 29–39. [Google Scholar] [CrossRef]
- Furuhashi, K. Biological routes to optically active epoxides. In Chirality in Industry; Collins, A.N., Sheldrake, G.N., Crosby, J.C., Eds.; John Wiley & Sons: NJ, USA, 1992; pp. 167–186. [Google Scholar]
- Li, K.; Frost, J. Synthesis of vanillin from glucose. J. Am. Chem. Soc. 1998, 120, 10545–10546. [Google Scholar] [CrossRef]
- Aresta, M.; Quaranta, E.; Liberio, R.; Dileo, C.; Tommasi, I. Enzymatic synthesis of 4-OH-benzoic acid from phenol and CO2: The first example of a biotechnological application of a carboxylase enzyme. Tetrahedron 1998, 54, 8841–8846. [Google Scholar] [CrossRef]
- Baldessari, A. Lipases as catalysts in synthesis of fine chemicals. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, NY, USA, 2012; Volume 861, pp. 445–448. [Google Scholar]
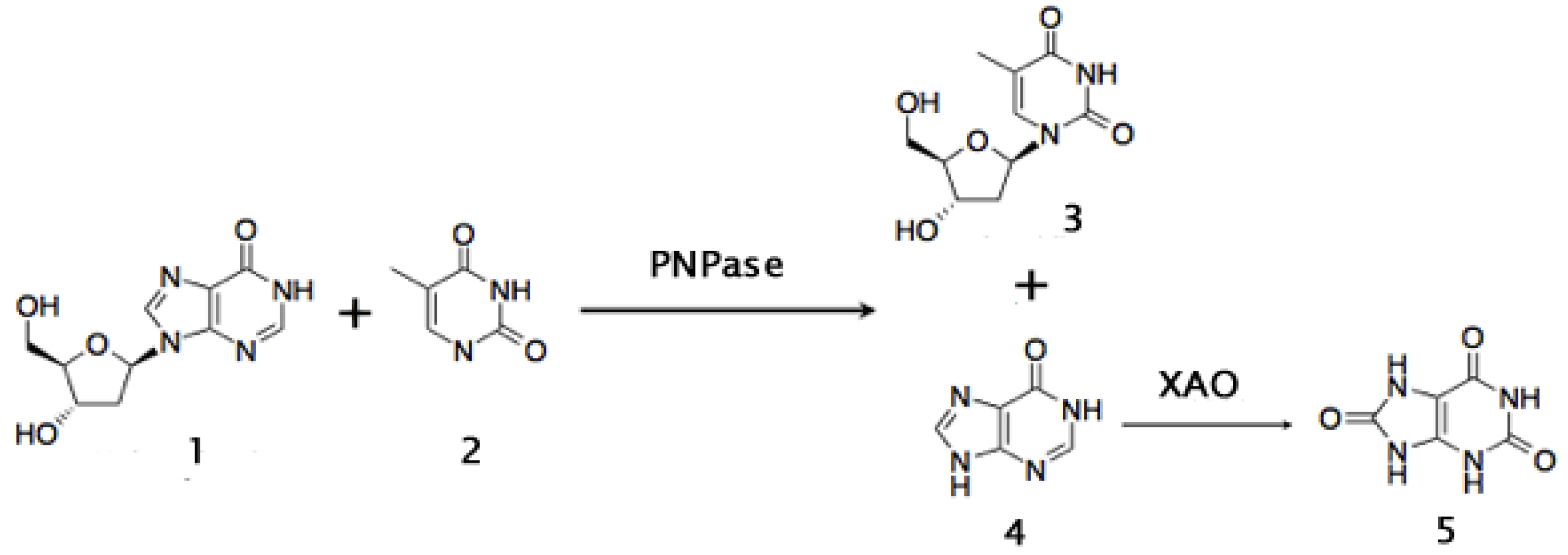
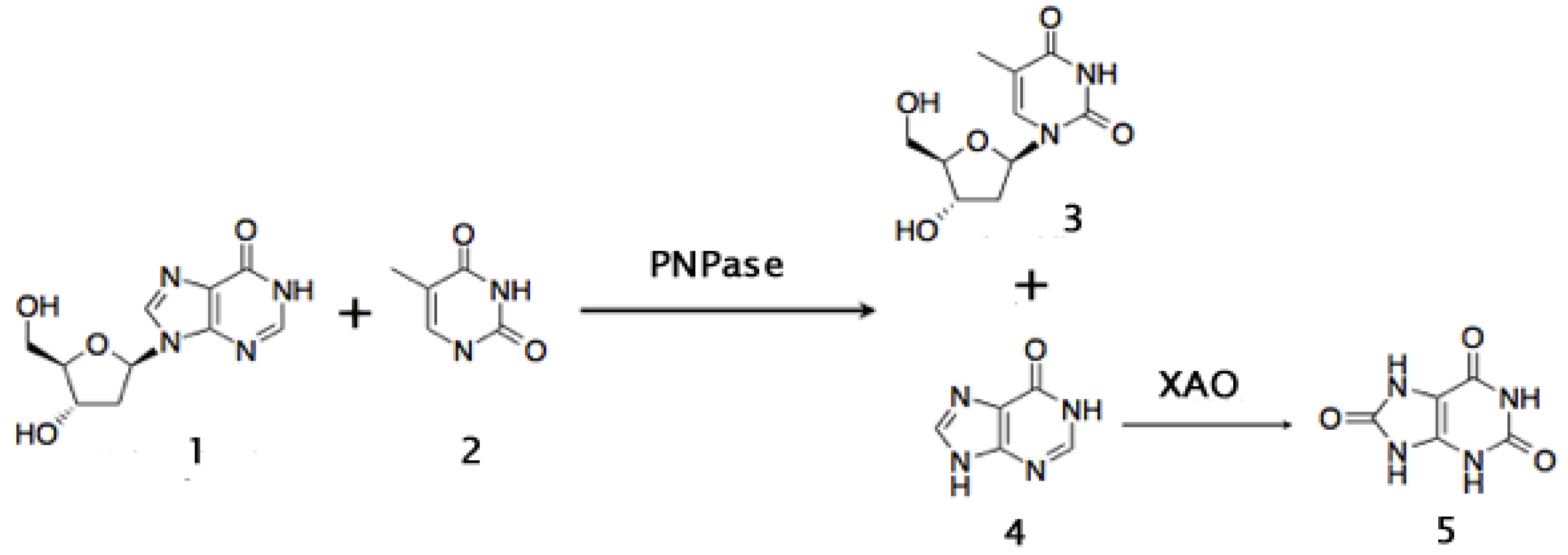
- Pal, S.; Nair, V. Enzymatic synthesis of thymidine using bacterial whole cells and isolated purine nucleoside phosphorylase. Biocatal. Biotransform. 1997, 15, 147–158. [Google Scholar] [CrossRef]
- Liese, A.; Villela-Filho, M. Production of fine chemicals using biocatalysis. Curr. Opin. Biotechnol. 1999, 10, 595–603. [Google Scholar] [CrossRef]
- Rivera, I.; Mateos, J.C.; Marty, A.; Sandoval, G.; Duquesne, S. Lipase from Carica papaya latex presents high enantioselectivity toward the resolution of prodrug (R,S)-2-bromophenylacetic acid octyl ester. Tetrahedron Lett. 2013, 54, 5523–5526. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Regil, R.; Sandoval, G. Biocatalysis for Biobased Chemicals. Biomolecules 2013, 3, 812-847. https://doi.org/10.3390/biom3040812
De Regil R, Sandoval G. Biocatalysis for Biobased Chemicals. Biomolecules. 2013; 3(4):812-847. https://doi.org/10.3390/biom3040812
Chicago/Turabian StyleDe Regil, Rubén, and Georgina Sandoval. 2013. "Biocatalysis for Biobased Chemicals" Biomolecules 3, no. 4: 812-847. https://doi.org/10.3390/biom3040812
APA StyleDe Regil, R., & Sandoval, G. (2013). Biocatalysis for Biobased Chemicals. Biomolecules, 3(4), 812-847. https://doi.org/10.3390/biom3040812