Biocatalysis for Biobased Chemicals


Unidad de Biotecnología Industrial, CIATEJ, A.C. Av. Normalistas 800, Col. Colinas de la Normal, Guadalajara, Jal, C.P. 44270, Mexico
*
Author to whom correspondence should be addressed.
Biomolecules 2013, 3(4), 812-847; https://doi.org/10.3390/biom3040812
Submission received: 3 September 2013
/
Revised: 8 October 2013
/
Accepted: 8 October 2013
/
Published: 17 October 2013
(This article belongs to the Special Issue Enzymes and Their Biotechnological Applications)
Abstract
:The design and development of greener processes that are safe and friendly is an irreversible trend that is driven by sustainable and economic issues. The use of Biocatalysis as part of a manufacturing process fits well in this trend as enzymes are themselves biodegradable, require mild conditions to work and are highly specific and well suited to carry out complex reactions in a simple way. The growth of computational capabilities in the last decades has allowed Biocatalysis to develop sophisticated tools to understand better enzymatic phenomena and to have the power to control not only process conditions but also the enzyme’s own nature. Nowadays, Biocatalysis is behind some important products in the pharmaceutical, cosmetic, food and bulk chemicals industry. In this review we want to present some of the most representative examples of industrial chemicals produced in vitro through enzymatic catalysis.
1. Introduction
It is becoming evident that in order to sustain the standard way of life of the developed and in-development world, it will be necessary to make some adjustments either to our consumption habits or to our sources of supplies of energy and materials. In the latter case, Biotechnology, as a diversified discipline in which chemistry, physics, biology, optics, electro-magnetism and thermodynamics converge, possess the knowledge tools to play a relevant and vital role to address the challenges of growth, ageing, employment, limited sources of raw materials, energy and water supply and living standard. Industrial biobased processes will increasingly become the de facto alternative to build a sustainable economy. It is estimated that, by 2030, the products of white biotechnology and bioenergy will account for 30% of industrial production worth €300 bn [1].
Conversion of biomass by Biocatalysis is likely to become standard technology which will contribute significantly to open up access to large feedstock supplies for bioprocesses and the production of transport fuels. On the health and ageing field, personalized nutrition, tailor-made medicine will become a reality in the coming years thanks to novel, specific biotech drugs and regenerative medicine obtained by more efficient and greener processes.
Biocatalysis refers to the transformation of substances of chemical or biological origin through the use of the enzymes produced by diverse living organisms. Enzymes may carry out reactions in a free, or immobilized form, or within the living cell in which they naturally live.
Isolated enzymes obtained as purified and concentrated extracts, whether used immobilized onto a support or in free form, may produce neater products than when the whole microorganism cell is used for the biotransformation. This is because of the absence of other kinds of enzymes from the internal cell machinery, which may subsequently modify the product, and the absence of internal cellular components during the lysis-extraction step. This advantage of isolated enzymes has a positive impact on yield and purification costs. However, isolated enzymes are known to be less stable in a purified form than within the cell. The lower stability of free enzymes has been improved to some extent by immobilization and crosslinking techniques, the results of which vary depending on the type of enzyme, pH memory, extent of crosslinking, immobilization support and the procedure itself.
Whole microorganism cells, obtained in sufficient amount by fermentation, are used instead of isolated enzymes when their enzymes become highly unstable or non-functional outside the inner environment of the cell, so catalysis is confined within the cell and products are later extracted from it.
Chemical synthesis in the food, feed, industrial and pharma sectors is the field to which Biocatalysis has contributed the most, but it has also contributed to the bioremediation sector, another field in which enzymes are having a significant role.
The relative success of Biocatalysis applied to Synthetic Chemistry has been due mainly to the high enantio- and regioselectivities that enzymes exhibit towards their substrates, because enzymes speed up chemical processes that would otherwise run very slowly or without selectivity, and because enzymes function under mild reaction conditions. These advantages have allowed chemists to avoid the burden of group-protecting procedures, saving time, materials (including the harsh, dangerous or toxic ones) and energy costs. Other advantages of enzymes are that they are easy to control and biodegradable. Thus, Biocatalysis has proved in many cases to be a superior pathway than conventional chemical synthesis pathways, not only in the simplicity of accomplishing the reactions, but also from an economical and environmental point of view.
An important landmark in Biocatalysis was the use of organic solvents as reaction media, as it was thought that enzymes may only work in aqueous medium, naturally. Organic solvents became an important aid in dissolving organic, hydrophobic molecules, which account for much of the library of organic chemicals used in Synthetic Chemistry. The possibility to carry out reactions in organic media favored the reactions rate precisely because of the better solubility of reactants. Purification steps became shorter and easier because the avoidance of surfactants needed in aqueous media and the use of low boiling point solvents easy to evaporate. Moreover, hydrolytic, secondary, water-dependent reactions responsible for lower product yields, and growth of microbes in containers and pipes, is avoided because of the absence of water and the harsh effects of solvents to living cells.
However, Biocatalysis still needs to overcome some limitations such as the enzyme costs of production and the relative narrow stability window of many enzymes under diverse reaction conditions, which restricts their applications onto broader industrial processes. Chemicals used in industrial settings are mostly derived from fossil oil, whereas active ingredients in pharmaceutical processes may be new and/or structurally complex. In general, a vast number of chemicals used in industry are artificial, new man-made chemicals for which enzymes did not evolve to work with and thus are not suitable for the synthetic needs in industry. So there are high discrepancies between the enzyme´s function in nature and the functions needed in the transformation industry. High throughput enzyme screening and protein engineering techniques, particularly Directed Evolution, are some of the tools being employed to advance faster in this aim to find enzymes for artificial substrates, and to create enzymes with new capabilities.
In this review we want to present some of the most representative examples of industrial chemicals produced in vitro through enzymatic catalysis, and hence, in vivo fermentation products are not included.
2. Enzymes in Food Industry
Applications of enzymes in the food industry are found in almost every sector of this industry: confectionary and sweeteners, fruit and vegetables, dairy, brewery and beverages, meat, dietary and the nutraceuticals niche [2]. However, most of these applications are of hydrolytic nature focused in debranching, improving solubility, clarification, and hydrolysis aimed at diverse goals depending on the food being treated.
2.1. Prebiotics and Sweeteners
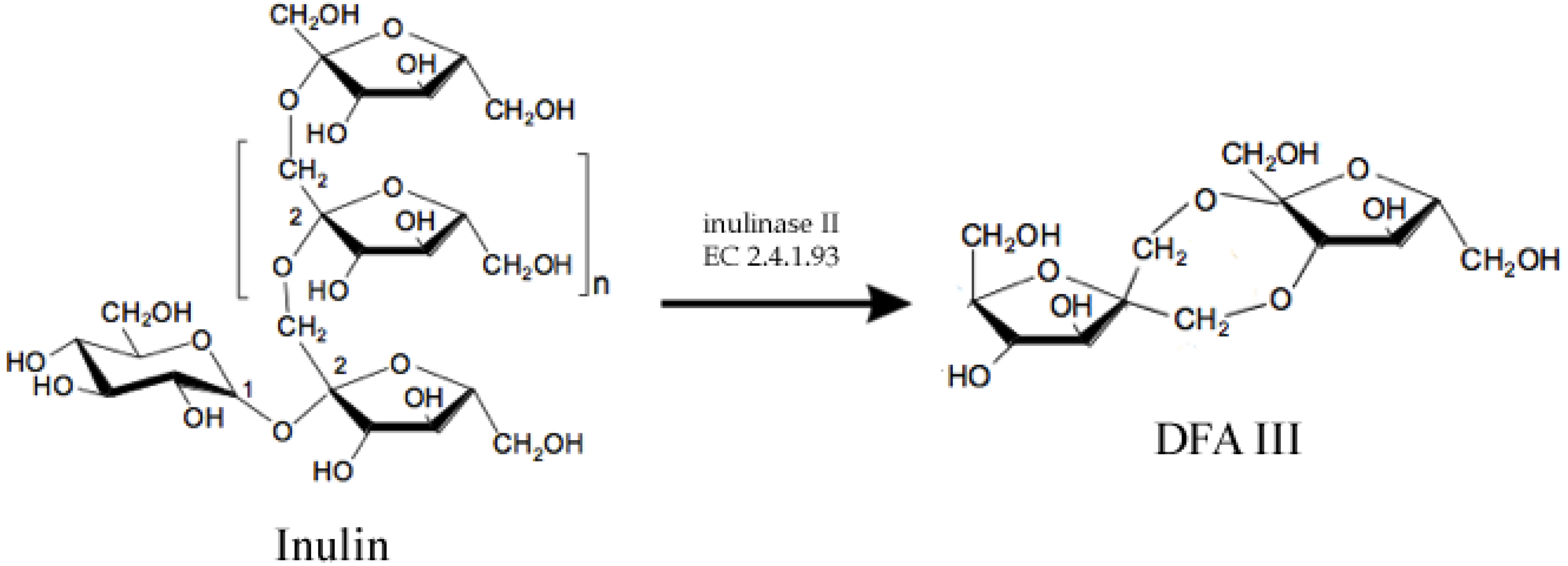
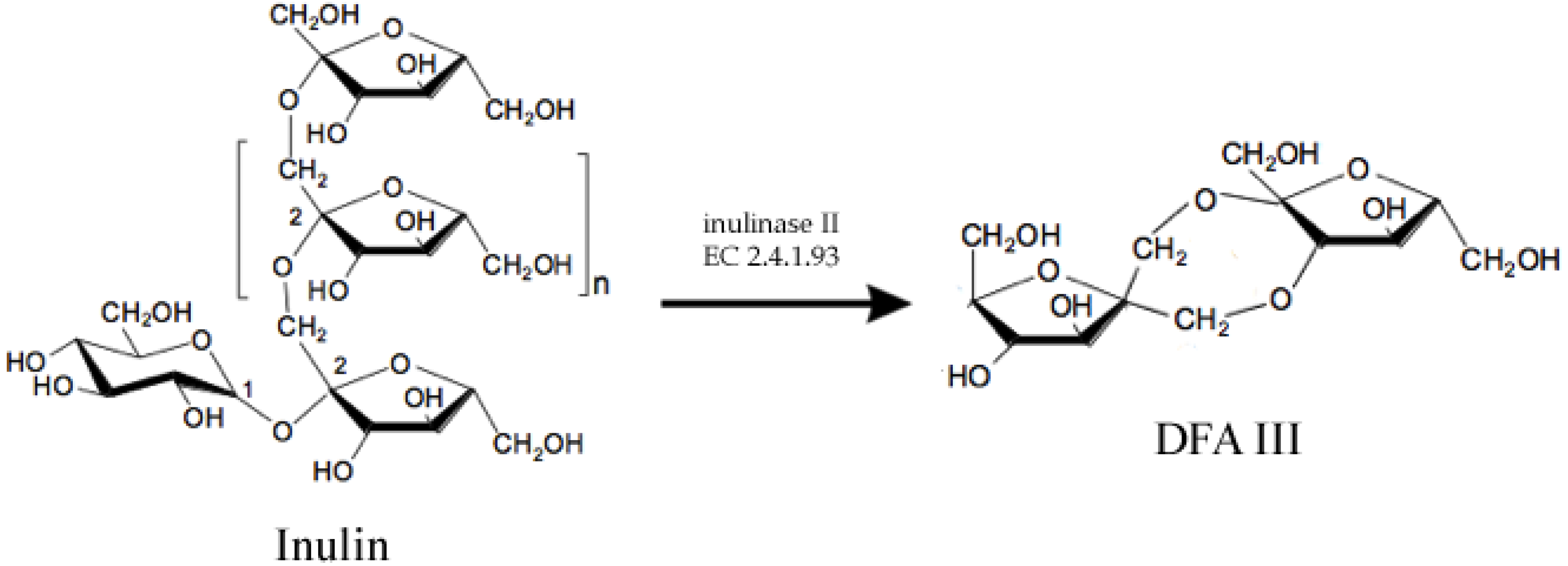
There are some examples of food chemicals being synthesized by enzymes. One of them is Disaccharide Difructose Anhydride III (DFA III), which is a non-reducing, non-cariogenic sweetener with probiotic properties [3]. DFA III can be synthesized by enzyme inulase II (EC 2.4.1.93), (Figure 1), which was first identified in Arthrobacter ureafaciens [4] and later in other bacteria [5,6,7]. However, native enzymes are not thermotolerant enough to resist high temperatures required for a large scale process. Thus, the ift-gene encoding for the inulase II from Buo141, a thermotolerant strain, was cloned and expressed into E. coli XL1-blue and further modified and immobilized to obtain the biocatalyst which was reported to have 1.7 × 106 U/L [3].
Prebiotics is another field in which enzymes are being used. Oligosaccharides are non-digestible saccharide polymers containing 3–10 monomeric sugar units that are found in low amounts in human milk [8,9], onion, garlic, banana, soybean and chicory [10]. They are classified as prebiotics because they selectively promote the growth of bifidobacteria and lactobacilli in the intestine, which are regarded to have a beneficial effect to human health [10,11,12,13,14].
Figure 1.
Enzymatic synthesis of Difructose Anhydride III (DFA III) from Inulin using enzyme inulase II, an inulin fructo-transferase.
Figure 1.
Enzymatic synthesis of Difructose Anhydride III (DFA III) from Inulin using enzyme inulase II, an inulin fructo-transferase.
Oligosaccharides composed either of fructose or galactose units are named fructo-oligosaccharides (FOS) and galacto-oligosaccharides (GOS) respectively.
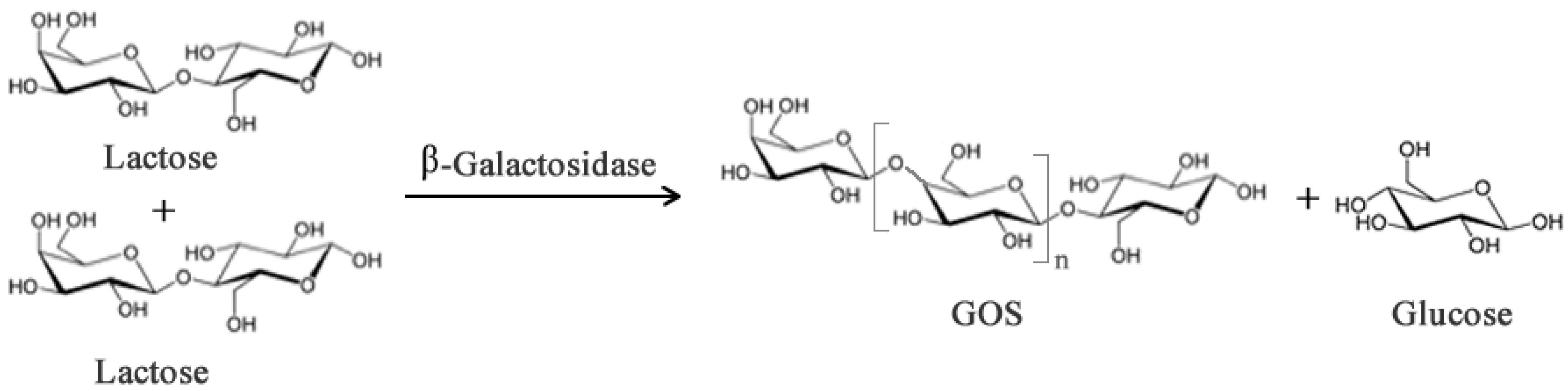
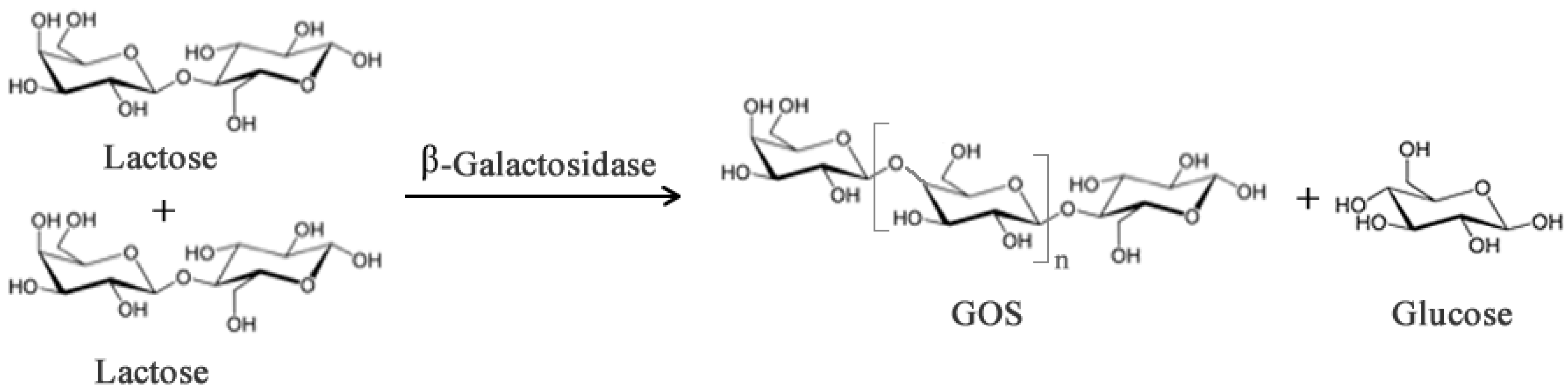
Galacto-oligosaccharides (GOS) can be synthesized from lactose when this sugar acts as the acceptor and transgalactosylation is catalyzed by β-galactosidase (Figure 2). In addition to the prebiotic activity, GOS have also been reported to contribute to (i) reduction of serum cholesterol and lipid level; (ii) synthesis of B-complex vitamins; (iii) enhance absorption of dietary calcium [14,15,16]; (iv) protection from infection and decrease of pathogenic bacteria; (v) stimulate absorption of some minerals [17]. However, these health promoting properties vary depending on the chemical composition, structure and degree of polymerization and these features depend, in turn, on the origin of the β-galactosidase [18].
Figure 2.
Simplified enzymatic reaction to produce Galacto-oligosaccharides (GOS) from lactose using β-galactosidase. Subscript n may range from 0–6 though most native enzymes produce GOS with n between 1–2.
Figure 2.
Simplified enzymatic reaction to produce Galacto-oligosaccharides (GOS) from lactose using β-galactosidase. Subscript n may range from 0–6 though most native enzymes produce GOS with n between 1–2.
The most recurring means of the synthesis of GOS is by enzymatic catalysis from lactose using glycosyltransferases (EC 2.4) or glycoside hydrolases (EC 3.2.1) [19]. Glycosyltransferases are not used in the synthesis of GOS due to their low availability, the need for nucleotide sugar substrates and prohibitive prices. Instead, glycoside hydrolases are used, despite the fact that they are less stereoselective than the former [20]. Glycoside hydrolases from archeas (Sulfolobus solfactaricum and Pyrococcus furiosus), bacteria (Saccharopolyspora sp., Bifidobacterium sp., Thermotoga sp., Thermus sp., Bacillus sp., Geobacillus sp., Caldicellulosiruptor sp., Lactobacillus sp., Streptococcus sp., Enterobacter sp., Escherichia sp.) and yeasts (Aspergillus sp., Penicillinum sp., Talaromyces sp., Trichoderma sp., Kluyveromyces sp., Sirobasidium sp., Sterigmatomyces sp., Rhodotorula sp., Sporobolomyces sp., Rhizopus sp.) have been tested for the synthesis of GOS [19,21].
Milk whey is a lactose-rich source which has been regarded in the past as a waste by-product from cheese production highly contaminant. So β-galactosidases have become a valued natural tool to produce a beneficial product out from a contaminant waste in a single step.
As a general finding, reports show that GOS yields average 30%–35% of total sugars [22,23] and are directly proportional to the initial lactose concentration. The major product is the trisaccharide accounting for as much as 80% of total GOS synthesized [12,24]. It was estimated that at least 3,500 tons of GOS are enzymatically synthesized from whey lactose, out of 30,000 tons of total world annual production [25]. They are commercialized to enrich mainly infant formulas.
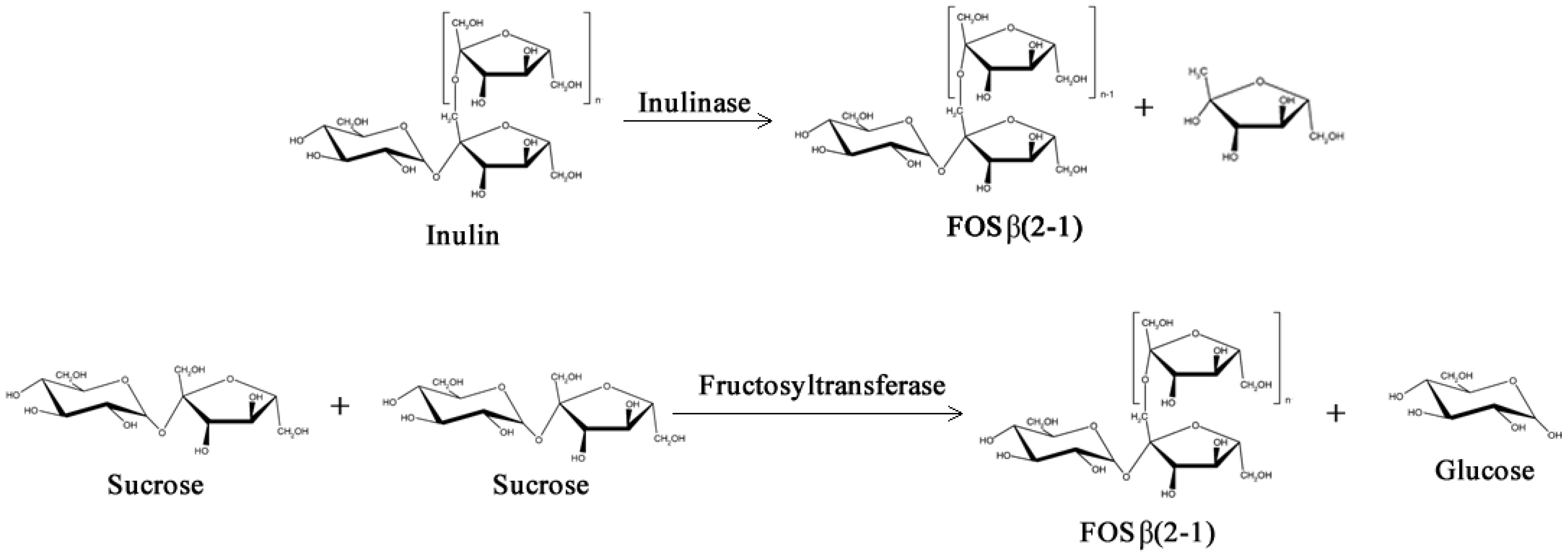
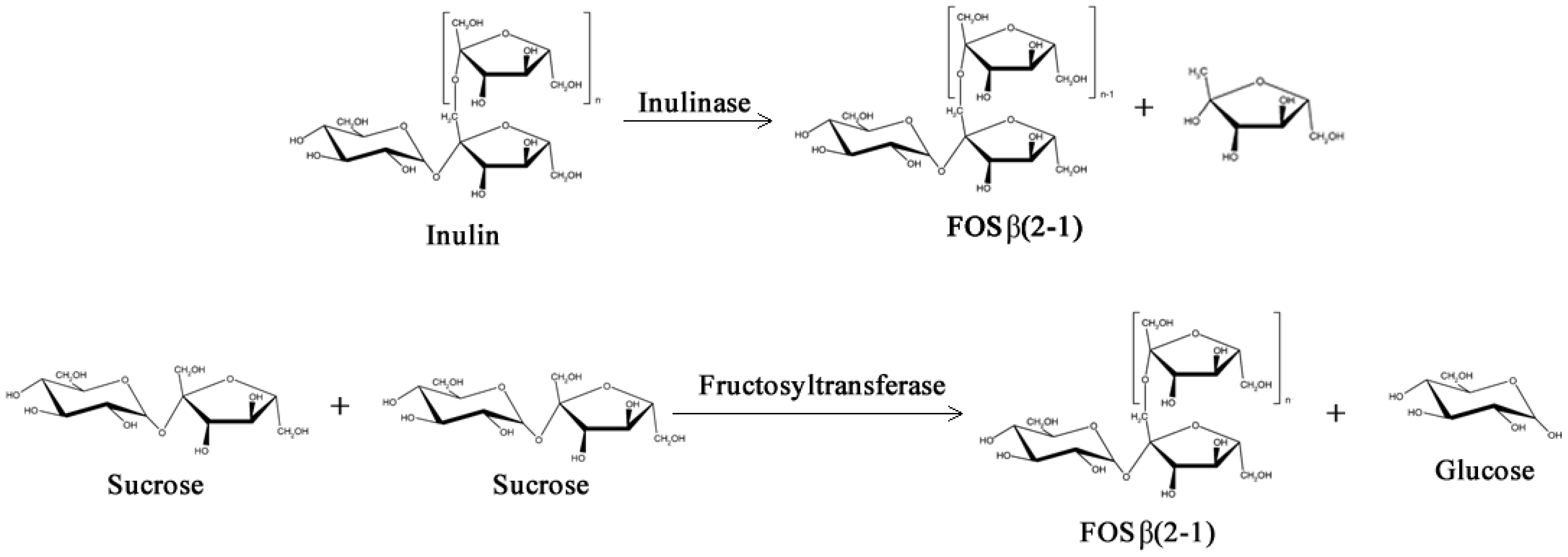
Just like GOS, Fructo-oligosaccharides (FOS) have low caloric value and have much the same prebiotic properties due to their indigestibility in the upper gastrointestinal tract: they are non-cariogenic and have ability to promote the activity of beneficial colonic lactic acid bacteria and to modulate the intestinal immune response [26,27,28]. However, unlike GOS, FOS are naturally occurring sugars biosynthesized by numerous plants such as asparagus, sugar beet, onion, artichoke, etc. [29,30]. Two main types of FOS can be found in nature, the inulin type, which are fructose polymers with linkages β(2–1) biosynthesized by inulosucrases (E.C. 2.4.1.9), and the levan type, which are fructose polymers with linkages β(2–6) biosynthesized by levansucrases (E.C. 2.4.1.10) [31,32,33].
Two different classes of FOS mixtures are produced commercially, one is based on inulin by controlled enzymatic hydrolysis, and the other is based on sucrose by a transfructosylation processes with fructosyltransferases, as shown in Figure 3 (β-fructofuranosidase, E.C. 3.2.1.26 or β-d-fructosyltransferase, EC 2.4.1.9) [34,35]. FOS from inulin hydrolysis contain longer fructose chains (Degree of Polymerization, DP 2–9) and may have either a fructose or glucose unit at the end of the chain. On the other hand, FOS from sucrose transfructosylation contain shorter polymer chains (DP 2–4) and always end with a glucose unit [35].
Figure 3.
Simplified enzymatic reactions used commercially to obtain Fructo-oligosaccharides (FOS).
A commercial enzyme Pectinex Ultra SP-L was used immobilized onto Eupergit C to synthesize FOS from a 600 g/L of sucrose solution. Reactions were carried out at 65 °C in pH 6.5 during 24 h with a yield of 57% FOS of total sugars [36].
Despite the big production numbers that prebiotics have, the overall European market for prebiotics is still at the beginning stage, with the $87-million fructan (inulin and fructo-oligosaccharide) segment as the most developed part. In US the prebiotics market is estimated to reach US$198.3 million in 2014 [37].
2.2. Structured Lipids
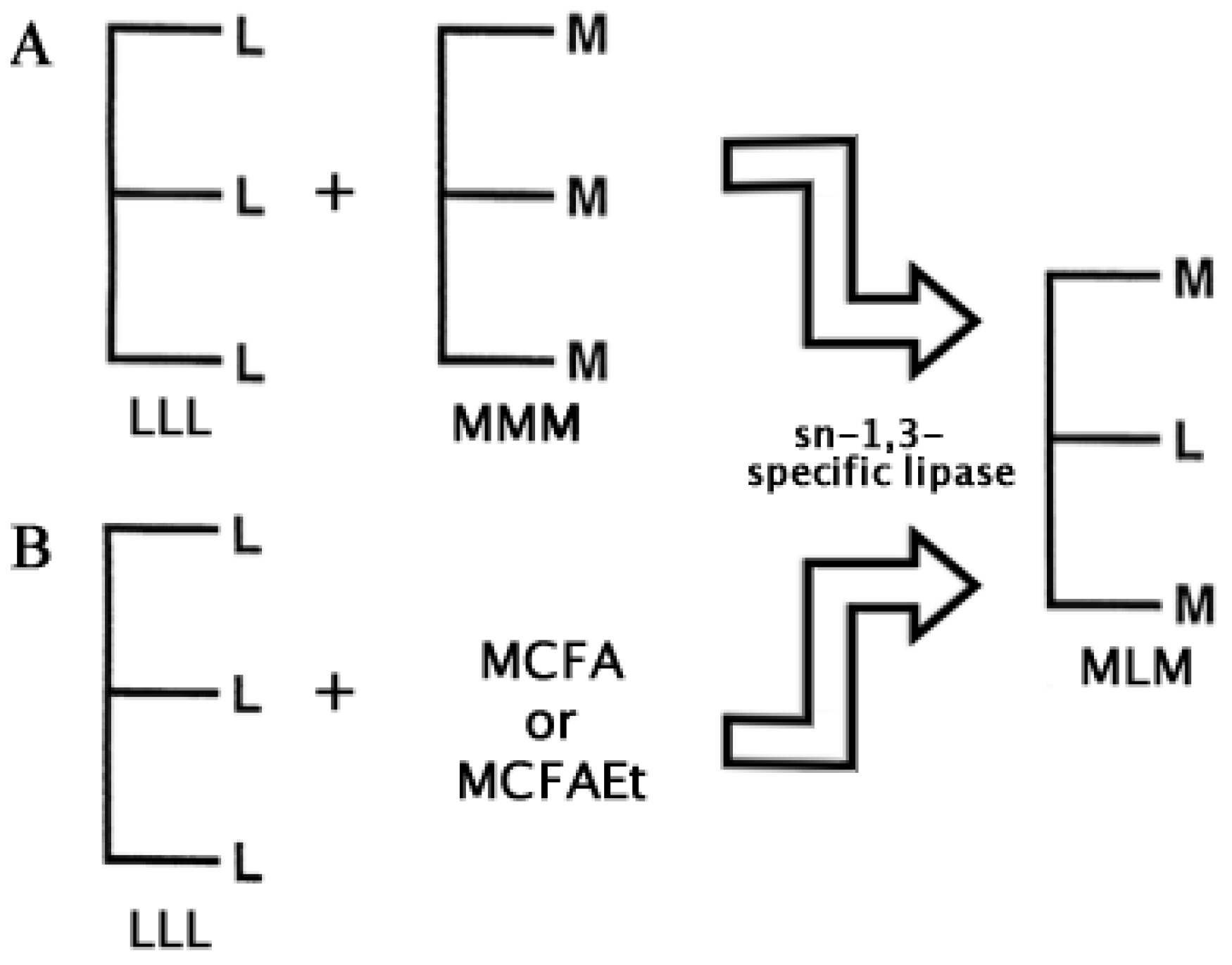
Specialty fats and oils are lipids with special functional or nutritional effects in the human body. Among these, structured lipids (SLs) have a predominant importance within this field [38,39]. SLs are tailor-made fats and oils which incorporate specific new fatty acids to triacylglycerols, or have different composition and positional distribution of existing fatty acids, within its glycerol backbone. Among the various types of SLs, the MLM (medium, long, medium fatty acid chain length esterified at the sn-1, sn-2 and sn-3 positions of the glycerol backbone respectively) have been a subject of great interest because of their ability to provide quick energy through the pancreatic lipase hydrolytic release of the medium chain fatty acids which become rapidly oxidized by the liver [40]. The remaining long chain sn-2-monoacylglyceride is left to be absorbed through the lymphatic system [41]. This long chain monoacylglyceride can be an omega-6 fatty acid or any other poly unsaturated fatty acid (PUFA) which will provide the already known health benefits to the body [42].
SLs are claimed to have a modulation effect on the immune system, to improve the lipid clearance in the blood [43], and as a fat for special nutritional feeding [44]. Fatty acids at the sn-2 position are more readily absorbed in vivo than those at the sn-1 or 3 positions, thus, SLs of the type MLM that include an essential fatty acid in the sn-2 position are desirable nutrition sources for people who suffer from malabsorption or a pancreatic condition [44,45].
SLs can be obtained either chemically or enzymatically using lipases [42]. The high regioselectivity of lipases makes Biocatalysis particularly well suited for the synthesis of SLs, avoiding the synthesis of unwanted by-products, which lower the yields [46]. However, enzymatic synthesis of SLs has yet to be optimized in order to avoid the high costs of large-scale purification of unreacted substrates (Free fatty acids, Triacyglicerides, Diacylgricerides, Esters) which must be removed upon completion of reaction [47].
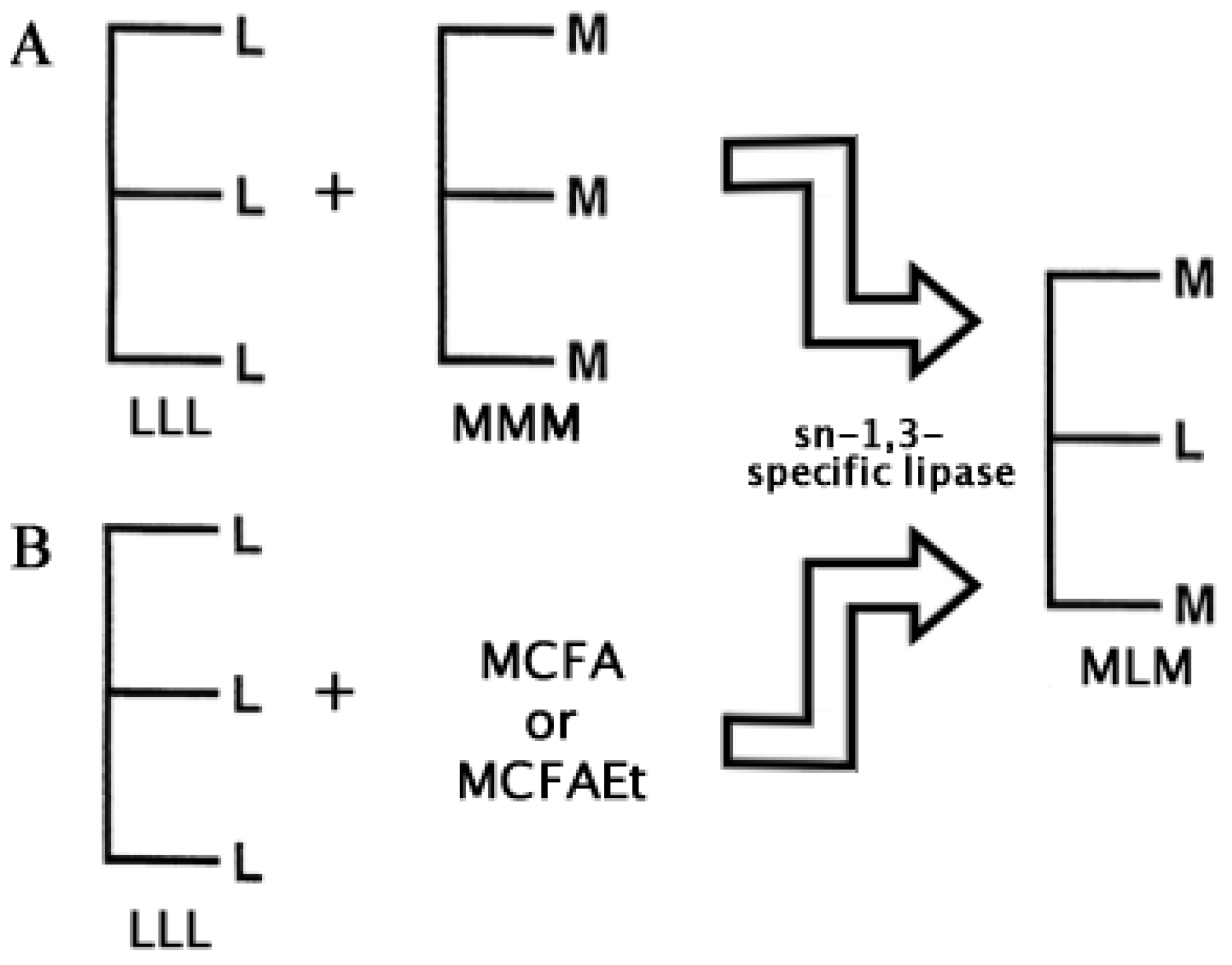
Two basic approaches have been followed to carry out the enzymatic synthesis of MLM SLs: (1) Transesterification and (2) Acidolysis [48]. Lipase transesterifications for the synthesis of MLM SLs have been performed using different vegetal and fish oils as source of essential long PUFAs, and coconut and palm kernel oil as a source of medium chain fatty acids (C6:0–C12:0) to be incorporated in the sn-1 and sn-3 positions [42,49]. Thus, to synthesize MLM SLs, sn-1 and sn-3, regiospecific lipases are favored. In Figure 4 these two approaches are depicted using a sn-1,3-specific lipase.
Casas-Godoy et al. [50] incorporated caprilyc and capric acid into olive oil using Yarrowia lipolytica immobilized on Accurel. Their optimized reaction allowed them to obtain an incorporation rate of 25% and 21% for capylic and capric acid respectively within 2 days at 40 °C and 5% (w/w) enzyme load.
Figure 4.
The main two approaches to synthesize MLM Structured Lipids enzymatically: (A) Transesterification and (B) Acidolysis. Subproducts like LML Structured lipids may also be obtained, but only the product of interest is shown. L = Long Chain; M = Medium Chain; MCFA = Medium Chain Fatty Acid; MCFAEt = Medium Chain Fatty Acid Ester (see also [48]).
Figure 4.
The main two approaches to synthesize MLM Structured Lipids enzymatically: (A) Transesterification and (B) Acidolysis. Subproducts like LML Structured lipids may also be obtained, but only the product of interest is shown. L = Long Chain; M = Medium Chain; MCFA = Medium Chain Fatty Acid; MCFAEt = Medium Chain Fatty Acid Ester (see also [48]).
In another work, castor oil was dehydrated, isomerized and then transesterified with methyl laurate using Thermomyces lanuginosa and Carica papaya. The incorporation and transesterification yields were 59% and 88% for T. lanuginosa and 44% and 67% for C. papaya, respectively. This was achieved in 72 h at 60 °C with a 10% (w/w) enzyme load. Transesterification in the sn-2 position was observed and accounted for less than 8% [51].
Palm oil trolein has been subject to enrichment in its sn-2 position with DHA and ARA through acidolysis using Novozym 435. The research Group achieved an incorporation of 11% of DHA at sn-2 position (17% total) and 5.5% of ARA at sn-2 position (7% total) in 24 h at 60 °C and 10% enzyme load [52]. In a similar work, Khodadadi et al. [49] transesterified palm oil with tricaprylin in order to modify the former. Their results showed that Linolenic acid chains in palm oil are more easily transesterified than Linoleic or Oleic. Their best results were around 20% incorporation of caprylic chains into sn-1, sn-2 and sn-3 positions of which around a half corresponded to sn-2 position. The experiments were carried out with Novozym 435, Lipozyme TL-IM, Lipozyme RM-IM and Amano DF, though the best results were with Novozym 435, 24 h of reaction time, 50 °C and 1% enzyme load. Tecelão et al. [53] recently achieved 21% and 8% of incorporation of oleic acid and DHA respectively, to tripalmitin using Carica papaya latex at 60 °C, in a solvent-free system during 24 h.
Nagao et al. [54] achieved almost 100% of fatty acid incorporation with a DHA-rich by-product waste from a tuna oil industrial, hydrolysis process employed to extract DHA. Their reaction conditions were 30 °C, 120 h, vacuum at 2 kPa and 10% of Novozym 435 load. The degree of esterification with DHA was 51% and 17% at sn-1,3 and sn-2 position respectively.
3. Enzymes in the Bulk Chemistry Industry
Although industrial biotransformations are mainly used for the production of fine chemicals, there are a few examples where Biocatalysis is also used to produce commodity chemicals such as acrylamide, and biodiesel. The use of biocatalysts employed to assist in synthetic routes to complex molecules of industrial interest is growing steadily [55,56,57].
3.1. Biodiesel
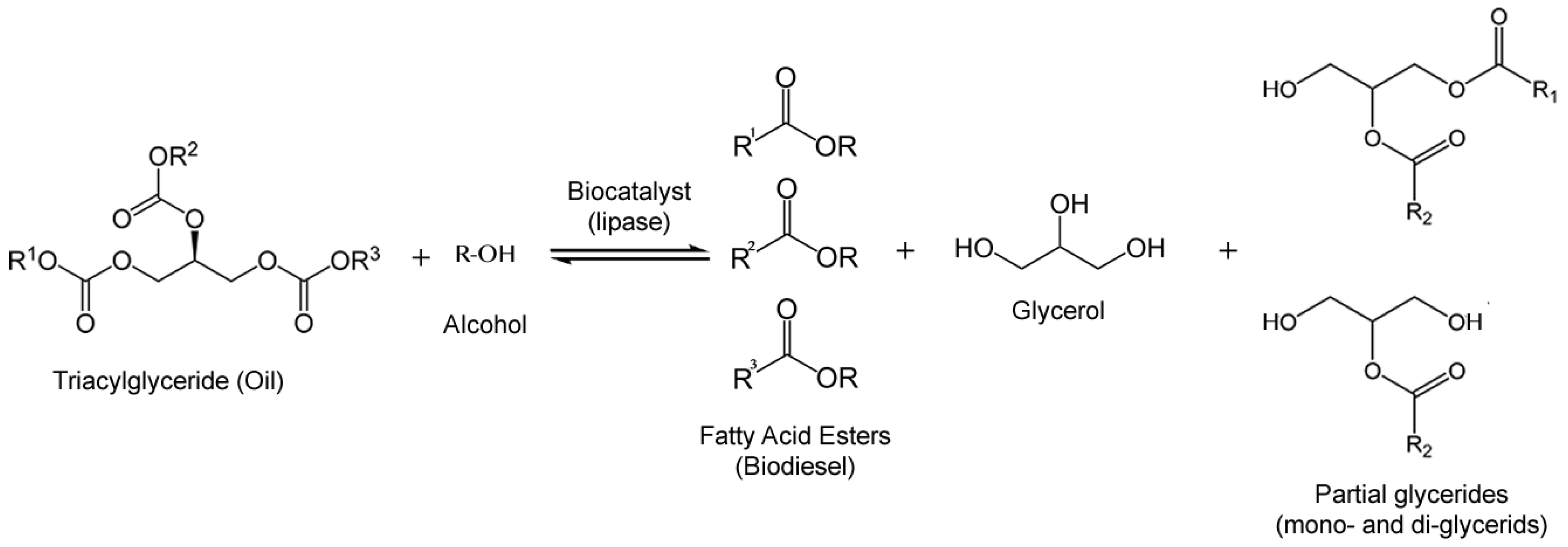
The lipase-catalyzed synthesis of biodiesel offers advantages over conventional methods for its production: lipases are able to work in gentle conditions and with a variety of triglyceride substrates, including waste oils and fats, corrosion problems present in chemical synthesis are avoided in the lipase-catalyzed synthesis, easy recovery of biocatalyst and glycerol, high levels of Free Fatty Acids (FFA) may also be esterified by lipases, and low environmental impact [58,59]. Furthermore, the separation and purification step of biodiesel is easier [60,61], resulting in a more attractive technical and environmental process. Numerous lipases have been studied for biodiesel production, with diverse triglyceride substrates and acyl acceptors. The use of edible oils is today not an option due to the food vs. fuel issue, which also makes them a more expensive raw material. Research studies so far have converted a series of non-edible fats and oils replacing the traditional alkaline transesterification. Process flow, reactor design and cost performance have been some of the main elements studied. It has been shown that high productivity, enzyme reuse and low reaction times can be achieved [62,63,64]. Nevertheless, enzymatic biodiesel still faces technical and economic challenges and has room for improvement to make enzymatic biodiesel a more attractive option for industrial production.
The most used method to produce enzymatic biodiesel is by transesterification of oil with an alcohol as acyl acceptor (Figure 5). Acyl acceptors, pose a challenge because the most available and cheap ones for industrial production and those who meet international specifications for biodiesel, are methanol and ethanol, which exert a strong denaturating action towards lipases [65]. Nevertheless, relatively successful efforts to overcome this inconvenience have emerged, like a pioneering work on stepwise alcohol addition [66], acyl acceptor alterations [67,68] and solvent engineering [69,70] which have sorted out this problem with relatively efficacy. Isopropanol [71], Isobutanol [72], 1-butanol [71], 2-butanol [72], 2-ethyl-1-hexanol [73], methyl acetate [74] and ethyl acetate [68] have been reported to be used as acyl acceptors. It was later documented that glycerol, a by-product of biodiesel synthesis, exerted lipase inhibition as well, which is more likely due to mass transfer limitation in the immobilized lipase [75].
Figure 5.
Enzymatic reaction of transesterification of oils with alcohol to produce fatty acid esters (biodiesel).
Figure 5.
Enzymatic reaction of transesterification of oils with alcohol to produce fatty acid esters (biodiesel).
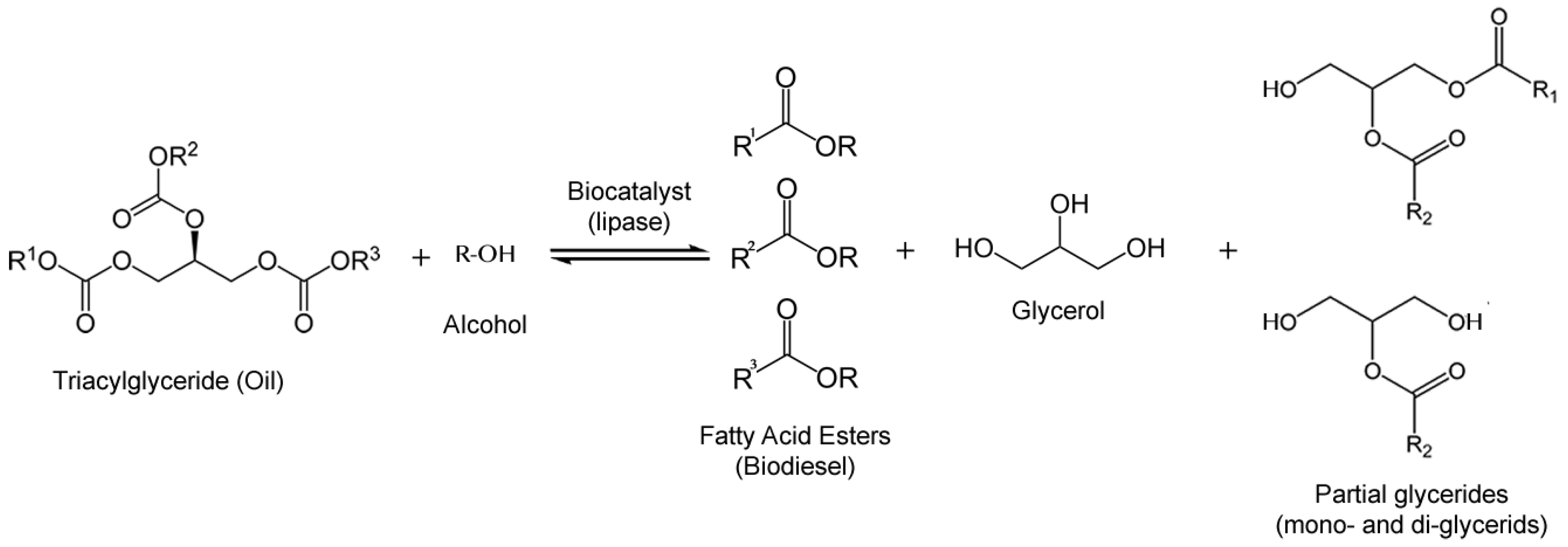
Free and immobilized lipases of fungal and yeast origin such as Mucor sp., Rhizopus sp., Candida sp., Aspergillus sp., Thermomyces sp., Penicillinum sp. and Pseudomonas sp. have shown to be good biocatalysts for biodiesel synthesis as yields for most of them are reported to be 80%–95% [58]. Enzyme immobilization is a cost which greatly influences the economic viability when scaling-up industrial biodiesel projects; thus, alternative methods of preparing the enzyme is a current active topic in the field [76].
Organic solvents have been used to solve problems like substrate solubility and mass transfer [63]. Hydrophobic organic solvents have been used because they improve trygliceride and fatty substrates solubility [77], but leave viscous glycerol insoluble, which poses mass transfer limitations as stated above. Despite the denaturing effects of hydrophilic organic solvents over enzymes [78,79], some solvents like 1,4-dioxane and tert-butanol have been proven to generate high transesterification yields [69,70,80]. Incubation of lipases in alcohols with carbon number ≥3 has been documented to improve enzymatic activity in the synthesis of methyl esters [81]. Acetone has also been tested; however, the maximum yields of biodiesel have reached 40% [82].
The use of waste animal fat from slaughterhouses as raw material for the production of biodiesel by transesterification with ethanol was recently evaluated by Rivera et al. [69]. Enzymes N435 and Lipozyme RM IM at 45 °C at a 2% (w/w) enzyme load, in an organic (tert-butanol) and solvent-free systems were used. The biodiesel yields were 80% for the solvent-free system after 2 days of reaction, and 65% for the organic system after 1 day of reaction.
Recently, solvents like supercritical carbon dioxide have been used and biodiesel yields accounted for as much as 81% at 60 °C with a 15% (w/w) enzyme load during 4 h [83]. Ionic liquids have also been used for biodiesel synthesis [84,85] and one of the best results was achieved after 12 h at 50 °C in 1-ethyl-3-methylimidazolium trifluoromethane-sulfonate ([C2mim][OTf]) with an 80 % yield [86].
Intracellular lipases in the form of whole-cell biocatalysts, present a feasible alternative for biodiesel production as well. Their main advantage is the lack of laborious and costly extraction and purification procedures needed when an isolated, free or immobilized, enzyme is used [87]. In order to use whole-cell Biocatalysis, immobilization is advocated. One of the most accepted whole-cell biocatalyst systems for industrial applications is the use of filamentous fungi immobilized onto biomass support particles (BSPs) [88,89,90]. Aspergillus sp. and Rhizopus sp., have been identified as robust for whole-cell immobilization, and thus have become the most widely studied fungi for biodiesel production [58]. Oils from yeasts and fungi are an alternative source of oils for biodiesel, as they can accumulate long-chain triacylglycerols with 16 and 18 carbons. The oil contents obtained from several yeast strains such as Cryptococcus, Lipomyces, and Rhodotorula species have reached 60%–70% of their dry weight [91].
3.2. Industrial Polymers
Biocatalysis has opened new synthetic strategies for organic chemists because it has been applied to synthetic chemistry in order perform stereochemical complex reactions in a relatively simple, straightforward way, which otherwise would require laborious protection-deprotection steps, toxic reactants and/or high-temperature/pressure procedures [92,93]. Thus, these advantages of using enzymes have been adopted in the field of polymer synthesis, in which most polymers are difficult to produce or to control by conventional chemical methods. Polymers with specific structures can be prepared enzymatically. In contrast, attempts to attain similar levels of polymer structural control by conventional chemical methods may prove to be a true challenge. From an environmental stand point, polymers derived from enzyme-mediated catalysis, whether polyesters, polyphenols, polysaccharides, proteins, or other polymers, are in most cases biodegradable, a feature highly sought after these days.
The most common type of enzymes which are capable to catalyze polymerization reactions are: Transferases, Oxidoreductases and Hydrolases.
3.2.1. Transferases
Transferases transfer a glycosyl group from a sugar nucleotide donor to specific nucleophilic acceptors. Despite they impose a high degree of regio- and stereochemical control to the glycosidic bond they form, and thus having a great synthetic potential for interesting polymeric materials, their use for synthesis in vitro is limited and even prohibitive by both, the availability of enzymes due to expression and solubility, and by the high costs of expensive activated donor sugars such as uridine diphosphate. They are very sensitive biocatalysts so their isolation and use on a larger scale is not practical, and they are mostly reserved for specialty research endeavors [94].
3.2.2. Oxidoreductases
On the other hand, Oxidoreductases and particularly Hydrolases are more robust enzymes, less sensitive and easier to obtain and use in vitro. Polymers typically produced by these three types of enzymes are shown in Table 1.

| Enzymes | Polymers synthesized |
|---|---|
| Oxidoreductases | |
| Peroxidases Laccases Tyrosinases Glucose oxidases | Polyphenols, polyanilines, vinyl polymers |
| Transferases | |
| Glycosyl transferases Acyl transferases | Polysaccharides, cyclic oligosaccharides, polyesters |
| Hydrolases | |
| Glycosidases Lipases Peptidases Proteases | Polysaccharides, polyesters, polycarbonates, polyamides, polythioesters, polyphosphates |
3.2.2.1. Polyaromatics
Polyaromatics are widely found in nature such as lignin, and flavonoid compounds. Much of their role in nature is as structural component conferring structural strength to living systems, in the case of lignin, and as a bioactive molecule in the case of flavonoids. Phenol is the single aromatic most important compound for industrial applications. Current polymeric materials produced commercially from phenolic compounds as industrial plastics include Bakelite and poly-2,6-dimethyl-1,4-phenylene oxide) (PPO) which bear good toughness and temperature-resistant properties. However, the oxidative polymerization of phenol with a conventional catalyst usually gives insoluble products with uncontrolled structure [95]. Moreover, as Bakelite and PPO are phenol-formaldehyde resins, the toxicity of formaldehyde has brought limitations on their industrial production.
On the other hand, an enzyme-catalyzed polymerization, offers not only the typical benefits of Biocatalysis, but the possibility to have a better control over polymerization [96].
In a recent work, Salvachúa et al. [97] used Versatil peroxidase (VP) to crosslink several monomeric lignans and peptides. Results show lignan oligomers of a maximum DP of 9 with a Number Average Molecular Weight Mn = 3,300. As per the reactions with peptides, peptide oligomers with a maximum DP of 11 and a Mn = 6,500 were obtained. Hetero-oligomers between lignans and peptides were also synthesized by VP. Oligomers of a DP = 6 and a Mn = 2,300 were obtained where the ratio peptide: lignan was 1:5. The reactions were done at room temperature with 1.5 U mL−1 of VP, 0.1 mM H2O2 and 0.1 mM Mn2+, during 24 h.
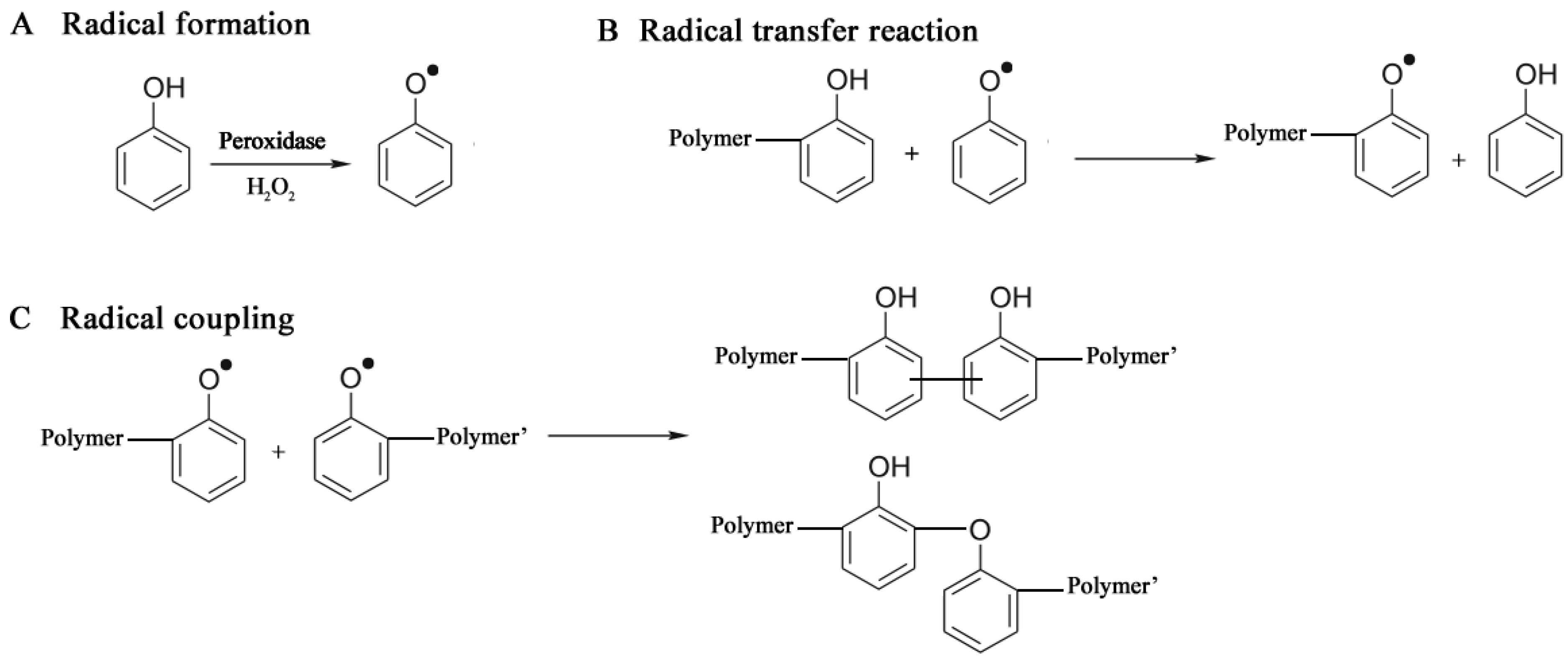
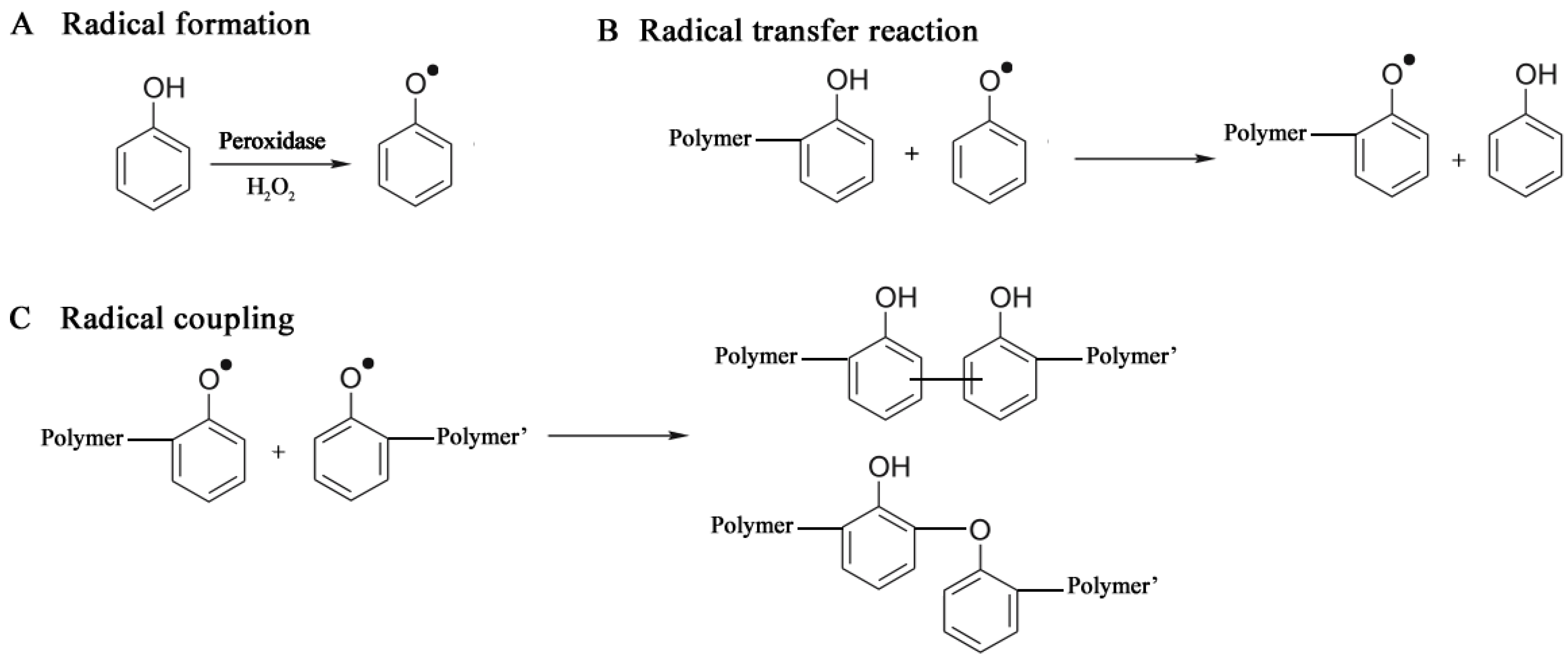
A peroxidase-catalyzed polymerization was performed under mild reaction conditions, using an aqueous buffer alcohol resulting in a soluble polyphenol with Mn of 3,000–6,000 [98,99]. The catalysts included horseradish peroxidase (HRP) and soybean peroxidase (SBP). The polymer solubility increased with increasing the oxyphenylene unit content (32%–59%) which was controlled by varying the methanol amount [100], and the solution pH [101]. Monodisperse polymer particles (250 nm diameter) were synthesized from phenol and subsituted phenols like m-cresol, p-cresol and polyphenylphenol, with HRP in a dispersion system 1,4-dioxane/buffer [102,103]. The mechanism of a peroxidase-catalyzed polymerization of phenols comprises three stages: Radical formation, Radical transfer and Radical coupling, shown in Figure 6.
Figure 6.
Mechanism of phenol polymer formation (see also [104]).
Figure 6.
Mechanism of phenol polymer formation (see also [104]).
It was revealed that meta-substituted phenol polymerizations catalyzed by HRP and SBP give rise both to higher yields when small and large-bulky substituents are used, respectively [105]. Cardanol, a natural phenol derived from cashew nut shell liquid (CNSL) with a C15 unsaturated alkyl meta-substituent, has good perspectives for industrial utilization such as resins, friction lining materials, and surface coatings. Therefore, an enzymatic approach to develop derivatives from this compound is attractive. Kobayashi et al. [106] obtained an artificial polymer structurally similar to “urushi”, a Japanese lacquer, through an SBP-polymerization of Cardanol. The polymer had a molecular weight between 2,000–4,000 Da., soluble in polar organic solvents and possessed a tough and hard property as a film with a glossy surface finish. Kim et al. [107] also synthesized polycardanol with SBP and found it had better anti-biofouling activity to Pseudomonas fluorescens compared to polypropylene.
Various other m-substituted phenols have been polymerized by HRP and SBP, for example meta-alkylphenols, meta-halogenated phenols, and meta-phenylphenol [108]. These enzymatically synthesized m-substituted polymers were applied to positive-type photoresists for printed wire boards, because of their high solubility toward alkaline solution and high thermal stability [108,109].
Ortho-substituted phenols have been polymerized with SBP affording a variety of oligophenols (dimers to pentamers) and some of their oxidation products, including quinones and demethylated quinones. Some of these are considered to serve as biologically important compounds with therapeutic potentials [110]. In separate reaction systems, it was possible to synthesize poly(phenylene oxide) (PPO) from two different o-substituted phenols, 2,6-dimethylphenol and 3,5-dimethoxy-4-hydroxybenzoic acid (syringic acid), using HRP, SBP and a laccase [111,112,113]. The polymers were formed exclusively of 2,6-dimethyl- or 2,6-dimethoxy-1,4-oxyphenylene units respectively, which are similar in structure to a widely used high-performance engineering plastic PPO.
Para-alkylphenols were polymerized by HRP in an aqueous 1,4-dioxane solution to synthesize polymers with Mn of several thousands, whereby the polymer yield increased as the n-alkyl chain length increased from 1 to 5 [102,114]. Oxidative polymerization of natural para-substituted phenols like tyrosine ethyl ester or methyl ester or phenol derivative hydroquinone-β-d-glucopyranoside (arbutin) [115] were carried out using HRP catalyst in an aqueous buffer solution. A mixture of phenylene and oxyphenylene units was observed in the tyrosine polymer, whereas the arbutin polymer, after degycosilation, consisted of C-C o-position hydroquinone units. Resulting polymers had a molecular weight from 1,500–4,000. Reactions catalyzed by HRP proceeded, involving radical species, yet took place chemoselectively. Para-phenylphenol is one well-studied para-substituted phenols in peroxidase-catalyzed polymerizations. Polymers up to 26,000 Da have been synthesized with HRP [99]. However, the polymerization of para-phenylphenol lacks precise structure control due to the existence of both stabilized ortho- and para-single electron radical species, causing different kinds of linkages and leading to a complex structural polymer [116].
Other numerous reports manage different aspects of the enzymatic polymerization of phenols: use of mixtures of aqueous-organic solvents with 1,4-dioxane, methanol, DMF [117], or total organic systems (isooctane) [118], ionic liquids [119], using cyclodextrins as a phenol solubilizing agent [120], incorporating PEG as a template aid [121], or carrying out the reaction in micelles and reverse micelles [122] or in capsules [123].
An important group of natural phenols widely distributed throughout the plant kingdom are flavonoids. Flavonoids are benzo-γ-pyrone compounds consisting of phenolic and pyrane rings. Their biological and pharmacological effects as antioxidant, antimutagenic, anticarcinogenic, antiviral and antiinflammatory have been extensively reviewed [124,125,126,127].
Flavanols or commonly known as catechins are the major group of polyphenols present in green tea. The main catechins in green tea are (+)-catechin, (−)-epicatechin (EC), (−)-epigallocatechin (EGC), (−)-epicatechin gallate (ECG), and (−)-epigal-locatechin gallate (EGCG). Poly(cathechin) has been synthesized by HRP in aqueous-organic solvents having yields of DMF-soluble polymers around 30% and molecular weights of 3,000 [128]. It has also been polymerized by a laccase derived from Myceliophthore (ML) in a mixture of acetone-buffer pH 5 rendering an 8,000 Da polymer [129]. In this case, as in other phenol polymerizations [100], the organic solvent/buffer ratio greatly affected the solubility of the synthesized polymer. The antioxidant properties of catechins and ECGC, not only remained, but increased in a concentration dependent manner in poly(catechin) and poly(ECGC) synthesized by HRP and ML [129,130]. Superoxide anion scavenging activity of poly(ECGC) was much superior than monomeric ECGC and poly(catechin) [130]. Inhibitory activity of Xanthine oxidase, which was negligible in monomeric catechin or ECGC, was markedly observed in poly(catechin) and in poly(ECGC), inclusive was higher than allopurinol, a commercial inhibitor frequently used for gout treatment [129,131]. Recently, a laccase was used to polymerize catechin in order to attach it to biomedical catheters in order to reduce bacterial biofilm formation [132].
Rutin has also been polymerized in organic-aqueous systems by Laccase ML-mediated catalysis. Yields superior to 70% were mostly observed and molecular weights between 7–9 kDa were registered [133].
Flavonols (quercetin) and isoflavones were also subject to an enzymatic oxidative polymerization by HRP and SBP in aqueous-organic solvent systems. Weight Average Molecular Weight Mw and Number Average Molecular Weight Mn for poly(quercetin) was 10,000 and 2,500 respectively, with a yield around 50%. Poly(rutin), poly(catechin), poly(daidzein), poly(formononetin) and others were also obtained in similar numbers [128].
3.2.3. Hydrolases
Hydrolases are the enzyme class with most widespread use and applications in the world. Around 75% of all commercialized industrial enzymes are hydrolytic in action. Despite proteases and carbohydrases are the most demanded subclass of enzymes accounting for around 40% of world’s enzyme sales, lipases remain as the biocatalyst subclass most widely used in organic chemistry [141,142].
3.2.4. Polyesters
Polyesters occupy the 4th place among the most important biomacromolecules in living systems. They are macromolecules with good biodegradability, biocompatibility and permeability, and thus are highly suitable for biomedical applications, such as implant biosorbable materials, for tissue engineering and delivery vehicles for drug or genes [147]. Polyesters are very valuable industrial materials that are broadly used like poly(ethylene terephthalate) (PET), poly(butylene succinate) (PBS), poly(ε-caprolactone) (PCL), and poly(lactic acid) (PLA).
Two major approaches can be taken to perform enzymatic polyester synthesis: (1) polycondensation and (2) ring-opening polymerization (ROP). PET and PBS are produced in large-scale by polycondensation whereas PCL and PLA are produced via ROP. Classical cationic, anionic or metallic chemical synthesis of polyesters may render them unsuitable for biomedical applications due to the toxicity of these reagents. Enzyme catalyzed synthesis of esters has circumvented this and other problems since enzymatic reactions do not require high energy input (temperature or pressure), most enzymes can carry out regio-, chemo- and enantiospecific reactions with no need of protecting groups, exhibit high catalytic activity, they may be reused and as a consequence of all this, reactions are environmentally and health safer than the conventional chemical route [148].
3.2.5. Polycondensations
3.2.5.1. Lipase-Catalyzed Polycondensations
In lipase-catalyzed polycondensations dicarboxylic acids are esterified with diols to produce polyesters. Reactions have been realized mostly in organic solvents, in solvent-free media and in aqueous systems. However, there have been studies in ionic liquids [149] and supercritical fluids as well [150]. In the early years of exploration of these systems, Okumura et al. [151] set a series of reactions with dicarboxylic acids and diols in aqueous systems using a lipase from Aspergillus niger and he obtained oligomers of 3–7 units. Extracting water molecules from the system, either with molecular sieves or applying vacuum, seemed to improve the polymer size up to a DP of 20 in an adipic acid-1,4-butanediol reaction [152], and up to a Mn = 77 kDa in an adipic acid-1,6-hexanediol reaction [153]. Organic solvents with high boiling points were identified to favor the polyester synthesis. In an aim to study natural hydroxy-acids, Gómez-Patiño et al. [154] obtained oligomers from tomato cuticle monomer, 10,16-dihydroxyhexadecanoic acid and its methyl ester. Candida antarctica B was the best performer among the 5 lipases tested in organic media reactions obtaining the largest oligomer with a DP = 3 and Mw = 1,206.
Reactions between dicarboxylic acids and diols conducted in water with Candida antarctica (N435), but with a continuous dehydrating process, are claimed to give rise to good yields [155].
Polycondensations with structurally different monomers have been realized, for example siloxane-containing diacids with diols (Mw = 20,000) [156], diacids with glycerol (Mw = 2,000–6,000) [157,158], thio-glycerol with diacids (Mw = 170,000) [159], diacids with a variety of sugars, sorbitol, erythritol, xylitol, ribitol, mannitol, glucitol, galactitol yielded polyesters with Mw = 10 kDa for galactitol and 75 kDa for Mannitol. The regioselecitivy of the sugar esterifications was mostly consistent in all the sugars to be on the primary hydroxyl groups [160].
Activation of dicarboxylic acids has been employed by means of esterification the carboxylic groups with methanol or ethanol [161], by esterification [162] or by vinyl activation [163]. Azim et al. [164], in an interesting strategy, firstly polymerized succinic acid with 1,4-butanediol to obtain a cyclic oligomer, which later was repolymerized by ring-opening to obtain a high molecular weight polymer (Mw = 130 kDa). Activation of dicarboxylic acids by means of esterification the carboxylic groups with halogenated alcohols like 2-chloroethanol, 2,2,2-trifluroethanol and 2,2,2-trichloroethanol lead to an increase of Mw by a factor of 5 [162]. However, the halogenated alcohols produced as a consequence of the reaction may damage the biocatalyst. Activation of carboxylic groups by vinyl esterification produced in most cases polyesters with Mw around 25 kDa [103,165,166].
Through reaction optimizations, Yao et al. [167] were able to achieve a polyester Mw = 16.6 kDa using 1,8-octanediol, adipic acid and l-malic acid as monomers. The incorporation of a sugar to be used as a third monomer is a likely means to modify the properties of conventional polymers, and an interesting strategy to prepare a variety of functional materials.
Recently it was reported the polymerization of β-alanine by a lipase catalyzed polycondensation reaction between β-alanine esters. Lipase B from Candida antarctica aggregated in CLEAs was the biocatalyst that converted β-alanine esters into polymers in high yield (up to 90%) and with high DP (up to 53 units). The best results were obtained by using methyl esters and methyl-tert-butyl ether (MTBE) as the solvent medium at 50 °C for 16 h [168].
3.2.5.2. Protease-Catalyzed Polycondensations
Proteases, like some other hydrolases, catalyze not only hydrolytic reactions (peptide hydrolysis in this case) but also peptide bond formation under appropriate conditions to give polypeptides. There exist several oligopeptides with varied biologically active properties that have been synthesized widely using proteases as catalysts via condensation reactions [169]. Some examples include, aspartame synthesized by thermolysin [170], oxytocin by papain, thermolysin, and chymotrypsin, and somatostatin by thermolysin, and chymotrypsin [171]. Various attempts to synthesize long-chain polyaminoamides were done by a protease-catalyzed polycondensation using chymotrypsin, trypsin, subtilisin and papain. However, only lower molecular weight polymers were obtained [172].
3.2.6. Polyamides
It was recently reported the synthesis of polyamides by lipase Novozym 435 in high yields (93%) and a short lapse of time (30 min). Authors attribute this high productivity to the use of “designed” monomers of the type: ω-amino-α-alkoxy acetic acid ethyl ester, which bears an ω-amino group and an oxygen atom in β position. The reactions were performed in bulk at 80 °C with an enzyme load of 45% (v/v) and the resultant polyamides had a DP = 15 (PDI = 1.6) and a Mw between 3,000–4,000 [173].
3.2.6. Vinyl Polymers
One field that has expanded tremendously in the past two decades is the enzyme-initiated radical polymerization of aromatic and vinyl monomers [149]. Vinyl monomers studied for enzymatic polymerization are basically classified into (meth)acrylic type and styryl type, which both involve the formation of a radical species. HRP and Laccase peroxidases have been reported to perform well in the meth(acrylic)-type polymerization of acrylamide. In a HRP-mediated free radical polymerization of vinyl monomers, the HRP-mediator system has three components: enzyme, oxidant (H2O2) and an initiatior, such as β-diketones which are commonly used. On the other hand, when several oxidoreductases, such as laccase, lipoxidase, and sarcosine oxidase, were used as catalysts, the acrylamide polymerization proceeded even without H2O2 or β-diketones. This was the case for laccase from Pycnoporus coccineus which induced the acrylamide polymerization in water at 50–80 °C under argon to produce poly(acrylamide) with Mn = 1 × 106 , Mw/Mn ≈ 2 for 4–24 h and up to 81% yields [174]. Since the laccase-mediator system (LMS) may induce the radical polymerization of vinyl monomer with no H2O2 and is environmentally benign, different conditions for polymerization with commercial laccase Myceliophthora thermophila were further examined and optimal conditions were found as slightly acidic reaction media at around 50 °C using β-diketones and O2 as the initiator and oxidant respectively. The molecular weight of poly(acrylamide) was Mn 6–28 × 104; Mw/Mn = 2.5–3.2 and could be controlled through the ratio of monomer to enzyme [175].
Poly(acrylamide) polymers of Mn of 1.5–4.6 × 105 with Mw/Mn 2.0–2.4, and acrylamide conversion between 70%–90% were synthesized by HRP in water at room temperature with 2,4-pentanedione as an initiator intermediate [176].
Enzyme-catalyzed vinyl polymerizations have been demonstrated in recent years with significant control of polymer molecular weight and yield depending on reaction conditions [149]. Resins of poly(sodium acrylate) are used as water-absorbent materials for cleaning surfaces, in water and oil conditioning, personal care products, and disposable materials for medical applications [177].
3.2.7. Nylons
Polyamides, also called nylons, display improved physical properties compared with polyolefins and polyesters due to the directionally specific inter-chain hydrogen bonds and significantly enhanced melting points [178]. Nylons can be synthesized either by polycondensation or by ROP. Ragupathy et al. [179] presented two and three step polycondensation and ROP methods to enzymatically produce nylon-8,10, nylon-8,13, nylon-6,13, and nylon-12,13. He used 1,8-diaminooctane (DAO) and diethyl sebacate (DES) for the polycondensations and DAO and lactone ethyleneridecanoate (ETD) for ROP. Novozym 435 in diphenyl ether at 150 °C carried out the polymerizations. Monomer conversion reached 97% and 90% and polyamides synthesized had an Mw of 5,000 and 8,000 for polycondensation and ROP reactions respectively.
3.2.8. Acrylate and Styrene Polymerization
Methyl-methacrylate (MMA) was polymerized by en enzyme-catalyzed reaction using the ternary system (enzyme, oxidant, and initiator) in water and water-miscible organic cosolvents such as DMF, acetone, dioxane, and THF. Soybean peroxidase in aqueous solution afforded a 48% yield of poly(methyl methacrylate) (PMMA) with Mn = 9.3 × 105 and Mw/Mn = 6.8. When HRP II (type II) was used, a 45% yield of PMMA was obtained with Mn = 6.3 × 105 and Mw/Mn = 3.0. It was found that yields of PMMA increased when cosolvents with low dielectric constant like dioxane and THF were used [180].
Respect to styrenic polymerizations, HRP in water was used to polymerize styrene and some derivatives. The polymerization was investigated using several β-diketones as initiators, and proceeded in a mixed solution of H2O/THF = 3 (v/v). Polystyrene synthesized using 2,5-cyclopentanedione as initiator resulted in 60% yield with Mn and Mw/Mn of 6.7 × 104 and 1.9 respectively. Polymer yield, Mn and Mw/Mn of the resulting polystyrene depended on the type of initiator used. Synthesis of polymers from styrene derivatives, 4-methylstyrene and 2-vinylnaphthalene were also investigated and afforded polystyrenes in a >90% yield of polymer with a Mn of 1.15 × 105 and Mw/Mn = 2.28 [181].
3.2.9. Ring-Opening Polymerizations (ROP)
Among the two major synthetic polymerization approaches, ring-opening polymerization (ROP) has been most extensively studied to polyester synthesis.
Polymerization of lactones via ring-opening catalysis carried out by lipases has been studied since the early 1990s. This catalytic activity for ROP of lactone monomers has been the research field on diverse lipases such as Pseudomonas fluorescens, Pseudomonas cepacia, Candida rugosa, Candida cylindracea, Candida antarctica, Aspergillus niger, Mucor miehei, Penicillium roqueforti, Rhizopus japanicus. Yarrowia lipolytica, Carica papaya and porcine pancreas lipase [182,183,184,185,186,187,188,189]. Cutinase from Humicola insolens (HiC) has also been reported to carry out ROP [190].
Poly(1,4-dioxan-2-one) (polyDO) is a desirable biocompatible polymer with good flexibility and tensile strength which might be considered for biomedical applications. This polyester was synthesized by ROP of 1,4-dioxan-2-one by lipase B from Candida antarctica at 60 °C, resulting in a polymer of Mw = 41,000 [191]. In another study, Jiang et al. [192] claimed to synthesize by ring-opening copolymerization a polyester from 1,4-dioxan-2-one (DO) with pentadecalactone (PDL) to give a copolyester of poly(DO-co-PDL) with Mw > 30,000. They used Candida antarctica B lipase in toluene or diphenyl ether at 70 °C for 26 h.
Lipase 2 from Yarrowia lipolytica and Carica papaya latex were recently employed to polymerize ε-CL in bulk. Lipase 2 from Y. lipolytica was found to be very efficient as catalyst for several reactions [193] and latex from C. papaya is a low-cost auto-immobilized biocatalyst. The polymerization yields were 74% and 40% with an Mw of 1,350 and 1,100 respectively. Reactions were realized at 150 °C and 6 h for Y. lipolytica and 70 °C and 24 h for C. papaya [182].
As to macrolides, lipase Pseudomonas fluorescens was able to polymerize 11-undecanolide (12-membered, UDL), 12-dodecanolide (13-membered, DDL), 15-pentadecanolide (16-membered, PDL), and 16-hexadecanolide (17-membered, HDL) [114,186,187,188,189,194,195,196]. Lipases from Candida cylindracea and Pseudomonas fluorescens catalyzed the ROP of UDL in bulk with quantitative yields affording polyesters of Mn of 23,000 and 25,000 [197,198]. In another study, ROP by lipases CA, lipase CC, lipase PC, lipase PF, or PPL in bulk at 45–75 °C for 5 days was done over HDL, the largest unsubstituted lactone monomer studied so far. The study showed the synthesis of polyHDL with Mn reaching to 5,800 in high yields [199]. It was discovered that a larger ring monomer, like PDL showed a greater reactivity towards polymerization than a smaller monomers like 1,4-polydioxan-2-one (DO) [192].
Ebata et al. obtained a poly(ε-CL) from a cyclic dimer of ε-CL (14-memberd) that was polymerized at 70 °C in toluene by lipase B from Candida antarctica, affording quantitatively a poly(ε-CL) with Mn of 18,000 [200]. This research Group, under the same strategy used to produce oligomers which are then subject to subsequent polymerization, claimed that a poly(butylene-succinate) (PBS) polyester of Mw 130,000 was obtained from cyclic dimers of butylene-succinate of Mn = 390. The reaction was carried out in toluene at 120 °C using 40% (w/w) of lipase Candida antarctica B for 24 h [201].
It was found that among ε-CL substituted monomers, ω-methyl ε-CL showed the least reactivity towards the lipase-catalytic ROP [202]. On the other hand, α-methyl or δ-methyl ε-CL afforded polymers of Mw = 8,400–11,000 with yields of 74% and 93% respectively [203].
An equilibrium phenomenon was observed between the synthesis of cyclic and linear polymers in a lipase-catalyzed ROP reaction of lactones. Cyclic oligomers in addition to major linear polyesters coexist in equilibrium [204,205]. This cyclic-linear equilibrium is solvent-dependent, for example Novozym 435 was used to catalyze the ROP of ε-CL at 60 °C in bulk or in an organic solvent. In bulk, the polymer products coexisted as a cyclic dimer of ε-CL and linear poly(ε-CL) in 2% and in 98% abundance, respectively. When the reaction was carried out in in acetonitrile, THF or 1,4-dioxane the same proportion was observed, as for example the equilibrium yields in acetonitrile were 70% for the cyclic oligomers (17% of dilactone and 53% of cyclic oligomers) and 30% for linear poly(ε-CL). When the reaction was carried out in isooctane, the proportion inverted to 3% and 97%, respectively, as in the bulk reaction [204].
Another hydrolase enzyme, a depolymerase, was found to catalyze ROP reactions in vitro. This depolymerase is a poly(hydroxybutyrate) (PHB) (EC 3.1.1.75), and was extracted from Pseudomonas stutzeri, Alcaligenes faecalis and Pseudomonas lemoignei. A. faecalis PHB depolymerase was assayed with a series of small, medium and large lactones and the best polimerization activity (93% yield) was obtained with β-butyrolactone, resulting in the formation of polyesters with a Mw = 16,000. On the other hand, medium and large lactones (ε-caprolactone, 11-undecanolide, and 12-dodecanolide), which are readily polymerized by lipases, were scarcely polymerized by PHB depolymerase [206].
3.3. Commodity Chemicals
One of the most noteworthy cases of biocatalytic production of a commodity chemical is the bioconversion of acrylonitrile to acrylamide [207]. This reaction, schematically simplified in Figure 7, is carried out by a nitrile hydratase whose catalytic activity transforms a cyanide group into an amide. Mitsubishi Rayon Co., Ltd. (Tokyo, Japan) currently produces over 20,000 tons annually of acrylamide using a third-generation biocatalyst, Rhodococcus rhodochrous J1, developed for commercial use by Nitto Chemical Industries. Acrylamide is produced from acrylonitrile in a continuous system of fixed-bed reactors at 10 °C with polyacrylamide-immobilized J1 cells. The process achieves conversion of acrylonitrile to acrylamide in ~99.9% yield, and the catalyst productivity is >7,000 g of acrylamide per g dry cell weight [208].
Figure 7.
Enzymatic conversion of acrylonitrile to acrylamide by means of a nitrile hydratase.
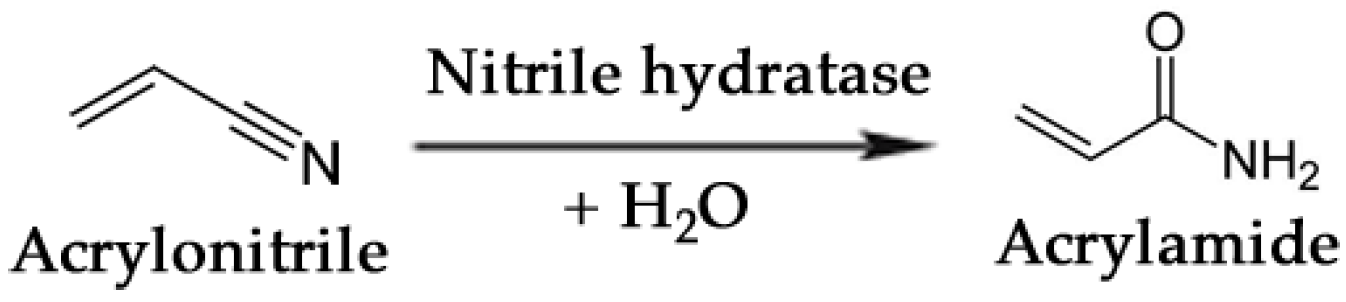
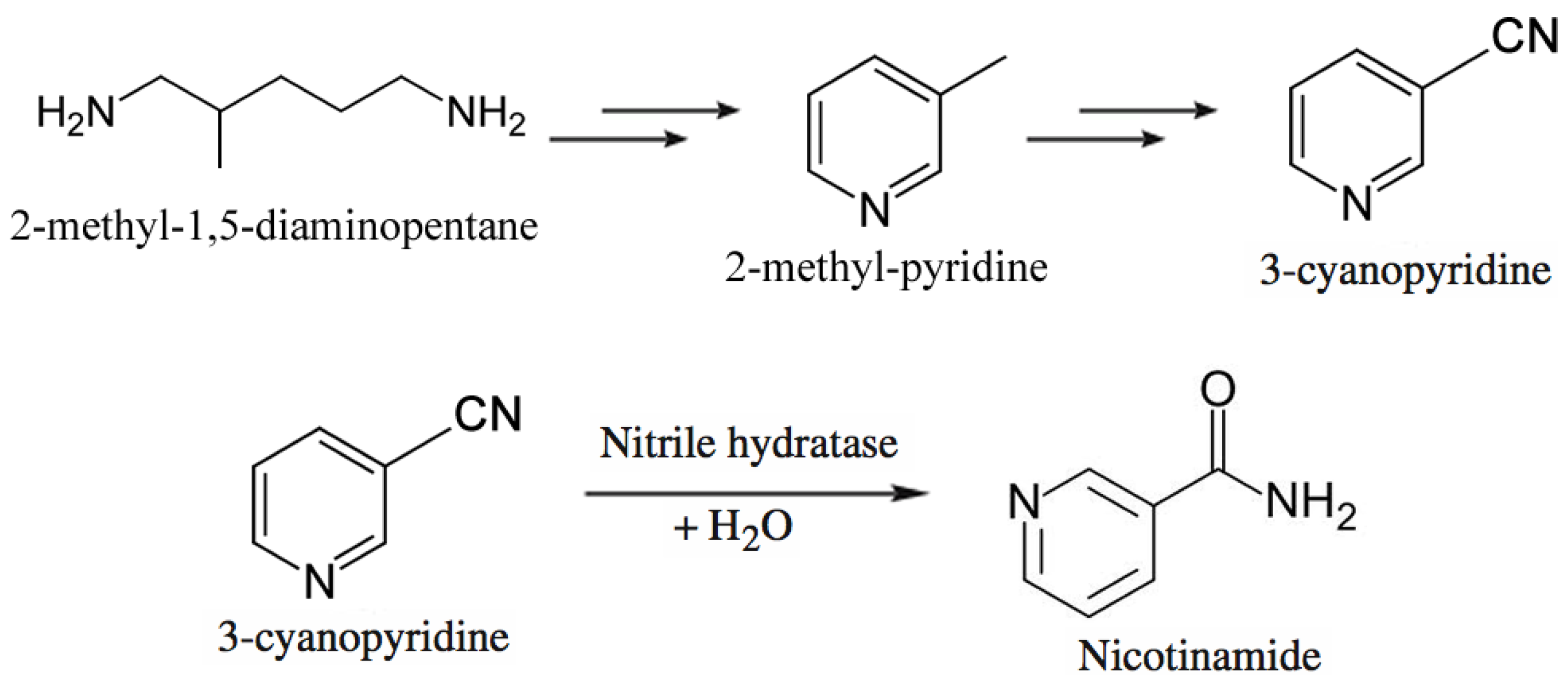
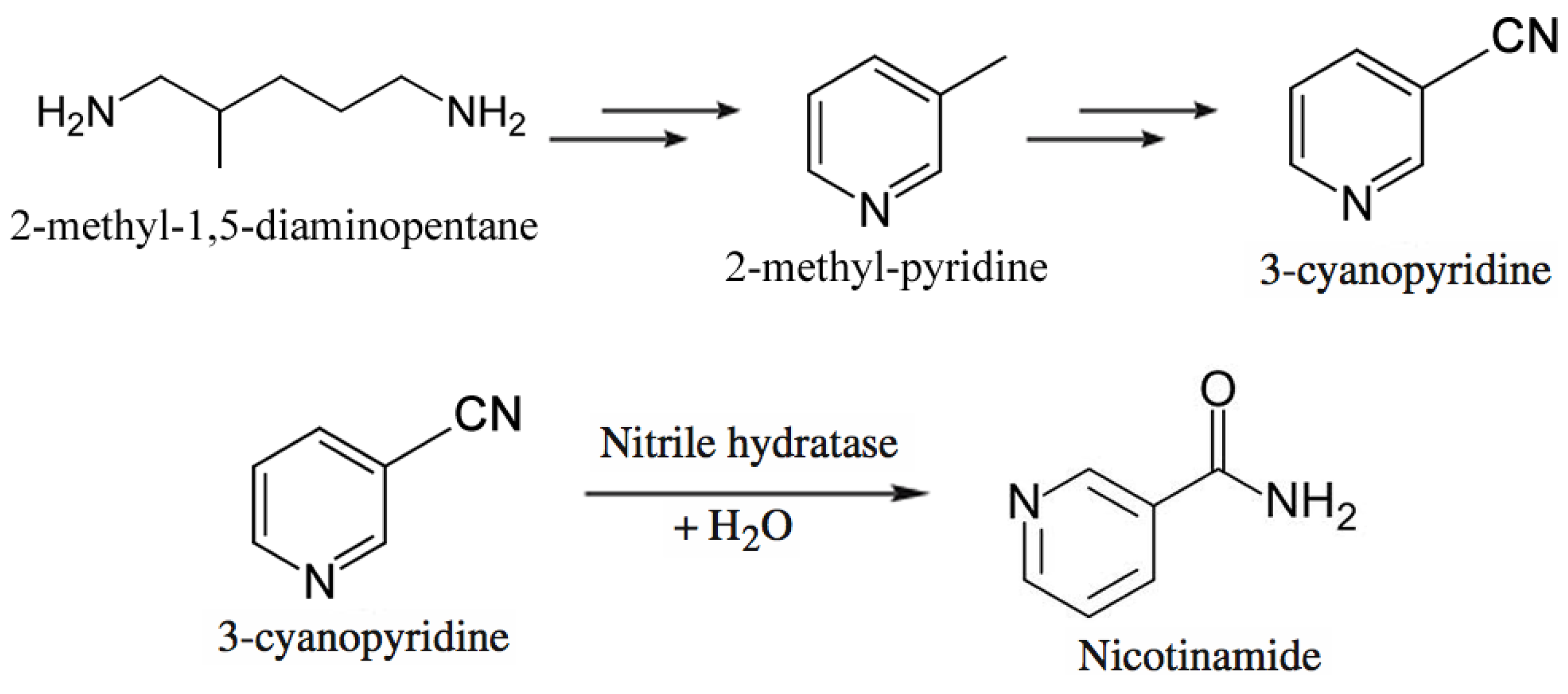
Lonza Guangzhou Fine Chemicals produces nicotinamide through a chemoenzymatic process shown in Figure 8 [209,210]. They start from 2-methyl-1,5-diaminopentane, a byproduct from production of nylon-6,6, which is converted to 3-methylpyridine, which in turn is ammoxidized. The resulting 3-cyanopyridine is hydrolyzed to nicotinamide using immobilized Rhodococcus rhodochrous J1 cells [211,212]. The Plant’s capacity is 3,400 tons per year of nicotinamide. The enzymatic process affords the desired amide at >99.3% selectivity at 100% conversion, whereas the chemical process requires caustic hydrolysis of 3-cyanopyridine but also produces 4% nicotinic acid as byproduct.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
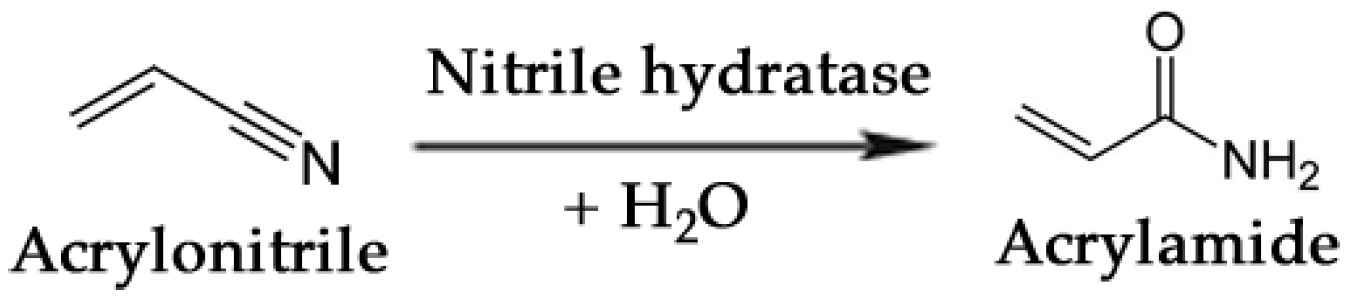{kind=link}
{kind=link}
{kind=link}
A commodity chemical which is aimed to substitute to some extent the universally oil-derived terephthalic acid is 2,5-furandicarboxylic acid (FDC) [213]. Terephthalic acid is one of the main raw materials to produce polyesters. FDC relies in the supply of 5-hydroxymethylfurfural (HMF), which is the product of the dehydration of hexoses. Oxidation of HMF yields FDC and may be realized by using heterogeneous or electrochemical catalysis [214,215]. However, two biocatalytic processes have been devised to help in having a reliable and sustainable supply for HMF: a whole-cell biocatalytic approach using a recombinant Pseudomonas putida hosting an oxidoreductase from Cupriavidus basilensis. This process is able to produce 30 g/L of 2,5-furandicarboxylic acid from HMF having a yield of 0.97 mol/mol [216]. The productivity of the whole process is 0.21 g/(L∙h) under aerobic fed-batch conditions. The second approach consists of the use of a chloroperoxidase and C. antarctica lipase B [217,218].
4. Enzymes in the Fine Chemicals Industry
The role of Biocatalysis in the pharmaceutical and fine-chemical industries is clearly expanding. There are estimated to be around 150 implemented biocatalytic processes in industry, and the majority of these are in the pharmaceutical sector [219]. As stereoisomerism is quite relevant in pharmaceutics and fine chemistry, and a single swap in hydrogen position may mean a great difference in the bioactive function of a compound, Biocatalysis fits well to meet these challenges. Besides the enantio-, regio- and chemoselectivity that enzymes exhibit, their ability to perform complex catalysis procedures in a simple step, which otherwise, under a chemical approach, might require laborious treatments, toxic reagents or high energy input, has provided a strong basis for their usage in the fine chemicals industry. Some examples of the contribution of Biocatalysis to this field are described below.
A precursor of aspartame, N-(benzyloxycarbonyl)-l-aspartyl-l,-phenylalanine methyl ester (Z-APM) has been synthesized from N-(benzyloxycarbonyl)-l-aspartic acid (Z-l-ASP) and l-phenylalanine methyl ester (l-PM) with thermolysin [220]. By reverting the peptide-bond hydrolytic nature of thermolysin, this precursor is synthesized on a multi-thousand ton per year scale by a DSM/Tosoh joint venture (Holland Sweetener Company, Geleen, The Netherlands).
Pyrethroids now constitute the majority of commercial household insecticides. A novel industrial application of a lyase enzyme is to produce a pyrethroid intermedieate, (S)-m-phenoxy-benzaldehyde cyanohydrin (sPBC). Hydroxynitrile lyase, is an enzyme that catalyzes the addition of HCN to aldehydes and ketones [221]. Thus, the production of sPBC from m-phenoxybenzaldehyde by hydroxynitril lyase has been carried out on a multi-ton scale by DSM.
l-carnitine is a quaternary ammonium compound biosynthesized from the amino acids lysine and methionine [222]. Within mammal cells, l-carnitine is required for the transport of fatty acids from the cytosol into the mitochondria during the lipids oxidation [223]. Due to this role in lipid oxidation, l-carnitine is often advertised to improve fat metabolism, reduce fat mass, and increase muscle mass, which is widely available as an over-the-counter nutritional. l-carnitine is produced by Lonza (Basel, Switzerland) on an industrial scale using an enzyme from strain Agrobacterium HK1349 in a whole cell biotransformation process [224]. The process actually comprises a dehydrogenation of γ-butyrobetaine to 4-(trimethylamino)-butenoic acid followed by selective addition of a water molecule by l-carnitine lyase.
l-tert-leucine is an important chiral building block and intermediate of many ligands in chemo-catalysis in the pharmaceutical industry [225]. Menzel et al. [226] presented a process to produce l-tert-leucine in a whole-cell Biocatalysis system using a recombinant E. coli host expressing the two enzymes required by the process: a leucine hydrogenase and a formate dehydrogenase. The former catalyzes the main reaction, reductive amination of an α-keto acid, and the latter serves as a NADH regenerator. A yield of 84% in 24 h and an enantiomeric purity of ee > 99% was obtained. The process has been scaled up to tons level. Degussa-Hüls has used a similar whole-cell Biocatalysis system to produce l-tert-leucine, which has taken to a commercial scale [227].
Lonza produces 5-methylpyrazine-2-carboxylic acid on a commercial scale from the p-xylene analogue 2,5-dimethylpyrazine. This compound is used as a blood-lowering drug of the sulfenyl urea class commercialized as Glycotrol. The synthesis is carried out in P. putida ATCC33015 as a whole-cell biocatalyst, expressing a series of enzymes (a monooxygenase and two dehydrogenases) [224].
Japan Energy (Saitama, Japan) produces alkyloxiranes and phenyloxiranes on an industrial scale, especially the chiral building block 2-(trifluoromethyl)oxirane from 3,3,3- trifluoropropene through an oxidative reaction, using a whole-cell biocatalyst from Nocardia corallina [228].
Vanillin, the main component of vanillin extract, a natural flavoring, whose demand has long exceeded the supply, should be synthesized chemically from guaiacol, in order meet demand. Nevertheless, a whole-cell biocatalytic synthesis of vanillin from glucose has now been elaborated. Glucose is transformed into vanillic acid by a recombinant E. coli biocatalyst under fed-batch fermentor conditions. Then reduction of vanillic acid to vanillin is carried out by aryl aldehyde dehydrogenase isolated from Neurospora crassa. The biocatalytic route avoids the use of carcinogenic reagents and non-renewable petroleum derivatives (guaiacol) [229].
Aresta et al. used a carboxylase in the synthesis of 4-hydroxy benzoic acid, which is an intermediate for the synthesis of preservatives. The carboxylase enzyme is extracted from the bacteria Thauera aromatica, and it acts over the phenyl-phosphate moeity carboxylating it with 100% selectivity and 90% yield at 300 °K, PCO2 = 0.1 MPa [230].
Alfuzosin, a quinazoline derivative, against Benign prostatic hyperplasia (BPH), is synthesized through intermediate compund tetrahydro-N-[3-(methylamino)-propyl]-2-furan-carboxamide. This intermediate was traditionally produced in a three-steps chemical route from 2-tetrahydrofuroic acid involving a series of chemicals and distillation. The enzyme-catalyzed reaction with lipase B from Candida antacrtica, afforded the intermediate in two steps in which the same enzyme catalyzed the two reactions: esterification and amidation. This biocatalytic procedure simplyfied and made more environmentally friendly the previous chemical synthesis [231].
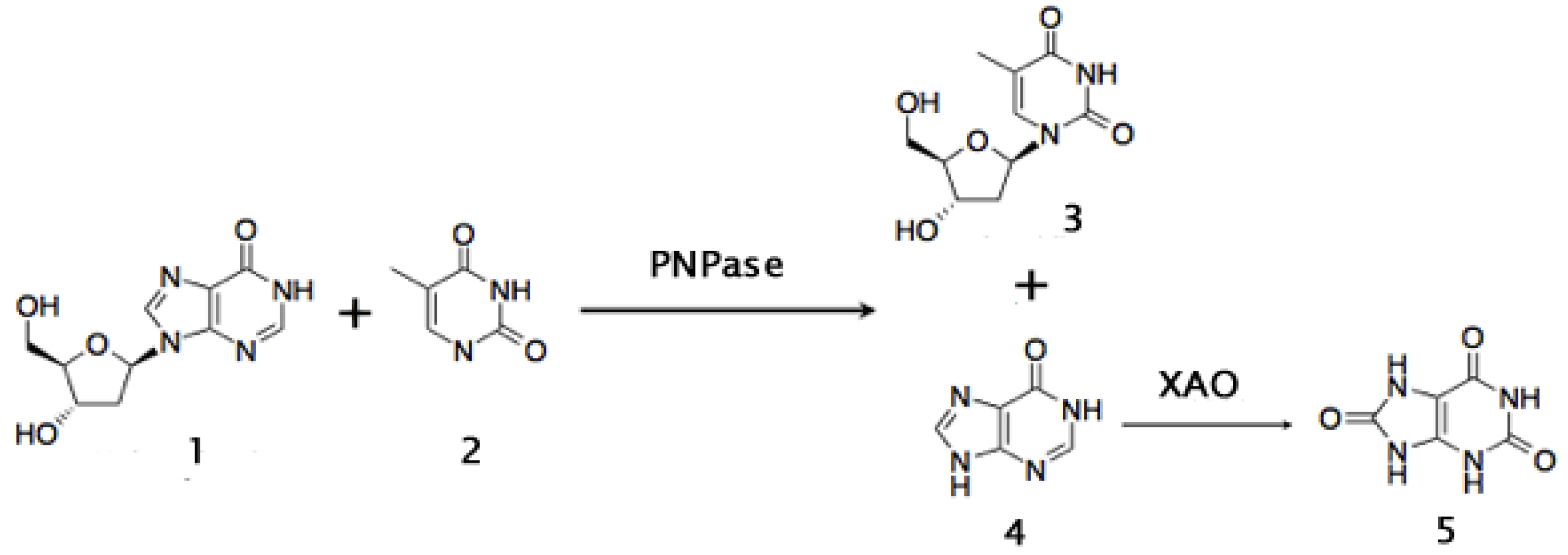
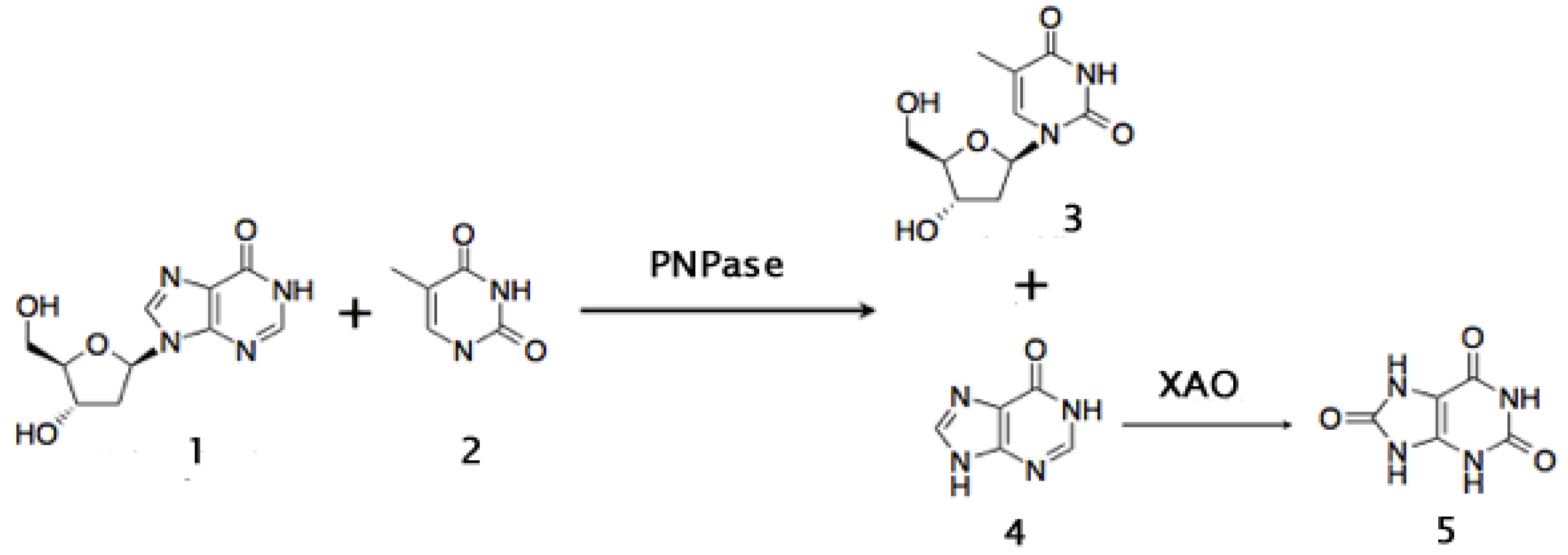
Another important intermediate in the synthesis of antiviral nucleosides such as 3'-deoxy-3'-azi-dothymidine (AZT), is thymidine. Production of thymidine is currently based in the hydrolysis of DNA from natural sources. A new biocatalytic approach for its production has been developed. It consists of transfering the 2'-deoxy-ribose moiety of 2'-deoxyinosine to thymine. The purine nucleoside phosphorylase of Bacillus stearothermophilus was used to carry out this reaction. A xantine oxidase is added in order to convert the concomitant hypoxantine produced to uric acid, and thus draw the thermodynamic equilibrium towards the synthesis of thymidine (Figure 9). The yield of thymidine of this whole-cell biocatalytic route reached 68% under mild conditions [232].
Figure 9.
Biocatalytic synthesis of thymidine using a Purine Nucleoside Phosphorilase (PNPase) and the biocatalytical removal of hypoxantine with a Xanthine Oxidase (XAO) to obtain uric acid. Compounds: 1: 2’deoxyinosine; 2: Thymine; 3: Thymidine; 4: Hypoxanthine; 5: Uric acid (see also [233]).
Figure 9.
Biocatalytic synthesis of thymidine using a Purine Nucleoside Phosphorilase (PNPase) and the biocatalytical removal of hypoxantine with a Xanthine Oxidase (XAO) to obtain uric acid. Compounds: 1: 2’deoxyinosine; 2: Thymine; 3: Thymidine; 4: Hypoxanthine; 5: Uric acid (see also [233]).
Many catalytic functions that enzymes perform in the pharmaceutical industry is in resolution of racemic mixtures, in which one enantiomer is acylated or hydrolyzed selectively affording an easier separation of both enantiomers. One recent example of this is the resolution of pro-drug (R,S)-2-bromophenylacetic acid octyl ester. Rivera et al. [234] accomplished its resolution with high enantioselectivity using lipases embedded in crude latex from Carica papaya. An enantioselectivity E-value E > 200 was achieved using decane as solvent, 50 mM of substrate and 50 mg/mL enzyme/reaction medium. Furthermore, the E-value doubled after purifying latex and removing proteases.
5. Conclusions
As outlined above, Biocatalysis is currently employed in a number of processes and products in diverse fields, and certainly new areas of application will be added. Advances in computational power have enabled the advent of bioinformatics, genomic sequencing and powerful analytic methods in physics, chemistry and molecular biology, which have allowed us to understand the dynamics, in vivo and in vitro, of biomacromolecules. The convergence of these tools has led to an improved access to more biocatalysts and to a deeper knowledge of them. In the case of enzymes, it has allowed us to explore their behavior, stability, specificity and even to begin to modify their very own nature through protein engineering. Larger availability of biocatalysts with superior qualities has been the main force behind the development of new industrial biocatalytic processes. The continued progress and interest in enzymes, serves as stimulus to make further efforts and ensure a steady success in meeting new synthetic challenges.
As per the environmental and economic benefits already reaped by the use of Biocatalysis in industry, it is possible to tell that the use of Biocatalysis is only expected to grow. This might be achieved with isolated enzymes but will probably be more successful using genetically engineered organisms where new pathways might be designed. The development of more robust, versatile, efficient enzymes via protein engineering, faster optimization of reaction conditions, microscale processing and better capabilities in design and machining new equipment will open new perspectives for the manufacture of many more products via Biocatalysis.
Future developments in Biocatalysis for industrial applications should be directed into the expansion of capabilities of current biocatalysts, to obtain new biocatalytic activities able to transform the vast amount of non-natural compounds found in industry and to achieve productivities comparable to current chemical processes. There is no doubt that modern biological tools, such as protein engineering and directed evolution will play an important role in meeting these challenges. As biodiversity remains largely unexplored, developing high-throughput screening methods to discover new efficient enzymes will also impact upon the development of biocatalyzed process for production of biobased chemicals.
Acknowledgments
We would like to thank to CONACYT for the scholarship provided to first author and for project CB 2008-01-104429.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Council of European Union. En Route to the Knowledge-Based Bio-Economy; German Presidency of the Council of European Union: Cologne, Germany, 2007. [Google Scholar]
- Sandoval, G.; Plou, F.G. Obtención enzimática de compuestos bioactivos a partir de recursos naturales iberoamericanos; Consejo Superior de Instigaciones Cientificas (CSIC): Madrid, Spain, 2012; pp. 1–336. [Google Scholar]
- Jahnz, U.; Schubert, M.; Baars, H.; Klaus, V. Process for producing the potential food ingredient DFA III from inulin-screening, genetic engineering, fermentation and immobilisation of inulase II. Int. J. Pharm. 2003, 256, 199–206. [Google Scholar] [CrossRef]
- Uchiyama, T.; Niwa, S.; Tanaka, K. Purification and properties of Arthrobacter ureafaciens inulase II. Biochim. Biophys. Acta 1973, 315, 412–420. [Google Scholar] [CrossRef]
- Kim, G.E.; Lee, S. Efficient production of DFA III (di-d-fructofuranose-dianhydride) from chicory root. In Abstracts of the World Congress on Biotechnology; DECHEMA: Berlin, Germany, 2000; Volume 272. [Google Scholar]
- Yokota, A.; Hirayama, S.; Enomoto, K.; Miura, Y.; Takao, S.; Tomita, F. Production of inulin fructotransferase (depolymerizing) by Arthrobacter sp. H65–7 and preparation of DFA III from inulin by the enzyme. J. Ferm. Bioeng 1991, 72, 258–261. [Google Scholar] [CrossRef]
- Kawamura, M.; Takahashi, S.; Uchiyama, T. Purification and some properties of inulin fructotransferase (depolymerizing) from Arthrobacter ilicis. Agric. Biol. Chem. 1988, 52, 3209–3210. [Google Scholar]
- Gosling, A.; Stevens, G.W.; Barber, A.R.; Kentish, S.E.; Gras, S.L. Recent advances refining galactooligosaccharide production from lactose. Food Chem. 2010, 121, 307–318. [Google Scholar] [CrossRef]
- Rastall, R.A.; Gibson, G.R.; Gill, H.S.; Guarner, F.; Klaenhammer, T.R.; Pot, B.; Reid, G.; Rowland, I.R.; Sanders, M.E. Modulation of the microbial ecology of the human colon by probiotics, prebiotics and synbiotics to enhance human health: An overview of enabling science and potential applications. FEMS Microbiol. Ecol. 2005, 52, 145–152. [Google Scholar] [CrossRef]
- Sanz, J.I. Production of Glactooligo-Sacchariddes from Lactose by Immobilized β-Galactosidase and Posterioir Chromatographic Spearation. Ph.D. Thesis, Ohio State University, Columbus, OH, USA, 2009. [Google Scholar]
- Gullón, B.; Gómez, B.; Martínez-Sabajanesb, M.; Yáñez, R.; Parajó, J.C.; Alonso, J.L. Pectic oligosaccharides: Manufacture and functional properties. Trends Food Sci. Technol. 2013, 30, 153–161. [Google Scholar] [CrossRef]
- Neri, D.F.M.; Balcão, V.M.; Costa, R.S.; Rocha, I.C.A.P.; Ferreira, E.M.F.C.; Torres, D.P.M.; Rodrigues, L.R.M.; Carvalho, L.B., Jr.; Teixeira, J.A. Galacto-oligosaccharides production during lactose hydrolysis by free Aspergillus oryzae β-galactosidase and immobilized on magnetic polysiloxane-polyvinyl alcohol. Food Chem. 2009, 115, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Delattre, C.; Michaud, P. Production of oligosaccharides as promising new food additive generation. Food Technol. Biotechnol. 2006, 44, 323–333. [Google Scholar]
- Gaur, R.; Pant, H.; Jain, R.; Khare, S.K. Galacto-oligosaccharide synthesis by immobilized Aspergillus oryzae β-Galactosidase. Food Chem. 2006, 97, 426–430. [Google Scholar] [CrossRef]
- Whisner, C.M.; Weaver, C.M. Galacto-oligosaccharides: Prebiotic effects on calcium absorption and bone health. In Nutritional Influences on Bone Health; Burckhardt, P., Dawson-Hughes, B., Weaver, C.M., Eds.; Springer: London, UK, 2013; pp. 315–323. [Google Scholar]
- Perugino, G.; Trincone, A.; Rossi, M.; Moracci, M. Oligosaccharide synthesis by glycosynthases. Trends Biotech. 2004, 22, 31–37. [Google Scholar] [CrossRef]
- Dias, L.G.; Veloso, A.C.A.; Correia, D.M.; Rocha, O.; Torres, D.; Rocha, I.; Rodrigues, L.R.; Peres, A.M. UV spectrophotometry method for the monitoring of galacto-oligosaccharides production. Food Chem. 2009, 113, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, B.; Poveda, A.; Jiménez, J.; Ballesteros, A.O.; Plou, F.J. Galacto-oligosaccharide synthesis from lactose solution or skim milk using the β-galactosidase from Bacillus circulans. J. Agric. Food Chem. 2012, 60, 6391–6398. [Google Scholar]
- Torres, D.; Gonçalves, M.D.P.F.; Teixeira, J.A.; Rodrigues, L. Galacto-oligosaccharides: Production, properties, applications, and significance as prebiotics. Compr. Rev. Food Sci. F. 2010, 9, 438–454. [Google Scholar] [CrossRef] [Green Version]
- Tzortzis, G.; Vulevic, J. Galacto-oligosaccharide prebiotics. In Prebiotics and Probiotics Science and Technology; Charalampopoulos, D., Rastall, R.A., Eds.; Springer: New York, NY, USA, 2009; pp. 207–244. [Google Scholar]
- Park, H.Y.; Kim, H.J.; Lee, J.K.; Kim, D.; Oh, D.K. Galactooligosaccharide production by a thermostable β-galactosidase from Sulfolobus solfataricus. World J. Microb. Biot. 2008, 24, 1553–1558. [Google Scholar] [CrossRef]
- Rodriguez, B.; de Abreu, M.A.; Fernandez, L.; de Beer, R.; Poveda, A.; Jimenez, J.; Haltrich, D.; Ballesteros, A.O.; Fernandez, M.; Plou, F.J. Production of galacto-oligosaccharides by the β-galactosidase from Kluyveromyces lactis: Comparative analysis of permeabilized cells versus soluble enzyme. J. Agric. Food Chem. 2011, 59, 10477–10484. [Google Scholar] [CrossRef]
- Hsu, C.A.; Lee, S.L.; Chou, C.C. Enzymatic production of galactooligosaccharides by β-galactosidase from Bifidobacterium longum BCRC 15708. J. Agric. Food Chem. 2007, 55, 2225–2230. [Google Scholar] [CrossRef]
- Hansson, T.; Adlercreutz, P. Optimization of galactooligosaccharide production from lactose using β-glycosidases from hyperthermophiles. Food Biotechnol. 2001, 15, 79–97. [Google Scholar]
- UBIC Europe Marketing Consulting. In The World GOS Market; UBIC Europe Press: Sierre, Switzerland, 2010.
- Padalino, M.; Perez-Conesa, D.; López-Nicolás, R.; Frontela-Saseta, C.; Berruezo, G. Effect of fructooligosaccharides and galactooligosaccharides on the folate production of some folate-producing bacteria in media cultures or milk. Int. Dairy J. 2012, 27, 27–33. [Google Scholar] [CrossRef]
- Fanaro, S.; Boehm, G.; Garssen, J.; Knol, J.; Mosca, F.; Stahl, B.; Vigi, V. Galacto-oligosaccharides and long-chain fructo-oligosaccharides as prebiotics in infant formulas: A review. Acta Paediatr. Suppl. 2005, 94, 22–26. [Google Scholar] [CrossRef]
- Watzl, B.; Girrbach, S.; Roller, M. Inulin, oligofructose and immunomodulation. Br. J. Nutr. 2005, 93, S49–S55. [Google Scholar] [CrossRef]
- Bornet, F.R.; Brouns, F.; Tashiro, Y.; Duvillier, V. Nutritional aspects of short-chain fructooligosaccharides: Natural occurrence, chemistry, physiology and health implications. Dig. Liver Dis. 2002, 34, S111–S120. [Google Scholar] [CrossRef]
- Yun, J.W. Fructooligosaccharides—Occurrence, preparation, and application. Enzyme Microb. Tech. 1996, 19, 107–117. [Google Scholar] [CrossRef]
- Arrizon, J.; Urias-Silvas, J.E.; Sandoval, G.; Mancilla-Margalli, N.A.; Gschaedler, A.C.; Morel, S.; Monsan, P. Production and bioactivity of fructan-type oligosaccharides. In Food Oligosaccharides: Production, Analysis and Bioactivity; Moreno, F.J., Sanz, M.L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; in press. [Google Scholar]
- Beine, R.; Morarua, R.; Nimtz, M.; Na’amniehc, S.; Pawlowskic, A.; Buchholz, K.; Seibel, J. Synthesis of novel fructooligosaccharides by substrate and enzyme engineering. J. Biotechnol. 2008, 138, 33–41. [Google Scholar] [CrossRef]
- Castillo, E.; López-Munguía, A. Synthesis of levan in water-miscible organic solvents. J. Biotechnol. 2004, 114, 209–217. [Google Scholar] [CrossRef]
- Vega, R.J.; Zúniga, M.E. Potential application of commercial enzyme preparations for industrial production of short-chain fructooligosaccharides. J. Mol. Catal. B Enzym. 2012, 76, 44–51. [Google Scholar] [CrossRef]
- Singh, R.S.; Singh, R.P. Production of fructooligosaccharides from inulin by endoinulinases and their prebiotic potential. Food Technol. Biotech. 2010, 48, 435–450. [Google Scholar]
- Tanriseven, A.; Aslan, Y. Immobilization of Pectinex Ultra SP-L to produce fructooligosaccharides. Enzyme Microb. Technol. 2005, 36, 550–554. [Google Scholar] [CrossRef]
- Guío, F.; Rodríguez, M.A.; Alméciga, C.J.; Sánchez, O.F. Recent trends in fructooligosaccharides production. Recent Pat. Food Nutr. Agric. 2009, 1, 221–230. [Google Scholar] [CrossRef]
- Jala, R.C.R.; Hu, P.; Yang, T.; Jiang, Y.; Zheng, Y; Xu, X. Lipases as biocatalysts for the synthesis of structured lipids. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, USA, 2012; Volume 861, Chapter 23. [Google Scholar]
- Xu, X.; Akoh, C.C. Enzymatic production of Betapol and other specialty fats. In Lipid Biotechnology; Marcel Dekker: New York, NY, USA, 2002; pp. 461–478. [Google Scholar]
- Odle, J. New insights into the utilization of medium-chain triglycerides by the neonate: Observations from a piglet model. J. Nutr. 1997, 127, 1061–1067. [Google Scholar]
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1987, 86, 439–472. [Google Scholar]
- Osborn, H.T.; Akoh, C. Structured lipids: Novel fats with medical, nutraceutical, and food applications. Compr. Rev. Food Sci. F. 2002, 1, 110–120. [Google Scholar] [CrossRef]
- Xu, X.; Hoy, C.E.; Balchen, S.; Adler-Nissen, J. Specific-Structured Lipid: Nutritional Perspectives and Production Potentials. In Proceedings of International Symposium on the Approach to Functional Cereals and Oils, CCOA, Beijing, China, 9–14 November 1997.
- Trivedi, R.; Singh, R.P. Modification of oils and fats to produce structured lipids. J. Oleo Sci. 2005, 54, 423–430. [Google Scholar] [CrossRef]
- Hellner, G.; Tőke, E.R.; Nagy, V.; Szakács, G.; Poppe, L. Integrated enzymatic production of specific structured lipid and phytosterol ester compositions. Process Biochem. 2010, 45, 1245–1250. [Google Scholar] [CrossRef]
- Lee, K.T.; Akoh, C.C. Characterization of enzymatically synthesized structured lipids containing eicosapentaenoic, docosahexaenoic, and caprylic acids. J. Am. Oil Chem. Soc. 1998, 75, 495–499. [Google Scholar] [CrossRef]
- Akoh, C.C. Structured lipids. In Food Lipids Chemistry, Nutrition, and Biotechnology; Akoh, C.C., Min, D.B., Eds.; Marcel Dekker: New York, NY, USA, 1998; pp. 699–727. [Google Scholar]
- Iwasaki, Y.; Yamane, T. Enzymatic synthesis of structured lipids. J. Mol. Catal. B Enzym. 2000, 10, 129–140. [Google Scholar] [CrossRef]
- Khodadadi, M.; Aziz, S.; St-Louis, R.; Kermash, S. Lipase-catalyzed synthesis and characterization of flaxseed oil-based structured lipids. J. Funct. Foods 2013, 5, 424–433. [Google Scholar] [CrossRef]
- Casas-Godoy, L.; Marty, A.; Sandoval, G.; Ferreira-Dias, S. Optimization of medium chain length fatty acid incorporation into olive oil catalyzed by immobilized Lip2 from Yarrowia lipolytica. Biochem. Eng. J. 2013, 77, 20–27. [Google Scholar] [CrossRef]
- Villeneuve, P.; Barouh, N.; Baréa, B.; Piombo, G.; Figueroa-Espinoza, M.C.; Turon, M.C.; Pina, M.; Lago, R. Chemoenzymatic synthesis of structured triacylglycerols with conjugated linoleic acids (CLA) in central position. Food Chem. 2007, 100, 1443–1452. [Google Scholar] [CrossRef]
- Nagachinta, S.; Akoh, C.C. Enrichment of palm olein with long chain polyunsaturated fatty acids by enzymatic acidolysis. LWT Food Sci. Technol. 2012, 46, 29–35. [Google Scholar] [CrossRef]
- Tecelão, C.; Rivera, I.; Sandoval, G.; Ferreira-Dias, S. Carica papaya latex: A low-cost biocatalyst for human milk fat substitutes production. Eur. J. Lipid Sci. Technol. 2012, 114, 266–276. [Google Scholar] [CrossRef]
- Nagao, T.; Watanabe, Y.; Maruyama, M.; Momokawa, Y.; Kishimoto, N.; Shimada, Y. One-pot enzymatic synthesis of docosahexaenoic acid-rich triacylglycerols at the sn-1(3) position using by-product from selective hydrolysis of tuna oil. New Biotechnol. 2011, 28, 7–13. [Google Scholar]
- Buchholtz, K.; Volker, K.; Borscheuer, U.T. Biocatalysts and Enzyme Technology; Wiley-VCH: Berlin, Germany, 2005; pp. 1–465. [Google Scholar]
- Bornscheuer, U.T.; Kazlauskas, R.J. Hydrolases in Organic Synthesis—Regio- and Stereoselective Biotransformations; Wiley-VCH: Berlin, Germany, 1999; pp. 1–355. [Google Scholar]
- Cao, L. Carrier-Bound Immobilized Enzymes; Wiley-VCH: Berlin, Germany, 2005; pp. 1–578. [Google Scholar]
- Gog, A.; Roman, M.; Toşa, M.; Paizs, C.; Irimie, F.D. Biodiesel production using enzymatic transesterification—Current state and perspectives. Renew. Energ. 2012, 39, 10–16. [Google Scholar] [CrossRef]
- Véras, I.C.; Silva, F.A.; Ferrão-Gonzales, A.D.; Moreau, V.H. One-step enzymatic production of fatty acid ethyl ester from high-acidity waste feedstocks in solvent-free media. Bioresour. Technol. 2011, 102, 9653–9658. [Google Scholar] [CrossRef]
- Fan, X.; Niehus, X.; Sandoval, G. Lipases as biocatalyst for biodiesel production. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, NY, USA, 2012; Volume 861, Chapter 27. [Google Scholar]
- Fukuda, H.; Kondo, A.; Noda, H. Biodiesel fuel production by transesterification of oils. J. Biosci. Bioeng. 2001, 92, 405–416. [Google Scholar]
- Maleki, E.; Aroua, M.K.; Sulaiman, N.M.N. Improved yield of solvent free enzymatic methanolysis of palm and jatropha oils blended with castor oil. Appl. Energ. 2013, 104, 905–909. [Google Scholar] [CrossRef]
- Hama, S.; Kondo, A. Enzymatic biodiesel production: An overview of potential feedstocks and process development. Bioresour. Technol. 2013, 135, 386–395. [Google Scholar] [CrossRef]
- Al-Zuhair, S.; Hasan, M.; Ramachandran, K.B. Kinetics of the enzymatic hydrolysis of palm oil by lipase. Process Biochem. 2003, 38, 1155–1163. [Google Scholar]
- Salis, A.; Pinna, M.; Monduzzi, M.; Solinas, V. Biodiesel production from triolein and short chain alcohols through biocatalysis. J. Biotechnol. 2005, 119, 291–299. [Google Scholar]
- Shimada, Y.; Watanabe, Y.; Samukawa, T.; Sugihara, A.; Noda, H.; Fukuda, H.; Tominaga, Y. Conversion of vegetable oil to biodiesel using immobilized Candida antarctica lipase. J. Am. Oil Chem. Soc. 1999, 76, 789–793. [Google Scholar]
- Li, Q.; Xu, J.; Du, W.; Li, Y.; Liu, D. Ethanol as the acyl acceptor for biodiesel production. Renew. Sust. Energ. Rev. 2013, 25, 742–748. [Google Scholar]
- Modi, M.K.; Reddy, J.R.; Rao, B.V.; Prasad, R.B. Lipase mediated conversion of vegetable oils into biodiesel using ethyl acetate as acyl acceptor. Bioresour. Technol. 2007, 98, 1260–1264. [Google Scholar]
- Rivera, I.; Villanueva, G.; Sandoval, G. Biodiesel production from animal grease wastes by enzymatic catalysis. Grasas Aceites 2009, 60, 468–474. [Google Scholar]
- Royon, D.; Daz, M.; Ellenrieder, G.; Locatelli, S. Enzymatic production of biodiesel from cotton seed oil using t-butanol as a solvent. Bioresour. Technol. 2007, 98, 648–653. [Google Scholar]
- Abigor, R.D.; Uadia, P.O.; Foglia, T.A.; Haas, M.J.; Jones, K.C.; Okpefa, E.; Obibuzor, J.U.; Bafor, M.E. Lipase-catalysed production of biodiesel fuel from some Nigerian lauric oils. Biochem. Soc. Trans. 2000, 28, 979–981. [Google Scholar]
- Nelson, L.; Foglia, T.; Marmer, W.N. Lipase-catalyzed production of biodiesel. J. Am. Oil Chem. Soc. 1996, 73, 1191–1195. [Google Scholar] [CrossRef]
- Linko, Y.Y.; Lamsä, M.; Huhtala, A.; Rantanen, O. Lipase biocatalysis in the production of esters. J. Am. Oil Chem. Soc. 1995, 72, 1293–1299. [Google Scholar]
- Du, W.; Xu, Y.; Liu, D.; Zeng, J. Comparative study on lipase-catalyzed transformation of soybean oil for biodiesel production with different acyl acceptors. J. Mol. Catal. B Enzym. 2004, 30, 125–129. [Google Scholar] [CrossRef]
- Watanabe, Y.; Shimada, Y.; Sugihara, A.; Noda, H.; Fukuda, H.; Tominaga, Y. Continuous production of biodiesel fuel from vegetable oil using immobilized Candida antarctica lipase. J.Am. Oil Chem. Soc. 2000, 77, 355–360. [Google Scholar]
- Zhang, B.; Weng, Y.; Xu, H.; Mao, Z. Enzyme immobilization for biodiesel production. Appl. Microbiol. Biotechnol. 2012, 93, 61–70. [Google Scholar] [CrossRef]
- Soumanou, M.M.; Bornscheuer, U.T. Lipase-catalyzed alcoholysis of vegetable oils. Eur. J. Lipid Sci. Technol. 2003, 105, 656–660. [Google Scholar]
- Zhu, X.; Zhou, T.; Wu, X.; Cai, Y.; Yao, D.; Xie, C.; Liu, D. Covalent immobilization of enzymes within micro-aqueous organic media. J. Mol. Catal. B Enzym 2011, 3–4, 145–149. [Google Scholar]
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 1985, 82, 3192–3196. [Google Scholar] [CrossRef]
- Iso, M.; Chen, B.; Eguchi, M.; Kudo, T.; Shrestha, S. Production of biodiesel fuel from triglycerides and alcohol using immobilized lipase. J. Mol. Catal. B Enzym. 2001, 16, 53–58. [Google Scholar] [CrossRef]
- Chen, J.W.; Wu, W.T. Regeneration of immobilized Candida antarctica lipase for transesterification. J. Biosci. Bioeng. 2003, 95, 466–469. [Google Scholar]
- Nie, K.; Xie, F.; Wang, F.; Tan, T. Lipase catalyzed methanolysis to produce biodiesel: optimization of the biodiesel production. J. Mol. Catal. B Enzym. 2006, 43, 142–147. [Google Scholar] [CrossRef]
- Ciftci, O.N.; Temelli, F. Enzymatic conversion of corn oil into biodiesel in a batch supercritical carbon dioxide reactor and kinetic modeling. J. Supercrit. Fluids. 2013, 75, 172–180. [Google Scholar] [CrossRef]
- Lin, Y.C.; Yang, P.M.; Chen, S.C.; Lin, J.F. Improving biodiesel yields from waste cooking oil using ionic liquids as catalysts with a microwave heating system. Fuel Process. Technol. 2013, 115, 57–62. [Google Scholar] [CrossRef]
- Earle, M.J.; Plechkova, N.V.; Seddon, K.R. Green synthesis of biodiesel using ionic liquids. Pure Appl. Chem. 2009, 81, 2045–2057. [Google Scholar]
- Ha, S.H.; Lan, M.N.; Lee, S.H.; Hwang, S.M.; Koo, Y.M. Lipase-catalyzed biodiesel production from soybean oil in ionic liquids. Enzyme Microb. Technol. 2007, 41, 480–483. [Google Scholar]
- Koda, R.; Numata, T.; Hama, S.; Tamalampudi, S.; Nakashima, K.; Tanaka, T; Ogino, C.; Fukuda, H.; Kondo, A. Ethanolysis of rapeseed oil to produce biodiesel fuel catalyzed by Fusarium heterosporum lipase-expressing fungus immobilized whole-cell biocatalysts. J. Mol. Catal. B Enzym. 2010, 68, 101–104. [Google Scholar]
- Adachi, D.; Koha, F.; Hamab, S.; Ogino, C.; Kondo, A. A robust whole-cell biocatalyst that introduces a thermo- and solvent-tolerant lipase into Aspergillus oryzae cells: Characterization and application to enzymatic biodiesel production. Enzyme Microb. Technol. 2013, 52, 331–335. [Google Scholar] [CrossRef]
- Fukuda, H.; Hama, S.; Tamalampudi, S.; Noda, H. Whole-cell biocatalysts for biodiesel fuel production. Trends Biotechnol. 2008, 26, 668–673. [Google Scholar] [CrossRef]
- Atkinson, B.; Black, G.M.; Lewis, P.J.S.; Pinches, A. Biological particles of given size, shape, and density for use in biological reactors. Biotechnol. Bioeng. 1979, 21, 193–200. [Google Scholar]
- Meng, X.; Yang, J.; Xu, X.; Zhang, L.; Nie, Q.; Xian, M. Biodiesel production from oleaginous microorganisms. Renew. Energ. 2009, 34, 1–5. [Google Scholar] [CrossRef]
- Turner, N.J.; Truppo, M.D. Biocatalysis enters a new era. Curr. Opin. Chem. Biol. 2013, 17, 212–214. [Google Scholar] [CrossRef]
- Woodley, J. New opportunities for biocatalysis: Making pharmaceutical processes greener. Trends Biotechnol. 2008, 26, 321–327. [Google Scholar] [CrossRef]
- Kittl, R.; Withers, S.G. New approaches to enzymatic glycoside synthesis through directed evolution. Carbohyd. Res. 2010, 345, 1272–1279. [Google Scholar] [CrossRef]
- Hay, A.S. Polymerization by oxidative coupling: Discovery and commercialization of PPO®. J. Polym. Sci. Pol. Chem. 1998, 36, 505–517. [Google Scholar] [CrossRef]
- He, F.; Li, S.; Garreau, H.; Vert, M.; Zhuo, R. Enzyme-catalyzed polymerization and degradation of copolyesters of ε-caprolactone and γ-butyrolactone. Polymer 2005, 46, 12682–12688. [Google Scholar] [CrossRef]
- Salvachúa, D.; Prieto, A.; Mattinen, M.L.; Tamminen, T.; Liitiä, T.; Lille, M.; Willför, S.; Martínez, A.T.; Martínez, M.J.; Faulds, C.B. Versatile peroxidase as a valuable tool for generating new biomolecules by homogeneous and heterogeneous cross-linking. Enzyme Microb. Technol. 2013, 52, 303–311. [Google Scholar] [CrossRef]
- Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Enzymatic synthesis of soluble polyphenol. Bull. Chem. Soc. Jpn. 2000, 73, 1389–1396. [Google Scholar] [CrossRef]
- Dordick, J.S.; Marletta, M.A.; Klibanov, A.M. Polymerization of phenols catalyzed by peroxidase in nonaqueous media. Biotech. Bioeng. 1987, 30, 31–36. [Google Scholar]
- Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Soluble polyphenol. Macromol. Rapid Commun. 1999, 20, 401–403. [Google Scholar] [CrossRef]
- Mita, N.; Oguchi, T.; Tawaki, S.; Uyama, H.; Kobayashi, S. Control of structure and molecular weight of polyphenols in enzymatic oxidative polymerization. Polymer Prepr. 2000, 41, 223–224. [Google Scholar]
- Kurioka, H.; Komatsu, I.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of alkylphenols. Macromol. Rapid Commun. 1994, 15, 507–510. [Google Scholar] [CrossRef]
- Uyama, H.; Kurioka, H.; Kobayashi, S. Preparation of polyphenol particles by dispersion polymerization using enzyme as catalyst. Chem. Lett. 1995, 24, 795–796. [Google Scholar]
- Uyama, H. Enzymatic polymerization. In Future Directions in Biocatalysis; Matsuda, T., Ed.; Elsevier Science: Cambridge, MA, USA, 2007; pp. 1–2. [Google Scholar]
- Reihmann, M.; Ritter, H. Synthesis of phenol polymers using peroxidases. Adv. Polym. Sci. 2006, 194, 1–49. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Ikeda, R. Artificial Urushi. Chem. Eur. J. 2001, 7, 4755–4760. [Google Scholar]
- Kim, Y.H.; An, E.S.; Song, B.K.; Kim, D.S.; Chelikani, R. Polymerization of cardanol using soybean peroxidase and its potential application as anti-biofilm coating material. Biotechnol. Lett. 2003, 25, 1521–1524. [Google Scholar] [CrossRef]
- Tonami, H.; Uyama, H.; Kobayashi, S.; Kubota, M. Peroxidase-catalyzed oxidative polymerization of m-substituted phenol derivatives. Macromol. Chem. Phys. 1999, 200, 2365–2371. [Google Scholar] [CrossRef]
- Kadota, J.; Fukuoka, T.; Uyama, H.; Hasegawa, K.; Kobayashi, S. New Positive-type photoresists based on enzymatically synthesized polyphenols. Macromol. Rapid Commun. 2004, 25, 441–444. [Google Scholar] [CrossRef]
- Antoniotti, S.; Santhanam, L.; Ahuja, D.; Hogg, M.G.; Dordick, J.S. Structural diversity of peroxidase-catalyzed oxidation products of o-methoxyphenols. Org. Lett. 2004, 6, 1975–1978. [Google Scholar] [CrossRef]
- Mita, N.; Tawaki, S.I.; Uyama, H.; Kobayashi, S. Laccase-catalyzed oxidative polymerization of phenols. Macromol. Biosci. 2003, 3, 253–257. [Google Scholar] [CrossRef]
- Ikeda, R.; Sugihara, J.; Uyama, H.; Kobayashi, S. Poly(2,6-dihydroxy-l,4-oxyphenylene synthesis of a new poly(phenylene oxide) derivative. Polym. Bull. 1997, 38, 273–277. [Google Scholar] [CrossRef]
- Ikeda, R.; Sugihara, J.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of 2,6-dimethylphenol. Macromolecules 1996, 29, 8702–8705. [Google Scholar] [CrossRef]
- Uyama, H.; Kurioka, H.; Sugihara, J.; Komatsu, I.; Kobayashi, S. Oxidative polymerization of p-alkylphenols catalyzed by horseradish peroxidase. J. Polym. Sci. Pol. Chem. 1997, 35, 1453–1459. [Google Scholar] [CrossRef]
- Wang, P.; Martin, B.D.; Parida, S.; Rethwisch, D.G.; Dordick, J.S. Multienzymic synthesis of poly(hydroquinone) for use as a redox polymer. J. Am. Chem. Soc. 1995, 117, 12885–12886. [Google Scholar] [CrossRef]
- Akkara, J.A.; Senecal, K.J.; Kaplan, D.L. Synthesis and characterization of polymers produced by horseradish peroxidase in dioxane. J. Polym. Sci. Pol. Chem. 1991, 29, 1561–1574. [Google Scholar] [CrossRef]
- Akita, M.; Tsutsumi, D.; Kobayashi, M.; Kise, H. Structural change and catalytic activity of horseradish peroxidase in oxidative polymerization of phenol. Biosci. Biotech. Bioch. 2001, 65, 1581–1588. [Google Scholar] [CrossRef]
- Angerer, P.S.; Studer, A.; Witholt, B.; Li, Z. Oxidative polymerization of a substituted phenol with ion-paired horseradish peroxidase in an organic solvent. Macromolecules 2005, 38, 6248–6250. [Google Scholar] [CrossRef]
- Eker, B.; Zagorevski, D.; Zhu, G.; Linhardt, R.J.; Dordick, J.S. Enzymatic polymerization of phenols in room-temperature ionic liquids. J. Mol. Catal. B Enzym. 2009, 59, 177–184. [Google Scholar] [CrossRef]
- Mita, N.; Tawaki, S.; Uyama, H.; Kobayashi, S. Enzymatic oxidative polymerization of phenol in an aqueous solution in the presence of a catalytic amount of cyclodextrin. Macromol. Biosci. 2002, 3, 127–130. [Google Scholar]
- Kim, Y.J.; Uyama, H.; Kobayashi, S. Regioselective synthesis of poly(phenylene) as a complex with poly(ethylene glycol) by template polymerization of phenol in water. Macromolecules 2003, 36, 5058–5060. [Google Scholar] [CrossRef]
- Kommareddi, N.S.; Tata, M.; Karayigitoglu, C.; John, V.T.; McPherson, G.L.; Herman, M.F.; Oconnor, C.J.; Lee, Y.S.; Akkara, J.A.; Kaplan, D.L. Enzymatic polymerizations using surfactant microstructures and the preparation of polymer-ferrite composites. Appl. Biochem. Biotechnol. 1995, 51–52, 241–252. [Google Scholar] [CrossRef]
- Ghan, R.; Shutava, T.; Patel, A.; John, V.T.; Lvov, Y. Enzyme-catalyzed polymerization of phenols within polyelectrolyte microcapsules. Macromolecules 2004, 37, 4519–4524. [Google Scholar]
- Marín, F.R.; Frutos, M.J.; Pérez-Alvarez, J.A.; Martinez-Sánchez, F.; del Río, J.A. Flavonoids as nutraceuticals: Structural related antioxidant properties and their role on ascorbic acid preservation. Stud. Nat. Prod. Chem. 2002, 26, 741–778. [Google Scholar] [CrossRef]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Flavonoids in Biology and Medicine III—Current Issues in Flavonoids Research; Das, N.P.; Cheeseman, K.H. (Eds.) Informa Healthcare: London, UK, 1991; Volume 14, pp. 77–78.
- Mejias, L.; Reihmann, M.H.; Sepulveda-Boza, S.; Ritter, H. New polymers from natural phenols using horseradish or soybean peroxidase. Macromol. Biosci. 2002, 2, 24–32. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Laccase-catalyzed synthesis and antioxidant property of poly(catechin). Macromol. Biosci. 2003, 3, 758–764. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Oxidative coupling of epigallocatechin gallate amplifies antioxidant activity and inhibits xanthine oxidase activity. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Kim, Y.J.; Uyama, H.; Kobayashi, S. Amplification of antioxidant activity and xanthine oxidase inhibition of catechin by enzymatic polymerization. Biomacromolecules 2003, 4, 469–471. [Google Scholar] [CrossRef]
- Gonçalves, I.; Matamá, T.; Cavaco-Paulo, A.; Silva, C. Laccase coating of catheters with poly(catechin) for biofilm reduction. Biocatal. Biotransform 2013, in press. [Google Scholar]
- Kurisawa, M.; Chung, J.E.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and antioxidant properties of poly(rutin). Biomacromolecules 2003, 4, 1394–1399. [Google Scholar] [CrossRef]
- Božič, M.; Gorgieva, S.; Kokol, V. Laccase-mediated functionalization of chitosan by caffeic and gallic acids for modulating antioxidant and antimicrobial properties. Carbohyd. Polym. 2012, 87, 2388–2398. [Google Scholar] [CrossRef]
- Brzonova, I.; Steiner, W.; Zankel, A.; Nyanhongo, G.S. Enzymatic synthesis of catechol and hydroxyl-carboxic acid functionalized chitosan microspheres for iron overload therapy. Eur. J. Pharm. Biopharm. 2011, 79, 294–303. [Google Scholar] [CrossRef]
- Fras-Zemljič, L.; Kokol, V.; Čakara, D. Antimicrobial and antioxidant properties of chitosan-based viscose fibres enzymatically functionalized with flavonoids. Text. Res. J. 2011, 81, 1532–1540. [Google Scholar] [CrossRef]
- Sousa, F.; Guebitz, G.M.; Kokol, V. Antimicrobial and antioxidant properties of chitosan enzymatically functionalized with flavonoids. Process Biochem. 2009, 44, 749–756. [Google Scholar] [CrossRef]
- Pina-Luis, G.; Rosquete-Pina, G.; Valdés, A.C.; Ochoa, A.; Rivero, I.; Díaz-García, M.E. Morin functionalized Merrifield’s resin: A new material for enrichment and sensing heavy metals. React. Funct. Polym. 2012, 72, 61–68. [Google Scholar] [CrossRef]
- Donato, L.; Chiappetta, G.; Drioli, E. Naringin-imprinted polymer layer using photo polymerization method. Sep. Sci. Technol. 2011, 46, 1555–1562. [Google Scholar] [CrossRef]
- Spizzirri, U.G.; Parisi, O.I.; Iemma, F.; Cirillo, G.; Puoci, F.; Curcio, M.; Picci, N. Antioxidant-polysaccharide conjugates for food application by eco-friendly grafting procedure. Carbohyd. Polym. 2010, 79, 333–334. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar]
- Sharma, R.; Chisti, Y.; Banerjee, U.C. Production, purification, characterization and applications of lipases. Biotechnol. Adv. 2001, 19, 627–662. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.; Liu, C.; Zhao, Z.; Yang, Y.; Li, Q. Lipase/esterase-catalyzed synthesis of aliphatic polyesters via polycondensation: A review. Process Biochem. 2012, 47, 1027–1036. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, Y.; Zhang, Y.; Liu, C.; Shi, W.; Li, Q. Lipase/esterase-catalyzed ring-opening polymerization: A green polyester synthesis technique. Process Biochem. 2011, 46, 1900–1908. [Google Scholar] [CrossRef]
- He, F.; Wang, Y.P.; Liu, G.; Jia, H.L.; Feng, J.; Zhuo, R.X. Synthesis, characterization and ring-opening polymerization of a novel six-membered cyclic carbonate bearing pendent allyl ether group. Polymer 2008, 49, 1185–1190. [Google Scholar] [CrossRef]
- Runge, M.; O’Hagan, D.; Haufe, G. Lipase-catalyzed polymerization of fluorinated lactones and fluorinated hydroxycarboxylic acids. J. Polym. Sci. Pol. Chem. 2000, 38, 2004–2012. [Google Scholar] [CrossRef]
- Zinck, P. One-step synthesis of polyesters specialties for biomedical applications. Rev. Environ. Sci. Biotechnol. 2009, 8, 231–234. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Uyama, H.; Takamoto, T.; Kobayashi, S. Enzymatic synthesis of polyesters in ionic liquids. Polym. J. 2002, 34, 94–96. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Beckman, E.J.; Russell, A.J. Rational control of polymer molecular weight and dispersity during enzyme-catalyzed polyester synthesis in supercritical fluids. J. Am. Chem. Soc. 1995, 117, 3728–3733. [Google Scholar] [CrossRef]
- Okumura, S.; Iwai, M.; Tominaga, T. Synthesis of ester oligomer by Aspergillus niger lipase. Agr. Biol. Chem. (Tokyo) 1984, 48, 2805–2813. [Google Scholar] [CrossRef]
- Binns, F.; Roberts, S.M.; Taylor, A.; Williams, C.F. Enzymic polymerization of unactivated diol/diacid system. J. Chem. Soc. Perkin Trans. 1 1993, 1, 899–904. [Google Scholar]
- Linko, Y.Y.; Seppala, J. Producing high molecular weight biodegradable polyesters. Chem. Tech. 1996, 26, 25–31. [Google Scholar]
- Gómez-Patiño, M.B.; Cassani, J.; Jaramillo-Flores, M.E.; Zepeda-Vallejo, L.G.; Sandoval, G.; Jimenez-Estrada, M.; Arrieta-Baez, D. Oligomerization of 10,16-dihydroxyhexadecanoic acid and methyl 10,16-dihydroxyhexadecanoate catalyzed by lipases. Molecules 2013, 18, 9317–9333. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Suda, S.; Namekawa, S. Dehydration polymerization in aqueous medium catalyzed by lipase. Chem. Lett. 1997, 26, 105–107. [Google Scholar] [CrossRef]
- Poojari, Y.; Palsule, A.S.; Cai, M.; Clarson, S.J.; Gross, R.A. Synthesis of organosiloxane copolymers using enzymatic polyesterification. Eur. Polym. J. 2008, 44, 4139–4145. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, W.; Cai, J.; Hou, Y.; Ouyang, S.; Xie, W.; Gross, R.A. Poly(oleicdiacid-co-glycerol): Comparison of polymer structure resulting from chemical and lipase catalysis. Macromolecules 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Korupp, C.; Weberskirch, R.; Muller, J.J.; Liese, A.; Hilterhaus, L. Scaleup of lipase-catalyzed polyester synthesis. Org. Process. Res. Dev. 2010, 14, 1118–1124. [Google Scholar] [CrossRef]
- Fehling, E.; Bergander, K.; Klein, E.; Weber, N.; Vosmann, K. Thiol-functionalized copolymeric polyesters by lipase-catalyzed esterification and transesterification of 1,12-dodecanedioic acid and its diethyl ester, respectively, with 1-thioglycerol. Biotechnol. Lett. 2010, 32, 1463–1471. [Google Scholar] [CrossRef]
- Hu, J.; Gao, W.; Kulshrestha, A.S.; Gross, R.A. Sweet polyesters: Lipase-catalyzed condensation polymerization of alditols. Macromolecules 2006, 39, 6789–6792. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. Enzymatic synthesis of polyesters via polycondensation. Adv. Polym. Sci. 2006, 194, 133–158. [Google Scholar] [CrossRef]
- Brazwell, E.M.; Filos, D.Y.; Morrow, C.J. Biocatalytic synthesis of polymers. III. Formation of a high molecular weight polyester through limitation of hydrolysis by enzyme-bound water and through equilibrium control. J. Polym. Sci. Pol. Chem. 1995, 33, 89–95. [Google Scholar] [CrossRef]
- Kobayashi, S. Recent developments in lipase-catalyzed synthesis of polyesters. Macromol. Rapid Commun. 2009, 30, 237–266. [Google Scholar] [CrossRef]
- Azim, H.; Dekhterman, A.; Jiang, Z.; Gross, R.A. Candida antarctica lipase B-catalyzed synthesis of poly(butylene succinate): Shorter chain building blocks also work. Biomacromolecules 2006, 7, 3093–3097. [Google Scholar] [CrossRef]
- Mesiano, A.J.; Beckman, E.J.; Russell, A.J. Biocatalytic synthesis of fluorinated polyesters. Biotechnol. Prog. 2000, 16, 64–68. [Google Scholar] [CrossRef]
- Uyama, H.; Yaguchi, S.; Kobayashi, S. Lipase-catalyzed polycondensation of dicarboxylic acid-divinyl esters and glycols to aliphatic polyesters. J. Polym. Sci. Pol. Chem. 1999, 37, 2737–2745. [Google Scholar]
- Yao, D.; Li, G.; Kuila, T.; Li, P.; Kim, N.H.; Kim, S.I.; Lee, J.H. Lipase-catalyzed synthesis and characterization of biodegradable polyester containing l-malic acid unit in solvent system. J. Appl. Polym. Sci. 2011, 120, 1114–1120. [Google Scholar] [CrossRef]
- Steunenberg, P.; Uiterweerd, M.; Sijm, M.; Scott, E.L.; Zuilhof, H.; Sanders, J.P.M.; Franssen, M.C.R. Enzyme-catalyzed polymerization of β-alanine esters, a sustainable route towards the formation of poly-β-alanine. Curr. Org. Chem. 2013, 17, 682–690. [Google Scholar]
- Kumar, D.; Bhalla, T.C. Microbial proteases in peptide synthesis: Approaches and applications. Appl. Microbiol. Biotechnol. 2005, 68, 726–736. [Google Scholar] [CrossRef]
- Kuhn, D.; Durrschmidt, P.; Mansfeld, J.; Ulbrich-Hofmann, R. Boilysin and thermolysin in dipeptide synthesis: A comparative study. Biotechnol. Appl. Biochem. 2002, 36, 71–76. [Google Scholar] [CrossRef]
- Bille, V.; Ripak, C.; Assche, I.; Forni, L.; Degelaen, J.; Searso, J. Semi-enzymic synthesis of somatostatin. In Proceedings of 21st European Peptide Symposium, Platja d’Aro, Spain, 2–8 September 1990.
- Gu, Q.M.; Maslanka, W.W.; Cheng, H.N. Enzyme-catalyzed polyamides and their derivatives. In Polymer Biocatalysis and Biomaterials II; ACS Symposium Series 999; Cheng, H.N., Gross, R.A., Eds.; Oxford University Press: Washington, DC, USA, 2008; pp. 309–319. [Google Scholar]
- Poulhès, F.; Mouysset, D.; Gil, G.; Bertrand, M.P.; Gastaldi, S. Speeding-up enzyme-catalyzed synthesis of polyamides using ω-amino-a-alkoxy-acetate as monomer. Polymer 2013, 54, 3467–3471. [Google Scholar] [CrossRef]
- Ikeda, R.; Tanaka, H.; Uyama, H.; Kobayashi, S. Laccase-catalyzed polymerization of acrylamide. Macromol. Rapid Commun. 1998, 19, 423–425. [Google Scholar] [CrossRef]
- Hollmann, F.; Gumulya, Y.; Tölle, C.; Liese, A.; Thum, O. Evaluation of the laccase from Myceliophthora thermophila as industrial biocatalyst for polymerization reactions. Macromolecules 2008, 41, 8520–8524. [Google Scholar] [CrossRef]
- Emery, O.; Lalot, T.; Brigodiot, M.; Maréchal, E. Free-radical polymerization of acrylamide by horseradish peroxidase-mediated initiation. J. Polym. Sci. Pol. Chem. 1997, 35, 3331–3333. [Google Scholar] [CrossRef]
- Mucientes, A.E.; Santiago, F.; Carrero, A.M.; Talavera, B. Superabsorbent hydrogels of poly(sodium acrylate) with crude and exfoliated vermiculites. J. Polym. Eng. 2013, 33, 61–69. [Google Scholar]
- Jones, N.A.; Atkins, E.D.T.; Hill, M.J.; Cooper, S.J.; Franco, L. Chain-folded lamellar crystals of aliphatic polyamides. comparisons between nylons 44, 64, 84, 104, and 124. Macromolecules 1996, 29, 6011–6018. [Google Scholar] [CrossRef]
- Ragupathy, L.; Ziener, U.; Dyllick-Brenzinger, R.; von Vacano, B.; Landfester, K. Enzyme-catalyzed polymerizations at higher temperatures: Synthetic methods to produce polyamides and new poly(amide-co-ester)s. J. Mol. Catal. B Enzym. 2012, 76, 94–105. [Google Scholar] [CrossRef]
- Kalra, B.; Gross, R.A. Horseradish peroxidase mediated free radical polymerization of methyl methacrylate. Biomacromolecules 2000, 1, 501–505. [Google Scholar] [CrossRef]
- Singh, A.; Ma, D.C.; Kaplan, D.L. Enzyme-mediated free radical polymerization of styrene. Biomacromolecules 2000, 1, 592–596. [Google Scholar] [CrossRef]
- Sandoval, G.; Rivera, I.; Barrera-Rivera, K.A.; Martínez-Richa, A. Biopolymer synthesis catalyzed by tailored lipases. Macromol. Symp. 2010, 289, 135–139. [Google Scholar] [CrossRef]
- Varma, I.K.; Albertsson, A.C.; Rajkhowa, R.; Srivastava, R.K. Enzyme catalyzed synthesis of polyesters. Prog. Polym. Sci. 2005, 30, 949–981. [Google Scholar] [CrossRef]
- Kobayashi, S. Enzymatic polymerization: A new method of polymer synthesis. J. Polym. Sci. Pol. Chem. 1999, 37, 3041–3056. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Ohmae, M. Enzymatic polymerization for precision polymer synthesis. Bull. Chem. Soc. Jpn. 2001, 74, 613–635. [Google Scholar] [CrossRef]
- Uyama, H.; Kikuchi, H.; Takeya, K.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization and copolymerization of 15-pentadecanolide. Acta Polym. 1996, 47, 357–360. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, K.; Kobayashi, S. Enzymatic ring-opening polymerization of lactones to polyesters by lipase catalyst: Unusually high reactivity of macrolides. Bull. Chem. Soc. Jpn. 1995, 68, 56–61. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, K.; Hoshi, N.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of 12-dodecanolide. Macromolecules 1995, 28, 7046–7050. [Google Scholar] [CrossRef]
- Bisht, K.S.; Henderson, L.A.; Gross, R.A. Enzyme-catalyzed ring-opening polymerization of ω-pentadecalactone. Macromolecules 1997, 30, 2705–2711. [Google Scholar] [CrossRef]
- Hunsen, M.; Azim, A.; Mang, H.; Wallner, S.R.; Ronkvist, A.; Xie, W.C.; Gross, R.A. A cutinase with polyester synthesis activity. Macromolecules 2007, 40, 148–150. [Google Scholar] [CrossRef]
- Nishida, H.; Yamashita, M.; Nagashima, M.; Endo, T.; Tokiwa, Y. Synthesis of metal-free poly(1,4-dioxan-2-one) by enzyme-catalyzed ring-opening polymerization. J. Polym. Sci. Pol. Chem. 2000, 38, 1560–1567. [Google Scholar] [CrossRef]
- Jiang, Z.; Azim, H. Lipase-catalyzed copolymerization of ω-pentadecalactone with p-dioxanone and characterization of copolymer thermal and crystalline properties. Biomacromolecules 2007, 8, 2262–2269. [Google Scholar] [CrossRef]
- Fickers, P.; Marty, A.; Nicaud, J.M. The lipases from Yarrowia lipolytica: Genetics, production, regulation, biochemical characterization and biotechnological applications. Biotechnol. Adv. 2011, 29, 632–644. [Google Scholar] [CrossRef]
- Uyama, H.; Kikuchi, H.; Takeya, K.; Hoshi, N.; Kobayashi, S. Immobilized lipase showing high catalytic activity toward enzymatic ring-opening polymerization of macrolides. Chem. Lett. 1996, 25, 107–108. [Google Scholar]
- Kobayashi, S.; Uyama, H. Precision enzymatic polymerization to polyesters with lipase catalysts. Macromol. Symp. 1999, 144, 237–246. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Namekawa, S. In vitro biosynthesis of polyesters with isolated enzymes in aqueous systems and organic solvents. Polym. Degrad. Stabil. 1998, 59, 195–201. [Google Scholar] [CrossRef]
- Kumar, A.; Kalra, B.; Dekhterman, A.; Gross, R.A. Efficient ring-opening polymerization and copolymerization of ε-caprolactone and ω-pentadecalactone catalyzed by Candida antartica lipase B. Macromolecules 2000, 33, 6303–6309. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Namekawa, S.; Hayakawa, H. Enzymatic ring-opening polymerization and copolymerization of 8-octanolide by lipase catalyst. Macromolecules 1998, 31, 5655–5659. [Google Scholar]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of 16-hexadecanolide. P. Jpn. Acad. B 1998, 74, 65–68. [Google Scholar] [CrossRef]
- Ebata, H.; Toshima, K.; Matsumura, S. Lipase-catalyzed transformation of poly(ε-caprolactone) into cyclic dicaprolactone. Biomacromolecules 2000, 1, 511–514. [Google Scholar] [CrossRef]
- Sugihara, S.; Toshima, K.; Matsumura, S. New strategy for enzymatic synthesis of high-molecular-weight poly(butylene succinate) via cyclic oligomers. Macromol. Rapid Commun. 2006, 27, 203–207. [Google Scholar] [CrossRef]
- Kikuchi, H.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of substituted lactones. Polym. J. 2002, 34, 835–893. [Google Scholar] [CrossRef]
- Küllmer, K.; Kikuchi, H.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of α-methyl-δ-valerolactone and α-methyl-ε-caprolacton. Macromol. Rapid. Commun. 1998, 19, 127–130. [Google Scholar] [CrossRef]
- Cordova, A.; Iversen, T.; Martinelle, M. Lipase-catalysed formation of macrocycles by ring-opening polymerisation of ϵ-caprolactone. Polymer 1998, 39, 6519–6524. [Google Scholar] [CrossRef]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening and copolymerization of β-propiolactione. Polym. J. 1996, 28, 730–731. [Google Scholar] [CrossRef]
- Suzuki, Y.; Taguchi, S.; Hisano, T.; Toshima, K.; Matsumura, S.; Doi, Y. Correlation between structure of the lactones and substrate specificity in enzyme-catalyzed polymerization for the synthesis of polyesters. Biomacromolecules 2003, 4, 537–543. [Google Scholar] [CrossRef]
- Nagasawa, T.; Shimizu, H.; Yamada, H. The superiority of the third-generation catalyst, Rhodococcus rhodochrous J1 nitrile hydratase, for industrial production of acrylamide. Appl. Microbiol. Biotechnol. 1993, 40, 189–195. [Google Scholar]
- Thomas, S.M.; DiCosimo, R.; Nagarajan, V. Biocatalysis: Applications and potentials for the chemical industry. Trends Biotechnol. 2002, 20, 238–242. [Google Scholar] [CrossRef]
- Chassin, C. A biotechnological process for the production of nicotinamide. Chim. Oggi. 1996, 14, 9–12. [Google Scholar]
- Nagasawa, T.; Matthew, C.D.; Mauger, J.; Yamada, H. Nitrile hydratase-catalyzed production of nicotinamide from 3-cyanopyridine in Rhodococcus rhodochrous J1. Appl. Environ. Microbiol. 1988, 54, 1766–1769. [Google Scholar]
- Robins, K.T.; Nagasawa, T. Process for Preparing Amides. PCT Int. Appl. US 7,666,635 B2, 23 February 2010. [Google Scholar]
- Heveling, J.; Armbruster, E.; Utiger, L.; Rhoner, M.; Dettwiler, H.R.; Chuck, R.J. Process for Preparing Nicotinamide. US Patent 5,719,045, 17 February 1998. [Google Scholar]
- Straathof, A.J.J. Transformation of biomass into commodity chemicals using enzymes or cells. Chem. Rev. 2013. [Google Scholar] [CrossRef]
- Vuyyuru, K.R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: Aerobic oxidation of 5-hydroxymethyl-2-furfural into 2,5-furandicarboxylic acid with gold nanoparticle catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef]
- Koopman, F.; Wierckx, N.; de Winde, J.H.; Ruijssenaars, H.J. Efficient whole-cell biotransformation of 5-(hydroxymethyl)furfural into FDCA, 2,5-furandicarboxylic acid. Bioresour. Technol. 2010, 101, 6291–6296. [Google Scholar] [CrossRef]
- Krystof, M.; Pérez-Sánchez, M.; Domínguez de María, P. Lipase-mediated selective oxidation of furfural and 5-hydroxymethylfurfural. ChemSusChem 2013, 6, 826–830. [Google Scholar] [CrossRef]
- Van Deurzen, M.P.J.; van Rantwijk, F.; Sheldon, R.A. Chloroperoxidase-catalyzed oxidation of 5-hydroxymethylfurfural. J. Carbohydr. Chem. 1997, 16, 299–309. [Google Scholar]
- Panke, S.; Wubbolts, M. Advances in biocatalytic synthesis of pharmaceutical intermediates. Curr. Opin. Chem. Biol. 2005, 9, 188–194. [Google Scholar] [CrossRef]
- Ooshima, H.; Mori, H.; Harano, Y. Synthesis of aspartame precursor by solid thermolysin in organic solvent. Biotechnol. Lett. 1985, 7, 789–792. [Google Scholar] [CrossRef]
- Griengl, H.; Klempier, N.; Pochlauer, P.; Schmidt, M.; Shi, N.Y.; Zabelinskaja-Mackova, A.A. Enzyme catalysed formation of (S)-cyanohydrins derived from aldehydes and ketones in a biphasic solvent system. Tetrahedron 1998, 54, 14477–14486. [Google Scholar]
- Bieber, L.L. Carnitine. Annu. Rev. Biochem. 1988, 57, 261–283. [Google Scholar] [CrossRef]
- Fritz, I.B. Action of carnitine on long chain fatty acid oxidation by liver. Am. J. Physiol. 1959, 197, 297–304. [Google Scholar]
- Meyer, H.P.; Kiener, A.; Imwinkelried, R.; Shaw, N. Biotransformations for fine chemicals production. Chimia 1997, 51, 287–289. [Google Scholar]
- Bommarius, A.S.; Schwarm, M.; Stingl, K.; Kottenhahn, M.; Huthmacher, K.; Drauz, K. Synthesis and use of enantiomerically pure tert-leucine. Tetrahedron Asymmetry 1995, 6, 2851–2888. [Google Scholar]
- Menzel, A.; Werner, H.; Altenbuchner, J.; Gröger, H. From enzymes to “designer bugs” in reductive amination: A new process for the synthesis of l-tert-leucine using a whole cell-catalyst. Eng. Life Sci. 2004, 4, 573–576. [Google Scholar] [CrossRef]
- Krix, G.; Bommarius, A.S.; Drauz, K.; Kottenhahn, M.; Schwarm, M.; Kula, M.R. Enzymatic reduction of α-keto acids leading to l-amino acids, d- or l-hydroxy acids. J. Biotechnol. 1997, 53, 29–39. [Google Scholar] [CrossRef]
- Furuhashi, K. Biological routes to optically active epoxides. In Chirality in Industry; Collins, A.N., Sheldrake, G.N., Crosby, J.C., Eds.; John Wiley & Sons: NJ, USA, 1992; pp. 167–186. [Google Scholar]
- Li, K.; Frost, J. Synthesis of vanillin from glucose. J. Am. Chem. Soc. 1998, 120, 10545–10546. [Google Scholar] [CrossRef]
- Aresta, M.; Quaranta, E.; Liberio, R.; Dileo, C.; Tommasi, I. Enzymatic synthesis of 4-OH-benzoic acid from phenol and CO2: The first example of a biotechnological application of a carboxylase enzyme. Tetrahedron 1998, 54, 8841–8846. [Google Scholar] [CrossRef]
- Baldessari, A. Lipases as catalysts in synthesis of fine chemicals. In Lipases and Phospholipases. Methods in Molecular Biology; Sandoval, G., Ed.; Springer-Humana Press: New York, NY, USA, 2012; Volume 861, pp. 445–448. [Google Scholar]
- Pal, S.; Nair, V. Enzymatic synthesis of thymidine using bacterial whole cells and isolated purine nucleoside phosphorylase. Biocatal. Biotransform. 1997, 15, 147–158. [Google Scholar] [CrossRef]
- Liese, A.; Villela-Filho, M. Production of fine chemicals using biocatalysis. Curr. Opin. Biotechnol. 1999, 10, 595–603. [Google Scholar] [CrossRef]
- Rivera, I.; Mateos, J.C.; Marty, A.; Sandoval, G.; Duquesne, S. Lipase from Carica papaya latex presents high enantioselectivity toward the resolution of prodrug (R,S)-2-bromophenylacetic acid octyl ester. Tetrahedron Lett. 2013, 54, 5523–5526. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
De Regil, R.; Sandoval, G. Biocatalysis for Biobased Chemicals. Biomolecules 2013, 3, 812-847. https://doi.org/10.3390/biom3040812
AMA Style
De Regil R, Sandoval G. Biocatalysis for Biobased Chemicals. Biomolecules. 2013; 3(4):812-847. https://doi.org/10.3390/biom3040812
Chicago/Turabian StyleDe Regil, Rubén, and Georgina Sandoval. 2013. "Biocatalysis for Biobased Chemicals" Biomolecules 3, no. 4: 812-847. https://doi.org/10.3390/biom3040812